

Abstract
The hepatitis C virus NS3 protease is responsible for the processing of the nonstructural region of viral precursor polyprotein in infected hepatic cells. NS3 has been considered a target for drug discovery for a long time. NS3 is a zinc-dependent serine protease. However, the zinc ion is not involved in the catalytic mechanism, because it is bound far away from the active site. Thus, zinc is essential for the structural integrity of the protein and it is considered to have a structural role. The first thermodynamic study on the conformational equilibrium and stability of NS3 and the effect of zinc on such equilibrium is presented here. In agreement with a previous calorimetric study on the binding of zinc to NS3, the global unfolding heat capacity is dominated by the zinc dissociation step, suggesting that the binding of zinc induces a significant structural rearrangement of the protein. In addition, contrary to other homologous zinc-dependent proteases, the zinc-free NS3 protease is not completely unstructured. It is apparent that the conformational landscape of hepatitis C virus NS3 protease is fairly complex due to its intrinsic plasticity, and to the interactions with its different effectors (zinc and the accessory viral protein NS4A) and their modulation of the population of the different conformational states.
Introduction
The hepatitis C virus (HCV) is the major etiologic agent of parenterally transmitted non-A, non-B hepatitis (1). HCV infected people amount to >200,000,000, with 80% of them becoming chronic patients, and many of them progressing to cirrhosis and hepatocellular carcinoma.
The viral RNA is directly translated in infected cells into a precursor polyprotein of ∼3000 residues, which must be processed for successful viral maturation. Host cellular proteases are implicated in the processing of the structural viral proteins, whereas two different proteolytic activities, nonstructural protein 2 (NS2) and nonstructural protein 3 (NS3), encoded in the viral polyprotein, are involved in the processing of the nonstructural region. The NS2-NS3 junction is cleaved intramolecularly by the NS2/3 protease activity. This protein comprises the NS2 C-terminal and the NS3 N-terminal domains and is a zinc-dependent protease (metallo- or cysteine-protease) (2–5). The rest of the nonstructural junctions downstream of NS3 (NS3-NS4A to NS5A-NS5B) are cleaved by the NS3 protease activity, which is a zinc-dependent serine protease comprising the NS3 N-terminal domain (6–9) (Fig. 1 A). The two viral proteolytic activities map to overlapping regions in the viral polyprotein, but they are different in terms of the catalytic residues, biochemical mechanism, and structural and conformational arrangements of the protein domains (10).
Figure 1.
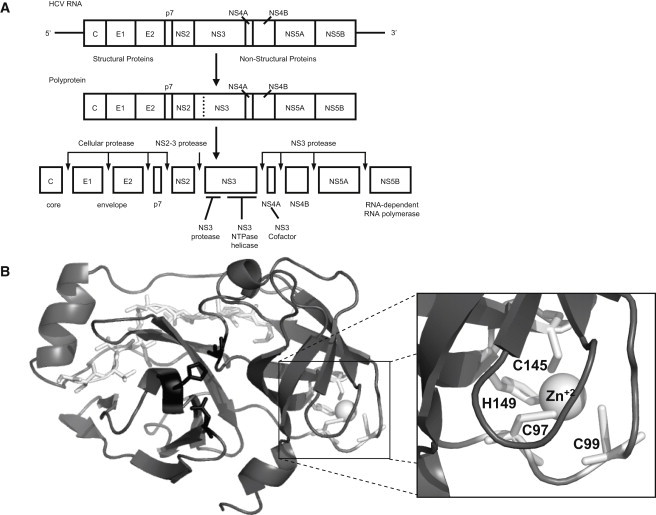
(A) Schematic representation of the processing of the viral polyprotein. (B) Structure of the NS3 protease. The zinc ion is shown in open representation (sphere); the coordinating residues are shown in shading (sticks) surrounding the zinc atom; the catalytic residues are shown in dark shading (sticks) in the groove between the two structural domains; and the accessory NS4A peptide is shown in open representation (sticks) along the N-terminal domain.
NS3 protease contains a zinc ion tetra-coordinated by three cysteine residues and a histidine in its C-terminal domain, though the histidine can be replaced by a water molecule (7) (Fig. 1 B). The zinc ion is essential for the proteolytic activity, because its removal leads to protein inactivation. However, the zinc ion is 20 Å from the catalytic triad. Therefore, the zinc ion is thought to have a structural role, stabilizing the protein conformation (6,11,12). Interestingly, extracellular homologous serine proteases (e.g., chymotrypsin) present several intramolecular disulfide bonds; in particular, one of the disulfide bonds is in a location equivalent to that of the zinc ion in NS3. Thus, the zinc ion is supposed to be involved in the folding mechanism or, alternatively, in the stabilization of the folded conformation of the enzyme, or both.
It has been previously suggested that both protease activities, NS2/3 and NS3, rely on the same zinc ion bound to NS3, but with the zinc ion having a catalytic role in NS2/3 protease and a structural role in NS3 protease (2,9,10,13). This is evidence for the functional and conformational plasticity of NS3 protein.
Since its identification, the NS3 protease active site has been considered a target for drug design. Although very few protease inhibitors have entered clinical trials, a broad variety of competitive inhibitors has been developed. Moreover, the NS4A binding site constitutes another weak point in the NS3 protease, which has not drawn much attention so far. Finally, the zinc-binding site has also been considered a valid target for drug design (11). Previously, the NS3-zinc interaction was studied directly by isothermal titration calorimetry (14). The main conclusion was that zinc removal elicits a conformational change associated with the loss of most NS3 protease structure.
The conformational stability of NS3, the modulation effect induced by zinc, and its structural consequences are investigated in the work presented here. Thermal denaturations followed by differential scanning calorimetry (DSC) and emission fluorescence spectroscopy were performed. To the best of our knowledge, this work is the first calorimetric study on the conformational stability of NS3 protease. Although the NS3 protease is the N-terminal domain of NS3 protein, it has been demonstrated that the isolated NS3 protease domain has a similar behavior (in terms of NS4A activation, proteolytic activity, and inhibition) to that of the NS3 protease domain in the full-length NS3 protein.
Finally, the characterization of the NS3 protease is appealing because:
-
1.
It is a small enzyme with a complex behavior rooted in its plasticity, as well as its interactions with effectors and substrates.
