

Abstract
The KCNE1 auxiliary subunit coassembles with the Kv7.1 channel and modulates its properties to generate the cardiac IKs current. Recent biophysical evidence suggests that KCNE1 interacts with the voltage-sensing domain (VSD) of Kv7.1. To investigate the mechanism of how KCNE1 affects the VSD to alter the voltage dependence of channel activation, we perturbed the VSD of Kv7.1 by mutagenesis and chemical modification in the absence and presence of KCNE1. Mutagenesis of S4 in Kv7.1 indicates that basic residues in the N-terminal half (S4-N) and C-terminal half (S4-C) of S4 are important for stabilizing the resting and activated states of the channel, respectively. KCNE1 disrupts electrostatic interactions involving S4-C, specifically with the lower conserved glutamate in S2 (Glu170 or E2). Likewise, Trp scanning of S4 shows that mutations to a cluster of residues in S4-C eliminate current in the presence of KCNE1. In addition, KCNE1 affects S4-N by enhancing MTS accessibility to the top of the VSD. Consistent with the structure of Kv channels and previous studies on the KCNE1-Kv7.1 interaction, these results suggest that KCNE1 alters the interactions of S4 residues with the surrounding protein environment, possibly by changing the protein packing around S4, thereby affecting the voltage dependence of Kv7.1.
Introduction
The voltage-sensing domain (VSD) is a protein subdomain that consists of four transmembrane helices (S1–S4) and imparts voltage sensitivity to voltage-gated potassium (Kv) channels (1). The potential across the membrane acts on highly conserved basic residues in the S4 segment (Fig. 1 A) to initiate a series of conformational changes that open the channel (2–4). Kv channels also contain conserved acidic residues in other VSD transmembrane segments (Fig. 1 A) that form electrostatic interactions with conserved basic residues in S4. These electrostatic interactions stabilize S4 during protein folding and membrane insertion (5,6) as well as during the gating process (7–10).
Figure 1.
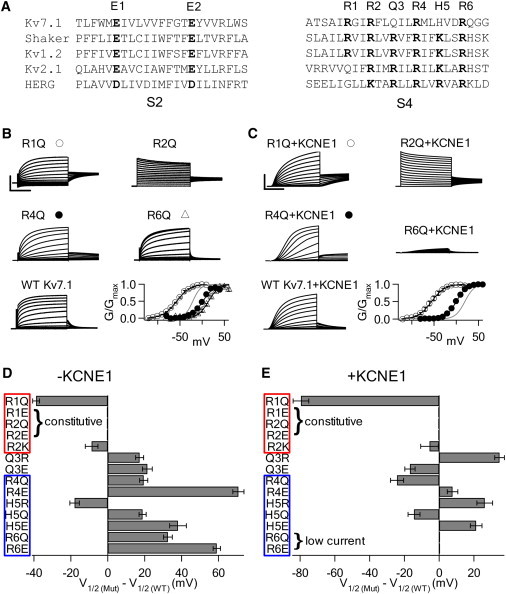
Mutations altering charge in S4 of Kv7.1 in the absence and presence of KCNE1 reveal the importance of charge in KCNE1 modulation. (A) Sequence alignment of S2 and S4 from various voltage dependent channels. Conserved acidic and basic residues in bold. (B) Representative currents from neutralization mutations of Arg in S4 of Kv7.1. Voltages were from −80 to 60 mV for activating R2Q, R4Q, R6Q, and WT Kv7.1 currents, and from −120 to 20 mV for R1Q. All were held at −80 mV and repolarized at −40 mV. Scale: R1Q 3 μA, R2Q 2 μA, R4Q 8 μA, R6Q 2 μA, and WT 4 μA; 2 s for all. In the G-V plots, black curves are fittings of the Boltzmann equation to the mutants and the gray curve is the G-V relation of WT Kv7.1. R2Q is voltage independent. (C) Representative currents from neutralization mutations of Arg in S4 of Kv7.1 coexpressed with KCNE1. Voltages were from −80 to 60 mV for activating R2Q+KCNE1, R4Q+KCNE1, R6Q+KCNE1, and WT Kv7.1+KCNE1 currents and from −120 to 20 mV for R1Q+KCNE1 currents. R1Q+KCNE1 was held at −120 mV for 32 s. R4Q+KCNE1 was held at −100 mV for 120 s between pulses. The rest were held at −80 mV for 32 s. All were repolarized at −40 mV. Scale: R1Q 15 μA, R2Q 40 μA, R4Q 40 μA, R6Q 4 μA, and WT 20 μA; 2 s for all. The symbols in G-V plots are similar to those in B. (D) Shift in the voltage of half-maximal activation for S4 mutations in Kv7.1 relative to WT Kv7.1. Red (top) and blue (bottom) boxes indicate charged residues that stabilize the resting state and activated state in WT Kv7.1, respectively. Error bars indicate standard error of the mean for this and all subsequent figures. (E) Shift in the voltage of half-maximal activation for S4 mutations in Kv7.1+KCNE1 relative to WT Kv7.1+KCNE1. Red (top) and blue (bottom) boxes indicate charged residues that are not and are significantly altered by KCNE1 coexpression, respectively.