-
2.
It is a pharmacological protein target, and its conformational behavior may give some valuable information on potential weak points for small-molecule ligand design, on the impact of resistance-associated mutations in its conformational stability, or on new methodologies for identifying noncompetitive conformational inhibitors that trap the enzyme in nonactive conformational states.
Materials and Methods
Expression and purification of the NS3 protease
NS3 protease from HCV genotype 1b J-strain is encoded in a pET7 plasmid (a kind gift from Dr. C. Steinkühler, Istituto di Ricerche Molecolare, “P. Angeletti”, Rome, Italy). The 180-residue protease sequence contains a solubilizing C-terminal tail (-SKKKK). Transformed BL21 Star (DE3) One Shot cells (Invitrogen, Carlsbad, CA) were grown in LB medium supplemented with ampicillin at 37°C. Induction of protein expression was initiated when optical density at 600 nm reached 0.6–0.8 by adding 0.4 mM IPTG (isopropyl β-D-1-thiogalactopyranoside) and zinc chloride 0.1 mM, and progressed for 3–5 h at 23°C. Cells were collected and resuspended in sodium phosphate 5 mM, pH 7.5, NaCl 150 mM. Then cells were collected again, and resuspended in Buffer A (Tris HCl 10 mM, dithiothreitol 5 mM, Glycerol 10%, zinc chloride 100 μM) and broken by sonication.
The supernatant was passed through a Q-Sepharose anion exchange column followed by an SP-Sepharose cation exchange column, in an ÄKTA Fast Performance Liquid Chromatography System (GE Healthcare Biosciences, Piscataway, NJ) at a 5 mL/min flow (with the absorbance monitored at 280 nm). Protein bound to the SP-Sepharose resin was eluted using two ionic strength gradients (0–15% and 15–50%) with Buffer A and NaCl 1 M added. For storage, protein was dialyzed in buffer MOPS 10 mM, pH 7, zinc chloride 10 μM. For sample preparation, a rapid buffer exchange process to sodium acetate 100 mM, pH 5, zinc sulfate 50 μM, was made, using Amicon centrifugal devices (Amicon, Houston, TX). Considering the affinity of NS3 for zinc (14), the final concentration of free zinc after several concentration-dilution steps by centrifugation must be close to 50 μM.
The apo-NS3 was obtained as described in Abian et al. (14). Briefly, purified NS3 was concentrated and diluted at least 10 times in a centrifugal device (Amicon) in the presence of 2 mM EDTA (ethylene-diamine tetra-acetic acid) at pH 5 (sodium acetate 100 mM). Because at pH 5 the affinity of EDTA for zinc binding is three orders-of-magnitude larger than that of NS3 for zinc binding at 15–25°C (and presumably to other temperatures, according to the heat capacities and enthalpies determined for zinc binding to EDTA and NS3 protease), all zinc ions were removed from the protein (14). Experimental evidences (NMR, fluorescence, and circular dichroism (CD) spectroscopies, and isothermal titration calorimetry) indicated that zinc-free NS3 protease loses most of its tertiary structure, but also retains most of its secondary one (14).
Differential scanning calorimetry
The heat capacity of NS3 as a function of temperature was measured with a high precision differential scanning VP-DSC microcalorimeter (MicroCal, Northampton, MA). Protein samples and reference solutions were properly degassed and carefully loaded into the cells to avoid bubble formation. Thermal denaturation scans were performed with freshly prepared buffer-exchanged protein solutions. The baseline of the instrument was routinely recorded before the experiments.
Experiments were performed in 100 mM sodium acetate, pH 5, at a scanning rate of 1°C/min. Experiments were carried out with 40 μM of protein and different total concentrations of zinc. Different concentrations of zinc were achieved by adding zinc or EDTA from calibrated reference stock solutions. Thermal denaturations were reversible (as judged by the agreement of the thermogram shapes and sizes between the first and the second scans), concentration independent (with regard to the protein), and scan-rate independent. Pre- and posttransition baselines were considered linear with temperature.
Intrinsic protein stability parameters were obtained by analyzing thermal scans for ligand-free protein considering a two-state unfolding model. A two-state protein in the presence of a ligand has been traditionally analyzed as a two-state protein with altered thermodynamic unfolding parameters (that is, considering just two states: native and unfolded). Thus, the ligand interaction and influence are only implicitly considered, so that the ligand-bound protein behaves as a protein with different unfolding parameters to those of the ligand-free protein, modulated by the ligand concentration. Therefore, the experimental unfolding of a ligand binding protein is always coupled to ligand dissociation, but that does not mean that the data analysis has always accounted for the ligand dissociation in an explicit manner.
In many published works, the analysis of the thermal unfolding of protein-ligand complexes has been performed by applying a two-state analysis without including the explicit dependence on ligand-binding affinity and ligand concentration. However, when a ligand is present, whether at equal or higher concentration than that of the protein, and irrespective of the ligand-binding affinity, the system must be considered as constituted by the protein, with intrinsic stability parameters equal to those of the ligand-free protein, and the ligand, with intrinsic binding parameters, modulating the protein stabilization energy. Thus, any perturbation in the system will involve the explicit coupling between two equilibria: conformational equilibrium and ligand binding equilibrium. In addition, the influence of the ligand on the conformational stability of the protein will be reflected through that coupling.