Because of the importance of the VSD in channel function, auxiliary subunits can modulate the VSD to regulate channel behavior (11). The β-subunits of BK-type voltage- and Ca2+-activated K+ channels contain two transmembrane segments that are located adjacent to and likely associated with the VSD (12–14), and their effects on the VSD have been shown by various measurements (15–18). The auxiliary protein DPP6, with a single membrane-spanning segment, has been shown to associate with the VSD of Kv4 channels to alter voltage-dependent activation of these channels (19,20). Moreover, the single transmembrane KCNE3 (MiRP2) peptide may also interact with S4 of Kv7.1 to stabilize it in an activated conformation, resulting in constitutively activated channels (21–23).
The KCNE1 (MinK) auxiliary subunit, a protein related to KCNE3, is a single transmembrane peptide that coassembles with the voltage-gated potassium channel Kv7.1 (KCNQ1/KvLQT1) to form the IKs channel complex (24,25). The IKs current is one of the major outward currents that contribute to the termination of the cardiac action potential (26). Congenital mutations in either Kv7.1 or KCNE1 that diminish or enhance IKs current cause long-QT (LQT) or short-QT (SQT) syndrome, respectively (27,28). KCNE1 modulates a number of Kv7.1 properties to give the IKs channel its unique characteristics, such as by slowing the activation kinetics, enhancing the current amplitude, suppressing inactivation, and shifting the voltage dependence of channel activation to more positive potentials (29). Previous studies have highlighted possible interactions between KCNE1 with the pore (30–34) and C-terminus (35–38). Because KCNE1 alters a diverse range of properties, there may be multiple interactions between the two partners. Since KCNE1 changes the voltage-dependent activation of Kv7.1, KCNE1 may also affect the VSD of Kv7.1. A number of SQT mutations located in S1 of the VSD exhibit abnormal phenotypes only in the presence of KCNE1 (39,40), providing a hint that KCNE1 modulates the VSD. Recently, several studies have positioned the extracellular end of the KCNE1 transmembrane segment proximal to S1 and S4 of the Kv7.1 VSD (22,41,42). Furthermore, computational studies that docked the NMR structure of KCNE1 onto closed- and open-state homology models of Kv7.1 placed KCNE1 in a cleft between two adjacent Kv7.1 subunits, which would allow it to contact both the VSD and the pore (43).
To understand how KCNE1 changes the VSD of Kv7.1, we systematically probed the VSD of Kv7.1 channels via site-directed mutagenesis, and then sought to determine how KCNE1 association affects the behavior of these mutants. In Kv7.1, basic residues in the N-terminal half of S4 (S4-N) stabilize the resting state of the channel, whereas basic residues in the C-terminal half of S4 (S4-C) stabilize the activated state of the channel. KCNE1 disrupts interactions in S4-C, which destabilizes the activated state. On the other hand, KCNE1 increases MTS accessibility to S4-N. The pattern that emerges from these results and a Trp scan of S4 suggests that the association of KCNE1 causes a repacking of the channel protein around S4, which disrupts S4 interactions (both steric and electrostatic) with other parts of the channel protein, thereby profoundly altering the function of the S4 segment.
Materials and Methods
Mutagenesis and oocyte preparation
Mutations were made via polymerase chain reaction as previously described (8). mRNA was made with the use of an mMessage mMachine T7 polymerase kit (Applied Biosystems, Carlsbad, CA). Defolliculated Xenopus laevis oocytes were injected with 46 ng of mRNA and incubated at 18°C in ND96 solution (in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES, pH 7.60) for 4–6 days before recordings were made.
Electrophysiology
Current recordings were obtained with a two-microelectrode voltage clamp (8). Oocytes were impaled with 3 M KCl-filled glass microelectrodes with resistances of 0.5–1 MΩ and clamped with a voltage clamp amplifier (CA-1B; Dagan, Minneapolis, MN). Oocytes were constantly bathed in ND96 at room temperature. Data acquisition was controlled by PULSE/PULSEFIT software (HEKA, Bellmore, NY). Data were analyzed using IGOR Pro 6 (WaveMetrics, Portland, OR). The Boltzmann function was used to fit current-voltage relationships: normalized Itail = 1/[1 + exp ((V1/2 − Vt)/k)].
[2-(Trimethylammonium)ethyl] MTS (MTSET+) and (2-sulfonatoethyl) MTS (MTSES−) (Toronto Research Chemicals) were made into aliquots of 100 mM stock solution in water. The aliquots were thawed and diluted with ND96 to a concentration of 400 μM immediately before use.
Biotinylation and Western blotting
Intact oocytes expressing Kv7.1 were incubated in ND96 containing 1 mg/mL of Sulfo-NHS-SS-Biotin (Thermo Fisher Scientific, Waltham, MA) to label membrane proteins. The oocytes were washed to remove unbound biotin and then homogenized. Lysates were incubated with NeutrAvidin beads (Thermo Fisher Scientific) to pull out biotin-bound proteins. The NeutrAvidin beads were collected via centrifugation and washed thoroughly. Lysates and beads were heated to 60°C in SDS-PAGE loading buffer. Samples were run on 4–20% gradient polyacrylamide gels (Bio-Rad, Hercules, CA). After electrophoresis, the samples were Western blotted with a 1:500 dilution of Kv7.1 primary (Santa Cruz Biotechnology, Santa Cruz, CA) and a 1:5000 dilution of goat anti-rabbit secondary antibody (Santa Cruz Biotechnology). Anti-Gβ antibody was used to probe for intracellular proteins (Santa Cruz Biotechnology).
Results
Basic residues in S4 of Kv7.1 stabilize different states
In a previous study, we showed that conserved basic residues in S4 of Kv7.1 interact with a countercharge in S2 to stabilize the VSD in different conformations (8). Likewise, it has been shown in other channels that conserved basic residues in S4 also interact with countercharges in other parts of the VSD (5,9,44). In this study, we systematically mutated S4 of Kv7.1 to examine whether charge at conserved positions interacts with other countercharges to affect voltage-dependent activation. S4 of Kv7.1 contains a Gln at the R3 position (Q3) and a His at the K5 position (H5) of Shaker, and thus contains fewer charges than the Shaker S4 (Fig. 1 A). Q3 is neutral and H5 has only a slight probability of carrying a positive charge at physiological pH. We individually mutated residues corresponding to basic residues in Shaker to either the neutral Gln or the negatively charged Glu. At Q3 and H5, we also increased charge by mutating these residues to Arg. We then recorded currents from these mutants both without and with KCNE1 (Fig. 1, B–E, and Fig. S1 and Fig. S2 in the Supporting Material).