Considering that the only protein conformational states are the native, the ligand-bound, and the unfolded state, the partition function of the three-state system, Q, is given by (15,16)
| (1) |
where K0 is the intrinsic unfolding equilibrium constant, Ka(T) is the intrinsic association constant, and [L] is the concentration of free ligand. The model takes into account the equilibrium between the native (N), ligand-bound native (NL), and unfolded (U) states: U ⇌ N+L ⇌ NL. The total concentration of native state ([N]+[NL]) has been selected as the reference state for normalizing in the definition of the partition function (15). The average enthalpy of the system, 〈ΔH〉, is obtained through the temperature derivative of the partition function,
| (2) |
where ΔH0 is the intrinsic unfolding enthalpy, ΔHB is the binding enthalpy, FU is the fraction of unfolded protein, FB is the fraction of protein bound to ligand, R is the gas constant, and T is the absolute temperature. Finally, the excess heat capacity, 〈ΔCP〉, is calculated as the temperature derivative of the enthalpy:
| (3) |
If we are interested in the parameters for the unfolded state, we get for the Gibbs energy, the unfolding enthalpy and the unfolding heat capacity,
| (4) |
which are valid in the case of ligand concentration much higher than the protein concentration, where ΔG0 is the intrinsic unfolding Gibbs energy, ΔCP0 is the intrinsic unfolding heat capacity, and ΔCPB is the intrinsic binding heat capacity. It is clear from the previous equations that the apparent unfolding parameters are a function of the intrinsic unfolding parameters and the intrinsic ligand binding parameters. The presence of the ligand modulates the apparent unfolding parameters. All the intrinsic parameters (for conformational and binding equilibria) are temperature-dependent. In order to evaluate numerically the excess heat capacity (Eq. 3) considering the limited total amount of ligand when both protein and ligand concentrations are comparable (15), the fraction of protein in the different conformational states must be calculated. The mass conservation for each species provides a set of equations,
| (5) |
which can be solved numerically or analytically. Here, N is the zinc-free native protein, and NL is the zinc-bound native protein. Once the free concentrations [N] and [L] are known, the concentration of each conformational state can be calculated. Finally, the excess heat capacity can be calculated at each experimental temperature from Eqs. 2 and 3. Data were analyzed using software developed in our laboratory implemented in ORIGIN 7 (Originlab, Northampton, MA).
The unfolding thermodynamic parameters for NS3 are not influenced by the ionization properties of the buffer as long as the pH of the experiment is close to the buffer pKa (4.76 for acetate), and the buffer ionization enthalpy is small (−0.1 kcal/mol for acetate) (17). Therefore, buffer-independent binding parameters are obtained from the calorimetric experiments.
Fluorescence spectroscopy
Fluorescence measurements were performed in a thermostated Chirascan fluorescence spectrophotometer (Applied Photophysics, Leatherhead, Surrey, UK), monitoring the intrinsic tryptophan fluorescence in NS3 protease, using an excitation wavelength of 280 nm, with excitation and emission bandwidths of 5 nm, and recording fluorescence emission spectra between 300 and 400 nm. Excitation at 295 nm yielded the same spectrum. All spectroscopic measurements were made in sodium acetate 100 mM, pH 5. Unfolding was reflected by red-shifted emission spectra and it was quantified by calculating the average emission energy from fluorescence emission spectra (18–21),
| (6) |
where Ii is the emission intensity at a certain wavelength λi. Average energy adds the possibility of focusing on the entire spectrum for quantifying global spectral changes, whereas other unfolding indexes, such as emission intensity at a certain wavelength or wavelength of maximal intensity may or not change significantly upon unfolding. Data were analyzed using software developed in our laboratory implemented in ORIGIN 7 (OriginLab). Briefly, chemical denaturation curves were analyzed using a two-state model for the native/unfolded equilibrium, according to the linear extrapolation model:
where ΔG is the stability Gibbs energy, ΔG0 is the stability Gibbs energy in the absence of chemical denaturant, and [U] is the denaturant concentration. The denaturation data from far-ultraviolet CD and fluorescence were fitted to the two-state equation (22),
| (7) |
where XN and XD are the intrinsic spectrum average emission energy of the folded (N) and unfolded states (U), respectively, for which a linear relationship with the denaturant (i.e., XN = αN + βN[U] and XU = αU + βU[U]) is admitted.
In thermal denaturations (see below), Eq. 7 was used, but ΔG was given by (23)
| (8) |
where ΔS(Tm) (with Tm as melting temperature or unfolding midtransition temperature) and ΔH(Tm) are the entropy and enthalpy of unfolding at the thermal denaturation midpoint, Tm, and ΔCp is the heat capacity of the transition.
Thermal unfolding experiments were performed at a scanning rate of 1°C/min. Blank measurements were carried out with the appropriate buffer. Experiments were carried out with 4 μM of protein and different total concentrations of zinc. Different concentrations of zinc were achieved by adding zinc or EDTA from calibrated reference stock solutions.
Chemical unfolding experiments were performed by adding increasing concentrations of urea. Blank measurements were carried out with the appropriate buffer. Samples were equilibrated for 2 h in the corresponding urea solutions before measurements. Experiments were carried out with 4 μM of protein and different total concentrations of zinc. Different concentrations of zinc were achieved by adding zinc or EDTA from calibrated reference stock solutions.
Results
Conformational stability of zinc-free NS3 protease
Fig. 2 shows the thermal denaturation for zinc-free NS3 protease. The conformational stability is low, with a broad transition and an apparent Tm at ∼40°C. Data analysis considering a two-state model provides an actual Tm of 30°C (Table 1). In addition, the van 't Hoff enthalpy, ΔHVH, is larger than the calorimetric enthalpy, ΔHcal (42 vs. 18 kcal/mol). The disagreement between the two unfolding enthalpies, and the real and the apparent Tm values, occurs in low-stability proteins when the unfolding enthalpy is small (18 kcal/mol) and the unfolding heat capacity is large (1.2 kcal/mol).
Figure 2.
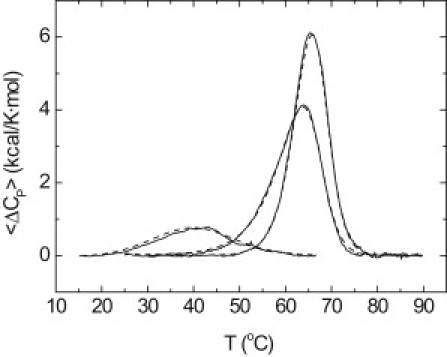
Thermal denaturation scans of NS3 protease followed by DSC in acetate buffer 100 mM, pH 5. Excess molar heat capacity is represented as a function of temperature. Protein concentration was 40 μM, and total zinc concentration was 0, 40, and 90 μM. Experimental traces (continuous line) and theoretical fits (dashed lines) are shown.
Table 1.