Charge neutralization by R1Q shifts the G-V leftward by 39 mV in the absence of KCNE1 (Fig. 1, B and D). A further charge reduction by R1E produces constitutively open channels (Fig. 1 B and Fig. S1), which could be due to the G-V shifting to hyperpolarizing voltages where the voltage-dependent range of activation is beyond the limit of our recordings. A reduction of charge at R1 either destabilizes the resting state or stabilizes the activated state, where the extent of such a perturbation depends on charge. Because we are mutating a conserved charge in the protein, it would seem more likely that the mutation disrupts interactions in the wild-type (WT) protein that stabilize the resting state, rather than establishes new interactions that stabilize the activated state. For this reason, these results suggest that the reduction of charge at R1 destabilizes the resting state. At the R2 position, both R2Q and R2E results in constitutively open, voltage-independent channels. In contrast, the charge-conserved mutant R2K remains voltage-dependent, with only a 9 mV leftward shift in G-V (Fig. 1, B and D; Fig. S1), demonstrating that a positive charge at this position is essential for the WT phenotype of voltage dependence. Both R1 and R2 are important factors in favoring the resting state of the voltage sensor in native channels, with R2 playing a more prominent role.
In WT Kv7.1, the neutral Q3 cannot participate in electrostatic interactions. Q3R shifts the G-V rightward by 17 mV. Q3E likewise shifts the G-V rightward by 21 mV (Fig. 1, B and D; Fig. S1). The presence of a charge at Q3, regardless of polarity, destabilizes the activated state of the voltage sensor.
Below Q3, there is a marked change in the stabilization pattern. R4Q shifts the G-V rightward by 19 mV, whereas a further reduction of charge by R4E shifts the G-V rightward by 71 mV (Fig. 1, B and D; Fig. S2). H5R increases the charge and shifts the G-V leftward by 18 mV. In contrast, reducing charge by H5Q shifts the G-V by 19 mV and H5E by 38 mV rightward (Fig. 1, B and D; Fig. S2). Finally, R6Q and R6E shift the G-V rightward by 33 and 59 mV, respectively (Fig. 1, B and D; Fig. S2). In this region, the degree by which mutations change voltage dependence is proportional to the charge of the mutation. Mutations that reduce charge at all three residues destabilize the activated state of the voltage sensor. In contrast, increasing charge at H5 favors the activated state. Therefore, R4, R6, and, to a lesser degree, H5 stabilize the activated state of the voltage sensor in WT channels.
Taken as a whole, S4 can be divided into two groups: 1), basic residues in the N-terminal half of S4 (S4-N) above Q3 (R1 and R2) stabilize the voltage sensor in the resting state, and 2), basic residues in the C-terminal half of S4 (S4-C) below Q3 (R4, H5, and R6) stabilize the voltage sensor in the activated state (Fig. 1 D). These properties may derive from the interactions of these S4 residues with countercharges in other parts of the VSD (8).
KCNE1 changes the stabilization pattern of basic residues in S4 of Kv7.1
To determine how KCNE1 affects S4, we coexpressed single mutations described above with KCNE1 and measured the shift in voltage dependence relative to WT Kv7.1+KCNE1. Mutating R1 or R2 to Gln or Glu shifts the voltage dependence to favor the activated state, resulting in either constitutively activated channels (R1E and R2Q/E) or a negative shift in voltage dependence (R1Q, −80 mV shift; Fig. 1, C and E; Fig. S1). R2K only shifts by −6 mV. This behavior is similar to that observed in Kv7.1 channels alone (Fig. 1 D). Of interest, the voltage-dependent shift caused by Q3 mutations seems to be correlated with charge in the presence of KCNE1. Q3R shifts the G-V rightward by 35 mV, whereas Q3E shifts the G-V leftward by 17 mV. As with R1 and R2, a positive charge at Q3 would favor the resting state (Fig. 1, C and E; Fig. S1).
In contrast, when coexpressed with KCNE1, charge mutations in S4-C affect voltage dependence differently than in Kv7.1 alone. Mutations to R6 (R6Q/E) express low currents identical to currents from oocytes injected with KCNE1 alone (Fig. 1, C and E; Fig. S2), suggesting that these are currents expressed by endogenous channels coassembling with KCNE1, and the mutant channel in the presence of KCNE1 does not tolerate charge reduction at R6. Likewise, mutations of R6 to Cys or His are linked to LQT (27), which also generates small currents in the presence of KCNE1 (45–47). Mutants that produce currents of normal magnitude shift voltage dependence differently than they do in the absence of KCNE1. For example, R4Q+KCNE1 shifts voltage dependence by −24 mV, whereas R4Q alone shifts +19 mV. Likewise, H5Q+KCNE1 produces a −14 mV shift, whereas H5Q alone shifts +19 mV. Of interest, mutating H5 to either oppositely charged Arg or Glu in the presence of KCNE1 shifts voltage dependence rightward by ∼20 mV. Also, R4E+KCNE1 produces only an 8 mV shift. Channels in the presence of KCNE1 activate at more negative voltages when R4 or H5 is substituted by a neutral residue, whereas charge at R4 or H5, regardless of polarity, favors the resting state. These results indicate that the association of KCNE1 alters the behavior of basic residues in S4-C.
KCNE1 alters E2 interactions with S4
One possible mechanism by which KCNE1 could alter the behavior of basic residues in S4 would be to disturb the interactions of the basic residues in S4 with residues in other parts of the protein. Other segments of the VSD contain conserved countercharges that may form electrostatic interactions with S4. Previous studies showed that a conserved Glu in the upper part of S2 (E1; Fig. 1 A) forms state-dependent electrostatic interactions with Args in S4, such that when S4 moves from the resting state to the activated state R1, R2 and then R4 sequentially engage in electrostatic interactions with E1 (8,9,48). R1 interacts with E1 in the resting state (8), whereas R4 interacts with E1 in the activated state of the VSD (48). KCNE1 does not significantly affect E1-S4 interactions. However, Kv7.1 contains two other conserved countercharges that have not been examined in detail. The crystal structure of the Kv1.2/2.1 paddle chimera (49) shows that basic residues in S4-C are in close proximity to a second conserved Glu in S2 (E2) as well as a conserved Asp in S3 (D3).
Charge reversal of E2 to Arg abolishes current in both the absence and presence of KCNE1 (Fig. 2 A). However, the Western blot of biotinylated proteins that are expressed in the membrane of Xenopus oocytes injected with the E2R mRNA shows that the E2R channel traffics to the membrane (Fig. 2 A), indicating that E2R channels are present in the membrane but are nonfunctional. This loss of function could be due to gating defects or a severe shift in voltage dependence to depolarizing voltages that are beyond the limits of our recordings. Bands at 280, 210, 140, and 70 kDa correspond to Kv7.1 tetramers, trimers, dimers, and monomers, respectively. As a control, we also probed for the intracellular Gβ protein to demonstrate that the biotinylation only targeted membrane proteins. A similar phenotype was observed with charge reversal mutations of E1 (8). When E2 is neutralized by mutation to Gln (E2Q), channels become voltage-independent and constitutively activated (Fig. 2 A). These results indicate that charge at the E2 position affects Kv7.1 activation, likely through electrostatic interactions. Consistently, the charge-conserved E2D remains voltage-dependent, with a G-V curve identical to that of WT Kv7.1 (Fig. 2 A). Strikingly, coexpressing E2Q with KCNE1 yields channels that are not only voltage-dependent but also display a phenotype that is nearly identical to WT Kv7.1+KCNE1 (Fig. 2 A), suggesting that the association of KCNE1 may change E2 interactions and reduce the perturbation induced by the E2Q mutation. E2D+KCNE1 is also voltage-dependent (Fig. 2 A).
Figure 2.