Intrinsic thermodynamic parameters for NS3 thermal unfolding and zinc binding at pH 5
| Tm (°C) | ΔH0 (Tm)(kcal/mol) | ΔCP0(kcal/K·mol) | ΔG0 (25°C)(kcal/mol) | |
|---|---|---|---|---|
| Unfolding | 30.1 ± 0.5 | 18 ± 1 | 1.2 ± 0.3 | 0.25 |
| Ka (25°C)(× 106 M−1) | ΔHB (25°C)(kcal/mol) | ΔCPB(kcal/K·mol) | ΔGB (25°C)(kcal/mol) | |
| Zinc binding | 9.8 ± 0.6 | 35 ± 2 | −1.8 ± 0.3 | −9.5 |
In that case, the analysis provides a value for the calorimetric unfolding enthalpy lower than the actual value, because the protein never populates completely the native state at low temperatures and there is always a significant fraction of unfolded protein, leading to a ratio ΔHVH/ΔHcal >1. According to Haynie and Freire (24), it is possible to correct the overestimation in ΔHVH and the underestimation in ΔHcal in a low-stability protein from a combination of the apparent calorimetric and the van 't Hoff enthalpies. This method provides an unfolding enthalpy of 33 kcal/mol. The low conformational stability in the absence of zinc was expected, because a significant destabilization and a loss of tertiary structure were previously observed upon zinc removal (14).
Traditionally, ΔHVH/ΔHcal ratios >1 have been associated with protein unfolding coupled to oligomeric native state dissociation. However, this is not necessarily the case. In proteins with low conformational stability, it is usual to observe a van 't Hoff enthalpy larger than the calorimetric enthalpy. The discrepancy between the two enthalpy values becomes larger for small unfolding enthalpies and large unfolding heat capacities. The reason is that in a low stability protein we do not observe all the expected heat effect (calorimetric enthalpy), because at low temperature there is a significant fraction of protein unfolded. For example, a monomeric two-state protein with the following parameters: Tm = 60°C, ΔH(Tm) = 70 kcal/mol, and ΔCP = 1 kcal/K·mol, will exhibit a calorimetric enthalpy of 69.9 kcal/mol and a van 't Hoff enthalpy of 70.6 kcal/mol, according to calculations; then, the ratio ΔHVH/ΔHcal is close to 1.
However, a monomeric two-state protein with the following parameters: Tm = 35°C, ΔH(Tm) = 30 kcal/mol, and ΔCP = 1 kcal/K·mol, will exhibit a calorimetric enthalpy of 28.3 kcal/mol and a van 't Hoff enthalpy of 36.5 kcal/mol, according to calculations; then, the ratio ΔHVH/ΔHcal is 1.3. The ΔHVH/ΔHcal ratio of 2.3 for NS3 protease seems to be a result of a low-stability highly-unstructured native zinc-free conformation.
Zinc-free NS3 protease does not self-associate, as indicated by several experimental lines of evidence:
-
1.
Thermal denaturation scans followed by DSC, fluorescence, and CD (spanning 1–40 μM concentration range) are not protein-concentration-dependent; and
-
2.
The hydrodynamic radius estimated through the translational diffusion coefficient determined by diffusion-ordered spectroscopy-NMR corresponds to the theoretical hydrodynamic radius of a monomeric NS3 protease (data not shown).
A similar behavior is observed when following the thermal denaturation by fluorescence (Fig. 3). In this case, the apparent Tm coincides with the actual Tm obtained from DSC data. Data analysis employing the two-state model described in Materials and Methods provides a Tm of 31°C and an unfolding enthalpy of 25 kcal/mol, in reasonable agreement with those parameters estimated by DSC.
Figure 3.
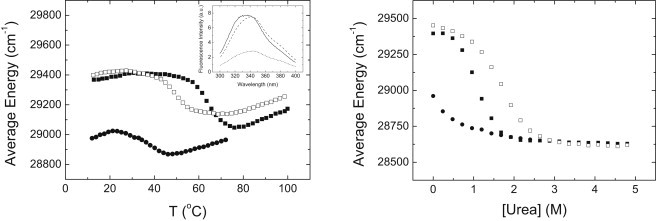
(Left) Thermal denaturation and (right) chemical denaturation of NS3 protease followed by fluorescence in acetate buffer 100 mM, pH 5. Average energy is represented as a function of temperature or urea concentration. Protein concentration was 4 μM, and total zinc concentration was 0 (solid circles), 90 μM (solid squares), and 5 mM (open squares). (Inset) Fluorescence emission spectra of NS3 protease at 25°C and 90 μM total zinc concentration (continuous line), at 25°C and zero total zinc concentration (dashed line), and at 70°C and zero total zinc concentration (dotted line).
Conformational stability of zinc-bound NS3 protease
The influence of zinc binding on the conformational stability of NS3 protease is shown in Fig. 2. The presence of zinc at increasing moderate concentrations enhances the thermal stability in terms of unfolding enthalpy, Tm and cooperativity. The stability increase indicates that zinc does not bind to the unfolded protein or that the binding affinity to the unfolded protein is negligible. It is interesting to point out that the analysis using a simple two-state unfolding model was not satisfactory. Therefore, a protein in the presence of ligand cannot be considered equivalent to a different protein with modified intrinsic unfolding parameters. Data analysis was performed considering a model for coupling between zinc binding and thermal unfolding. From that analysis, intrinsic conformational parameters and intrinsic zinc-binding parameters at 25°C were estimated (Table 1).
These estimated values differ somewhat from those estimated by calorimetric titration experiments, although those parameters obtained by isothermal titration calorimetry,
are apparent binding parameters that must be corrected by the folding of the protein (14), because 40% of NS3 protease is unfolded at 25°C. Considering those corrections, the agreement is closer,
and some discrepancies remain mainly due to different approximations assumed in the model and in the data analysis (e.g., temperature-independent heat capacities and errors when extrapolating along the temperature range).
The intrinsic binding heat capacity is much larger than the intrinsic folding heat capacity (−1.8 kcal/K·mol vs. −1.2 kcal/K·mol), suggesting that the structural ordering induced by zinc binding on the native free protein is considerably larger than that induced by folding on the unfolded protein in the absence of zinc.
The unfolding of the NS3 protease in the presence of zinc shows a ΔHVH/ΔHcal >1, but lower than in the absence of zinc (1.3–1.5, compared to 2.7 in the absence of zinc). Again, this is not an indication of self-associating native state. As it is discussed below, the overall unfolding process is dominated by the ligand interaction, which is characterized by a marked temperature dependency of the binding enthalpy dominating the overall energetics. According to our calculations, this would give rise to a ΔHVH/ΔHcal ratio >1, even for proteins with medium-high conformational stability exhibiting a two-state thermal unfolding.
Similar behavior is observed following the thermal denaturation by fluorescence (Fig. 3) and also when denaturations were followed by CD using 10–20 μM of protein concentration, which has allowed us to explore a wider range of protein concentrations (data not shown). For example, the apparent Tm at 90 μM total concentration of zinc is 65°C, in agreement to that obtained by DSC.
The transition peak observed at equimolar concentration of zinc is somewhat asymmetric. A protein that unfolds in a two-state fashion may show more than one transition peak or an asymmetric transition peak in the presence of ligand (15,25). If the binding affinity is large enough and the ligand concentration is lower than that of the protein, two peaks may appear along the thermal denaturation: the peak with lower Tm and small unfolding enthalpy corresponds to the ligand-free protein (with low stability) and the peak with higher Tm and large unfolding enthalpy corresponds to the ligand-bound protein (with high stability).
If the binding affinity is not large enough and the ligand concentration is lower or similar to that of the protein, partial overlapping of the two transitions will occur, so that a single asymmetric apparent transition peak appears. In this last case, an additional phenomenon occurs: as some protein-ligand complexes become unfolded along the thermal denaturation process, the free concentration of ligand increases; then, the stabilization energy provided by the ligand (which depends on the binding affinity and the free ligand concentration) increases, therefore further stabilizing those remaining folded protein-ligand complexes. This phenomenon contributes to amplify the observed asymmetry.
Effect of high zinc concentration on NS3 stability
Although the presence of zinc at moderate concentrations results in enhanced thermal stability, an unexpected destabilization occurs at high zinc concentrations (>200 μM), as observed by DSC and emission fluorescence (see Figs. 3 and 4). The same behavior is apparent even when unfolding is followed by CD (data not shown). Substitution of zinc sulfate by zinc chloride equally destabilized the protein, but substitution by sodium chloride did not destabilize the protein, pointing to the divalent cation as the one responsible for such destabilization. This unfolding behavior might be due to additional low affinity zinc-binding sites in the unfolding state, which become populated at higher zinc concentrations. Those additional zinc-binding sites in the unfolded state, although they show lower affinity than that of the zinc-binding site in the native protein (otherwise, there would not be protein stabilization at low zinc concentration), may have a larger influence at high zinc concentration due to their higher stoichiometry.
Figure 4.
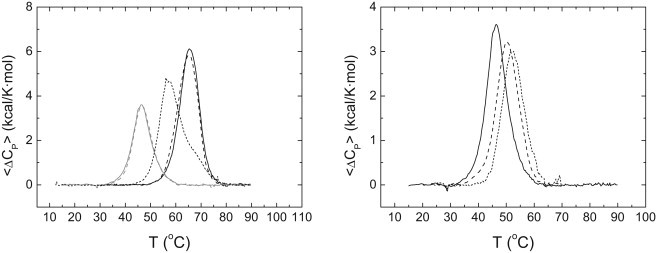
(Left) Thermal denaturation scans of NS3 protease followed by DSC in acetate buffer 100 mM, pH 5. Excess molar heat capacity is represented as a function of temperature. Protein concentration was 40 μM, and zinc concentration was: 90 μM total zinc concentration (black continuous line); 90 μM total zinc concentration and 5 mM total sodium chloride (black dashed line); 1 mM total zinc concentration (black dotted line); 5 mM total zinc concentration (gray continuous line); and 5 mM total zinc (chloride) concentration (gray dashed line). (Right) Thermal denaturation scans of NS3 protease followed by DSC in acetate buffer 100 mM, pH 5. Excess molar heat capacity is represented as a function of temperature. Total zinc concentration was 5 mM, and protein concentration: 40 μM (continuous line), 30 μM (dashed line), and 10 μM (dotted line).
Chemical denaturation experiments either followed by fluorescence (Fig. 2) and by CD (data not shown) do not exhibit such phenomenon, and they show the expected trend: higher zinc concentrations induce larger stabilization (Fig. 3). It might be possible that the competition between denaturant and unfolded protein for zinc, or between denaturant and zinc for the unfolded protein, may hinder zinc binding to the unfolded protein and, therefore, preclude protein destabilization.
Thermal denaturation experiments done at different protein concentrations at 5 mM zinc concentration (Fig. 4) show that the apparent unfolding Tm decreases when increasing the protein concentration, suggesting that protein oligomerization occurs upon unfolding, with zinc controlling such process. In addition, thermal denaturations at high zinc concentration proved to be fully reversible.
Although at high zinc concentration thermal and chemical denaturations are not equivalent, because zinc influence is markedly different, below a certain zinc concentration threshold the two types of denaturations are equivalent. If an apparent two-state unfolding analysis is performed, considering the linear extrapolation model for the urea effect on conformational equilibrium, a stabilization Gibbs energy of 2.7 kcal/mol is obtained for NS3 protease in the presence of 90 μM total zinc concentration.
We recognize that this analysis is not rigorous because it does not explicitly consider the coupling between ligand dissociation and unfolding. However, because we do not know the dependence of the zinc affinity on denaturant concentration, we presently have no other choice. If a two-state unfolding analysis is performed for the DSC experiment (again, such calculation is not adequate, because we are not considering explicitly the ligand dissociation coupled to unfolding, but it is carried out for comparison purposes), a stabilization Gibbs energy of 2.5 kcal/mol is obtained under the same conditions, in agreement with the value obtained from the chemical denaturation.
Discussion
Specific metal ion binding to a protein is often accompanied by a considerable structural reorganization of the protein. The associated conformational change is even larger than those elicited by the binding of bulkier ligands. The amount of protein surface area buried from the solvent upon binding is large compared to that corresponding to the binding of small organic ligands with bulky molecular volumes. Then, the binding heat capacity (per ligand volume or surface area) is usually very large, compared to that of the binding of small organic ligands. As a consequence, specific metal ion binding to proteins is one of the key mechanisms for controlling protein conformation and function.
Zinc is one of the most commonly bound transition metals in proteins (26). Zinc-binding sites in proteins can be classified as:
-
1.
Sites with a catalytic role.
-
2.
Sites with a structural role.