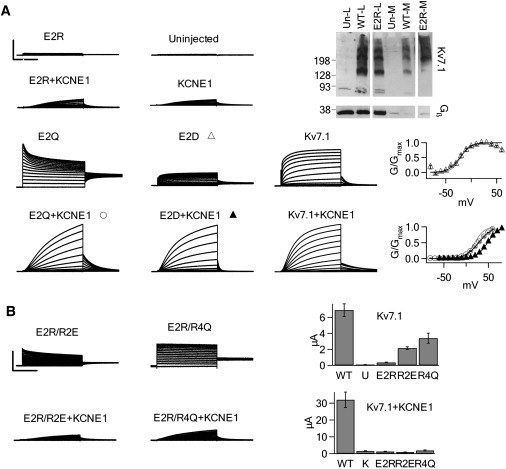
KCNE1 changes electrostatic interactions between E2 with S4. (A) Left: Currents generated from E2R, E2Q, E2D, WT, and endogenous channels. E2R does not express currents beyond the background oocyte currents. Except for E2D+KCNE1, currents were elicited by depolarization from −80 to 60 mV. E2D+KCNE1 was depolarized from −60 to +80 mV. Scale: E2R 4 μA, E2R+KCNE1 4 μA, uninjected 4 μA, KCNE1 4 μA, E2Q 5 μA, E2Q+KCNE1 7 μA, E2D 4 μA, E2D+KCNE1 7 μA, Kv7.1 4 μA, Kv7.1+KCNE1 20 μA; 2 s for all. Top right: Western blot probing for Kv7.1 and Gβ in the whole cell lysate and biotinylated membrane fraction from oocytes. Gβ is a cytoplasmic protein. E2R is expressed on the membrane. Middle right: G-V relationship of E2D relative to WT Kv7.1 (gray line). G-V of E2D is superimposed on WT Kv7.1. Bottom right: G-V relationship of E2Q+KCNE1 and E2D+KCNE1 relative to WT Kv7.1+KCNE1 (gray line). KCNE1 restores voltage dependence to E2Q. (B) Left: Currents from S4 mutations to Glu paired with E2R without (top) and with (bottom) KCNE1 in response to depolarized voltages from −80 mV to +60 mV. Scale: 4 μA; 2 s. Right: Peak current amplitudes at +60 mV were averaged for each mutant. R2E and R4Q restore current to E2R in the absence of KCNE1 but not in the presence of KCNE1. In the bar graph, U indicates currents from uninjected oocytes, and K indicates currents from oocytes injected with KCNE1 only.
Positively charged residues in the transmembrane segments of Kv7.1 are located only in S4. To determine whether these basic residues interact with E2, we utilized an intragenic suppression technique to mutate S4 residues on the background of E2R to identify mutations that restore current (Fig. 2 B). Two mutations (R2E and R4Q) restored currents from E2R in Kv7.1 channels, suggesting that R2 and R4 may interact with E2. In the presence of KCNE1, neither R2E nor R4Q restored current to E2R (Fig. 2 B); thus, KCNE1 association may disrupt interactions between E2 and S4. We attempted to use Cys substitution and disulfide cross-linking to study E2-S4 interactions, but were unable to obtain useful data because certain mutants remained constitutively open regardless of the chemical treatment used.
Of interest, D3R behaves similarly to E2Q (data not shown) in that it is constitutively open in the absence of KCNE1 but becomes voltage-dependent with KCNE1 coexpression. However, we could not conclusively determine whether D3 interacts with S4 because all of the mutations involving D3R remained constitutively open, which prevented us from using an intragenic suppression or mutant cycle approach to study the mutant.
KCNE1 changes the effect of Trp mutations in S4
The above experiments suggest that the association of KCNE1 alters the electrostatic interaction of basic residues in S4 with other parts of the VSD, including E2 in the S2 segment. These results may derive from a KCNE1-induced change in the environment or packing of S4 in the channel protein. To further detect such a change, we performed a Trp scan of the entire S4 segment in both the absence and presence of KCNE1, and observed how the mutations affect macroscopic current amplitude- and voltage-dependent activation (Fig. 3, Fig. S3, and Fig. S4). It has been shown that introducing Trp to a tightly packed protein environment causes drastic changes in protein function (50,51). On the other hand, Trp tends to cause minor disruptions if it is positioned in a loosely packed protein environment or a lipid-exposed environment. In a previous study, a Trp scan of S4 in Kv7.1 demonstrated changes in gating caused by Trp (52). In our study, we used the same technique to highlight mutants that show KCNE1-dependent changes in Trp perturbation, which was not shown in the previous study.