In this latter case, the effect in proteins can vary from the assisting in the folding of a completely natively-unfolded protein (27), to enhancing protein stability (28). Furthermore, zinc has been shown to shift the equilibrium between two different well-folded protein conformations (29).
It has been reported that the NS3 zinc ion is required for both NS2/3 and NS3 proteolytic activities (with catalytic and structural function, respectively), made possible by different protein conformations and zinc coordinating cages. Then, such plasticity is expected to be reflected in the energetics of the NS3-zinc interaction and the conformational equilibrium of NS3 protease. Other homologous chymotrypsin-like cysteine proteases from picornaviruses require a zinc ion bound, with coordinating residues in topological positions similar to those of NS3 (30,31). Similar results for the structural role of zinc were reported previously for the rubella virus nonstructural protein (32) and the human rhinoviral protease 2A (30), where zinc is required for the structural integrity of these enzymes, but it is not directly involved in their catalytic mechanism. In these proteins, conformational changes involving loss of structure are elicited by zinc removal. In particular, human rhinoviral protease 2A does not show any thermal unfolding transition in the absence of zinc, which would indicate that the zinc-free protein adopts either a completely unstructured or a molten globule conformation.
The intrinsic unfolding heat capacity can be related to what it has been previously called “weak coupling” (14,33), that is, conformational equilibrium coupled to ligand binding with conformational state populations changing significantly along the experimental temperature range in the absence of ligand, so that both ligand binding and temperature may shift the conformational equilibrium (and this results in a nonlinear temperature-dependence of the apparent binding enthalpy).
On the other hand, the intrinsic binding heat capacity can be related to what has been previously called “strong coupling” (14,34), which is associated with intrinsically (partially) unstructured proteins. This means that the conformational equilibrium is coupled to ligand binding with conformational state populations remaining practically constant along the experimental temperature range in the absence of ligand, so that only ligand binding may shift the conformational equilibrium (and this results in a linear temperature-dependence of the apparent binding enthalpy). This last situation is usually termed “ligand-induced” fit (33).
Because the heat capacity is the thermodynamic parameter exhibiting the best correlation with changes in solvent-exposed surface area in proteins (35), the relative contribution of each intrinsic heat capacity to the global unfolding heat capacity (ΔCP = ΔCP0 − ΔCPB, when FB = 1) gives a measure of the structural rearrangements associated with each equilibrium regarding the complete overall folding process. Therefore, when the NS3 protease undergoes structural ordering from unfolded to zinc-bound native protein, most of the structural rearrangements (60%, because −ΔCPB/ΔCP = 0.6) are due to zinc binding, as observed in a previous work (14).
In fact, zinc dissociation causes a red-shift in the fluorescence emission spectrum, as was also observed previously (14), whereas thermal denaturation does not contribute significantly to such red shift (Fig. 4). The only two tryptophans in NS3 protease (W53 and W85) are located at the N-terminal domain, >15 Å away from the zinc-binding site, in the C-terminal domain. Therefore, zinc binding has a global impact on the NS3 protease conformation, with an effect on tryptophan solvent-exposure larger than the thermal unfolding of the zinc-free native protein. The loss of structure induced by zinc removal in NS3 protease can be considered at two levels:
-
1.
Loss of structure due to zinc dissociation (NL-to-N conformational change), which would reflect the strong coupling or the ligand-induced fit.
-
2.
The loss of structure due to protein unfolding (N-to-U conformational change), which would reflect the weak coupling, because of the low stability of the native zinc-free state.
It is interesting to explore further the effect of ligand binding in a strong coupling-biased scenario. The populations of the different conformational states may be calculated by using the estimated intrinsic parameters for folding and binding (Fig. 5). It is evident that, in the absence of zinc, the protein never populates the native state in a fraction higher than 75%. An equimolar concentration of zinc is enough to raise the native state fraction up to 100%. Increasing the total concentration of zinc (from 40 to 90 μM) hardly has an additional effect, even though there is a considerable increase in free zinc concentration. The temperature dependency of the apparent Gibbs energy associated with the unfolded state, calculated from the intrinsic thermodynamic parameters or from the population of each conformational state,
is shown in Fig. 5.
Figure 5.
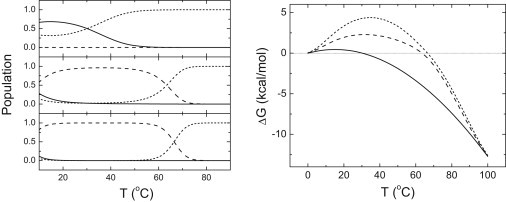
(Left) Population of the different conformational states as a function of temperature at pH 5 for 40 μM NS3 protease in the presence of 0 μM (top), 40 μM (middle), and 90 μM (bottom) total zinc concentration is shown. Three different conformational states are considered: zinc-free native (N, continuous line), zinc-bound native (NL, dashed line), and unfolded (U, dotted line). (Right) Apparent Gibbs energy of unfolding (or stabilization) as a function of temperature at pH 5 for 40 μM NS3 protease in the presence of 0 μM (continuous line), 40 μM (dashed line), and 90 μM (dotted line) total zinc.
The intersection with the x axis corresponds to the unfolding Tm at each zinc concentration. The stability at high temperature is very similar, where the total concentration of zinc is 40–90 μM. However, there is a considerable difference in the stability at moderate temperature. In addition, at low temperature there is no additional stability induced by zinc binding.
The main cause for these observations is the strong temperature dependency of the intrinsic NS3-zinc interaction. Because the intrinsic binding heat capacity is very large, the intrinsic binding affinity reaches a maximal value at a temperature at ∼35°C (where the intrinsic binding enthalpy is zero) and goes to zero very quickly as soon as the temperature goes below or above 35°C. Therefore, the effect of ligand binding on the stability is restricted to a limited temperature range. An inflection point is observed at low temperature (at ∼10°C), which would not appear if the intrinsic binding heat capacity were smaller.
The average energy has been used as the observable for following spectroscopically the unfolding of NS3 protease. This quantity has several advantages; in particular, contrary to the fluorescence intensity at a certain wavelength, it is concentration-independent, which is important when several protein samples must be compared. If the wavelength for maximal fluorescence intensity is used as the observable for following unfolding (which is another concentration-independent quantity), similar results are obtained (data not shown): a transition between 334 nm and 342 nm at 65°C, and 46°C for 90 μM and 5 mM total zinc concentration, respectively. However, no transition and a roughly constant wavelength of 342 nm for maximal fluorescence intensity are observed in the absence of zinc. Then, the unfolding of the zinc-free NS3 protease would be overlooked.
All these results may be summarized as follows:
-
1.
Zinc-free native NS3 protease maintains some level of (secondary) structure, and exhibits low stability.
-
2.
Zinc-free native NS3 protease (N) is considerably unstructured compared to zinc-bound protease (NL) and, in addition, it never populates completely the partially folded zinc-free native state.
-
3.
Zinc binding elicits a significant structural stabilization.
-
4.
Zinc binding is responsible for most (60%) of the structural ordering in the overall transition from unfolded to ligand-bound protein.
-
5.
The coupling between the conformational and binding equilibria is a combination of weak and strong coupling, but with a higher contribution from strong coupling (ligand-induced fit).
-
6.
The ligand stabilization effect appears mainly at moderate temperatures, due to the strong dependence of the intrinsic binding parameters with temperature.
-
7.
Chemical and thermal denaturation are not equivalent at high zinc concentration—the latter possibly affected by protein oligomerization upon unfolding.
The oligomerization mechanism upon thermal unfolding at high zinc concentration requires further insight. The three zinc-coordinating cysteines, as well as another two cysteines very close in the three-dimensional structure (C47 and C52), might be involved in thermal denaturation in the presence of high zinc concentration.
Even though NS3 protease is a small monomeric protein, it exhibits a complex behavior. NS3 protease acts as an allosteric protein with two effectors, NS4A and zinc, enhancing the catalytic efficiency on the substrate. Although NS3 protease presents some basal level of proteolytic activity in the absence of NS4A, it has no activity in the absence of zinc. Binding of NS4A induces structural rearrangements on the N-terminal domain in protease NS3, having an effect on the configuration of the catalytic triad. In addition, the binding of NS4A affects the zinc-binding site, and, therefore, a cooperative interaction exists in the binding of both NS4A and zinc ion (11).
Now it is clear that, if zinc is not bound, most of the protein structure protease is lost and whatever remains shows low thermodynamic stability, and, therefore, binding of NS4A would have a considerable energetic penalty. The activation effects of both NS4A and zinc are associated to concomitant conformational changes elicited by the binding of both effectors. Thus, the conformational landscape of NS3 is fairly intricate, involving at least six different conformational states:
-
1.
Unfolded protein,
-
2.
Zinc-free native protein,
-
3.
Zinc-bound protein,
-
4.
NS4A-zinc-bound protein,
-
5.
Zinc-substrate-bound protein.
-
6.
NS4A-zinc-substrate-bound.
Other conformational states, such as NS4A-bound zinc-free protease, are less important because of their low population due to energetic penalties.
Acknowledgments
Financial support was received from grant No. CP07/00289 (to O.A.) from the Instituto de Salud Carlos III (Spanish Ministry of Science and Innovation); grants No. SAF2004-07722 (to A.V.-C.), and No. SAF2008-05742-C02-01 and No. CSD2008-00005 (to J.L.N.) from the Spanish Ministry of Science and Innovation; grants No. PI044/09 and PI078/08 from Diputación General de Aragón, Spain (to A.V.-C.); grant No. ACOMP2010-114 from Generalitat Valenciana (to J.L.N.); grant No. 36557/06 from FIPSE Foundation (to J.L.N.); and grant No. UZ2009-BIO-05 (A.V.-C.) from Universidad de Zaragoza. O.A. was supported by a Miguel Servet Research Contract from the Instituto Carlos III. A.V.-C. was supported by a research contract from Fundación ARAID (Diputación General de Aragón, Spain).
Contributor Information
Olga Abian, Email: omabian.iacs@aragon.es.
Adrian Velazquez-Campoy, Email: adrianvc@unizar.es.
References
- 1.Choo Q.L., Kuo G., Houghton M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244:359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 2.Hijikata M., Mizushima H., Shimotohno K. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol. 1993;67:4665–4675. doi: 10.1128/jvi.67.8.4665-4675.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grakoui A., McCourt D.W., Rice C.M. A second hepatitis C virus-encoded proteinase. Proc. Natl. Acad. Sci. USA. 1993;90:10583–10587. doi: 10.1073/pnas.90.22.10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pieroni L., Santolini E., La Monica N. In vitro study of the NS2-3 protease of hepatitis C virus. J. Virol. 1997;71:6373–6380. doi: 10.1128/jvi.71.9.6373-6380.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welbourn S., Green R., Pause A. Hepatitis C virus NS2/3 processing is required for NS3 stability and viral RNA replication. J. Biol. Chem. 2005;280:29604–29611. doi: 10.1074/jbc.M505019200. [DOI] [PubMed] [Google Scholar]
- 6.De Francesco R., Urbani A., Tramontano A. A zinc binding site in viral serine proteinases. Biochemistry. 1996;35:13282–13287. doi: 10.1021/bi9616458. [DOI] [PubMed] [Google Scholar]
- 7.Love R.A., Parge H.E., Hostomska Z. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell. 1996;87:331–342. doi: 10.1016/s0092-8674(00)81350-1. [DOI] [PubMed] [Google Scholar]
- 8.Kim J.L., Morgenstern K.A., Thomson J.A. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell. 1996;87:343–355. doi: 10.1016/s0092-8674(00)81351-3. [DOI] [PubMed] [Google Scholar]
- 9.Stempniak M., Hostomska Z., Hostomsky Z. The NS3 proteinase domain of hepatitis C virus is a zinc-containing enzyme. J. Virol. 1997;71:2881–2886. doi: 10.1128/jvi.71.4.2881-2886.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tedbury P.R., Harris M. Characterization of the role of zinc in the hepatitis C virus NS2/3 auto-cleavage and NS3 protease activities. J. Mol. Biol. 2007;366:1652–1660. doi: 10.1016/j.jmb.2006.12.062. [DOI] [PubMed] [Google Scholar]