Figure 3.
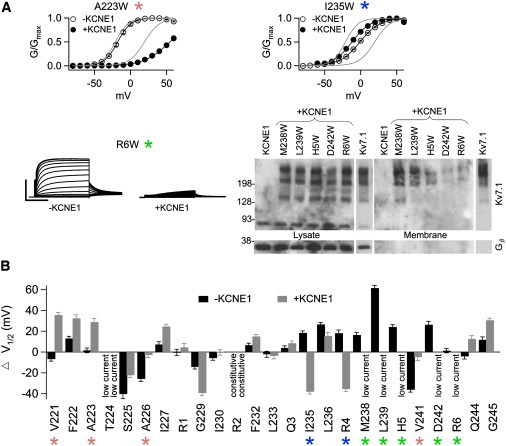
Effect of Trp substitutions on voltage dependence and current amplitude in the absence and presence of KCNE1. (A) Sample G-V relationships or currents from three different representative Trp mutations that affect KCNE1 modulation. Currents were elicited by depolarizing from −80 to 60 mV in 10 mV increments for 5 s. Scale: R6W 4 μA, R6W+KCNE1 1 μA. In the G-V plots, black curves are fittings of the Boltzmann equation to the mutants and the gray curves are G-V relations of WT Kv7.1 (left) and Kv7.1+KCNE1 (right). Right, Western blot probing for Kv7.1 and Gβ in the whole cell lysate and biotinylated membrane fraction from oocytes. (B) Shift in the voltage of half-maximal activation for Trp mutations in S4 relative to WT Kv7.1 in the absence or presence of KCNE1, respectively. Green identifies mutations that express low currents only in the presence of KCNE1 (M238, L239, H5, D242, R6). Blue identifies mutations that shift voltage dependence in opposing directions with and without the presence of KCNE1 (I235, R4). Pink identifies mutations that shift voltage dependence in either in the presence or absence of KCNE1 (V221, A223, A226, V241).
In contrast to WT channels, two mutants (V221W and A223W) shift voltage-dependent activation in the presence of KCNE1 but do not cause much of a shift in the absence of KCNE1 (Fig. 3, A and B). Conversely, two mutants (A226W and V241W) shift voltage dependence in the absence of KCNE1 but not in its presence. All four of these residues are labeled pink in Fig. 3 B. Another category of mutants (I235W and R237W) shifts voltage dependence both with and without KCNE1, but the shift is in opposite directions (Fig. 3, A and B). In effect, these two mutants ablate the characteristic KCNE1-induced rightward shift in voltage dependence. These mutants are labeled blue in Fig. 3 B).