- 11.Urbani A., Bazzo R., Barbato G. The metal binding site of the hepatitis C virus NS3 protease. A spectroscopic investigation. J. Biol. Chem. 1998;273:18760–18769. doi: 10.1074/jbc.273.30.18760. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y.M., Lim C. Physical basis of structural and catalytic Zn-binding sites in proteins. J. Mol. Biol. 2008;379:545–553. doi: 10.1016/j.jmb.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 13.Wu Z., Yao N., Weber P.C. Mechanism of autoproteolysis at the NS2-NS3 junction of the hepatitis C virus polyprotein. Trends Biochem. Sci. 1998;23:92–94. doi: 10.1016/s0968-0004(98)01180-3. [DOI] [PubMed] [Google Scholar]
- 14.Abian O., Neira J.L., Velazquez-Campoy A. Thermodynamics of zinc binding to hepatitis C virus NS3 protease: a folding by binding event. Proteins. 2009;77:624–636. doi: 10.1002/prot.22475. [DOI] [PubMed] [Google Scholar]
- 15.Robert C.H., Gill S.J., Wyman J. Quantitative analysis of linkage in macromolecules when one ligand is present in limited total quantity. Biochemistry. 1988;27:6829–6835. doi: 10.1021/bi00418a027. [DOI] [PubMed] [Google Scholar]
- 16.Straume M., Freire E. Two-dimensional differential scanning calorimetry: simultaneous resolution of intrinsic protein structural energetics and ligand binding interactions by global linkage analysis. Anal. Biochem. 1992;203:259–268. doi: 10.1016/0003-2697(92)90311-t. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg R.N., Kishore N., Lennen R.M. Thermodynamic quantities for the ionization reactions of buffers. J. Phys. Chem. Ref. Data. 2002;31:231–370. [Google Scholar]
- 18.Fernando T., Royer C.A. Unfolding of Trp repressor studied using fluorescence spectroscopic techniques. Biochemistry. 1992;31:6683–6691. doi: 10.1021/bi00144a007. [DOI] [PubMed] [Google Scholar]
- 19.Royer C.A., Mann C.J., Matthews C.R. Resolution of the fluorescence equilibrium unfolding profile of Trp aporepressor using single tryptophan mutants. Protein Sci. 1993;2:1844–1852. doi: 10.1002/pro.5560021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Royer C.A. Fluorescence spectroscopy. Methods Mol. Biol. 1995;40:65–89. doi: 10.1385/0-89603-301-5:65. [DOI] [PubMed] [Google Scholar]
- 21.Cremades N., Velazquez-Campoy A., Sancho J. The flavodoxin from Helicobacter pylori: structural determinants of thermostability and FMN cofactor binding. Biochemistry. 2008;47:627–639. doi: 10.1021/bi701365e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Clarke J., Fersht A.R. Engineered disulfide bonds as probes of the folding pathway of barnase: increasing the stability of proteins against the rate of denaturation. Biochemistry. 1993;32:4322–4329. doi: 10.1021/bi00067a022. [DOI] [PubMed] [Google Scholar]
- 23.Privalov P.L. In: Protein Folding. Creighton T.E., editor. Freeman; New York: 1992. pp. 83–125. [Google Scholar]
- 24.Haynie D.T., Freire E. Estimation of the folding/unfolding energetics of marginally stable proteins using differential scanning calorimetry. Anal. Biochem. 1994;216:33–41. doi: 10.1006/abio.1994.1004. [DOI] [PubMed] [Google Scholar]
- 25.Shrake A., Ross P.D. Biphasic denaturation of human albumin due to ligand redistribution during unfolding. J. Biol. Chem. 1988;263:15392–15399. [PubMed] [Google Scholar]
- 26.Holm R.H., Kennepohl P., Solomon E.I. Structural and functional aspects of metal sites in biology. Chem. Rev. 1996;96:2239–2314. doi: 10.1021/cr9500390. [DOI] [PubMed] [Google Scholar]
- 27.Yi S., Boys B.L., Choy W.Y. Effects of zinc binding on the structure and dynamics of the intrinsically disordered protein prothymosin α: evidence for metalation as an entropic switch. Biochemistry. 2007;46:13120–13130. doi: 10.1021/bi7014822. [DOI] [PubMed] [Google Scholar]
- 28.Park P.S., Sapra K.T., Muller D.J. Stabilizing effect of Zn2+ in native bovine rhodopsin. J. Biol. Chem. 2007;282:11377–11385. doi: 10.1074/jbc.M610341200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerasoli E., Sharpe B.K., Woolfson D.N. ZiCo: a peptide designed to switch folded state upon binding zinc. J. Am. Chem. Soc. 2005;127:15008–15009. doi: 10.1021/ja0543604. [DOI] [PubMed] [Google Scholar]
- 30.Voss T., Meyer R., Sommergruber W. Spectroscopic characterization of rhinoviral protease 2A: Zn is essential for the structural integrity. Protein Sci. 1995;4:2526–2531. doi: 10.1002/pro.5560041209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sommergruber W., Casari G., Skern T. The 2A proteinase of human rhinovirus is a zinc containing enzyme. Virology. 1994;204:815–818. doi: 10.1006/viro.1994.1599. [DOI] [PubMed] [Google Scholar]
- 32.Zhou Y., Tzeng W.P., Yang J.J. A cysteine-rich metal-binding domain from rubella virus non-structural protein is essential for viral protease activity and virus replication. Biochem. J. 2009;417:477–483. doi: 10.1042/BJ20081468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bruzzese F.J., Connelly P.R. Allosteric properties of inosine monophosphate dehydrogenase revealed through the thermodynamics of binding of inosine 5′-monophosphate and mycophenolic acid. Temperature dependent heat capacity of binding as a signature of ligand-coupled conformational equilibria. Biochemistry. 1997;36:10428–10438. doi: 10.1021/bi9708040. [DOI] [PubMed] [Google Scholar]
- 34.Kouvatsos N., Meldrum J.K., Thomas N.R. Coupling ligand recognition to protein folding in an engineered variant of rabbit ileal lipid binding protein. Chem. Commun. (Camb.) 2006;44:4623–4625. doi: 10.1039/b610130e. [DOI] [PubMed] [Google Scholar]
- 35.Robertson A.D., Murphy K.P. Protein structure and the energetics of protein stability. Chem. Rev. 1997;97:1251–1268. doi: 10.1021/cr960383c. [DOI] [PubMed] [Google Scholar]