It is most striking that mutants labeled green in Fig. 3 produce low currents with KCNE1 but generate currents with normal amplitude in the absence of KCNE1 (e.g., R6W; Fig. 3, A and B). When coexpressed with KCNE1, these mutants have currents similar in amplitude, morphology, and voltage dependence to currents produced by oocytes injected with KCNE1 alone. Therefore, the small currents observed in our recordings are likely generated by endogenous oocyte channels coassembling with KCNE1; mutant channels do not generate any additional current on top of this background current. These residues may become more tightly packed against protein when KCNE1 is present, thus becoming less tolerant of Trp substitution. We conducted biotinylation experiments as described in Fig. 2 A to determine whether the mutations affect trafficking of the channel to the membrane (Fig. 3 A). M238W+KCNE1 and L239W+KCNE1 are both strongly expressed on the membrane. H5W+KCNE1 has less expression relative to M238W+KCNE1 but is still present in the membrane. Thus, for these mutants (M238W, L239W, and H5W) with KCNE1, the lack of current could be due to channels present in the membrane having gating defects or having an extreme positive shift in voltage dependence that prevents the channels from opening within the tested voltage range. On the other hand, both D242W+KCNE1 and R6W+KCNE1 have very low membrane expression. Because Kv7.1 multimers are still found in the lysate in large quantities, protein synthesis does not appear to be affected, but trafficking or membrane insertion and retention may be compromised. Therefore, the lack of current observed in D242W and R6W with KCNE1 is likely due primarily to a reduction in the surface expression of Kv7.1. The rest of the mutants that affect the properties of the channel similarly both with and without KCNE1 are not highlighted (Fig. 3 B). Of interest, mutants that possess KCNE1-dependent changes largely cluster in S4-C (Fig. 3 B), in similarity to the results in Fig. 1, suggesting that the environment of this region is significantly altered by KCNE1.
KCNE1 also alters the environment of the extracellular part of S4
Mutations to the basic residues in S4 and to E2 in S2, and the Trp scan highlight the changes that KCNE1 induces in S4-C (Figs. 1–3). The Trp scan also indicates that residues in S4-N are affected (Fig. 3). To further test whether S4-N is affected by KCNE1, we measured the accessibility of R2C to extracellular MTS reagents to determine whether KCNE1 changes solvent exposure (Fig. 4 A). In the absence of KCNE1, extracellular MTSET+ application does not result in any changes in current. On the other hand, MTSET+ modifies R2C+KCNE1 to cause decay in current, suggesting that KCNE1 increases R2C accessibility to the extracellular solution. These results are analogous to our previous finding that E1C accessibility to MTSES− dramatically increases with KCNE1 coexpression (Fig. 4 B) (8). On the other hand, Q3C and Q3C+KCNE1 are insensitive to MTSET+, suggesting that Q3C is not exposed to the extracellular milieu (Fig. 4 A). Although we cannot rule out the possibility that Q3C is modified but shows no functional change, it is unlikely that attaching a charged compound to a highly mobile and vital region of the channel like S4 would result in no changes in current. Moreover, as previously described, mutation of Q3 to Arg shifts the G-V curve both with and without KCNE1 (Fig. 1). These results suggest that KCNE1 increases solvent accessibility to the S4-N.
Figure 4.
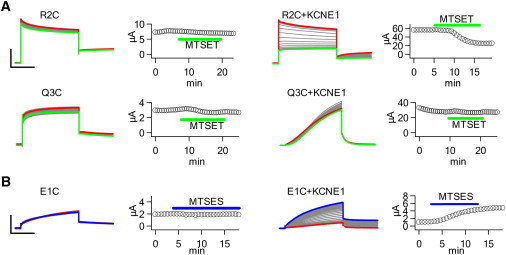
KCNE1 association enhances extracellular MTS accessibility to the VSD. (A) MTSET+ modification of R2C and Q3C in the absence and presence of KCNE1. Oocytes were held at −80 mV for 32 s and repeatedly pulsed to +40 mV for 5 s, and repolarized at −40 mV for 3 s. MTSET+ reduces R2C current only in the presence of KCNE1. Q3C current is unchanged by MTSET+. Scale: R2C 4 μA, R2C+KCNE1 30 μA, Q3C 1.5, Q3C+KCNE1 15 μA; 2 s. The peak current amplitude after each voltage pulse is plotted against time. (B) MTSES− modification of E1C in the absence and presence of KCNE1 using the voltage protocol in A. MTSES− enhances E1C current only in the presence of KCNE1. Scale: E1C 2 μA, E1C+KCNE1 4 μA; 2 s.
Previous studies and our data suggest that because of the lack of charge in S4, Kv7.1 has a propensity to adopt a constitutively open, voltage-independent conformation upon further reduction of charge in S4 (53). We took advantage of this unique property of Kv7.1 to further test whether KCNE1 affects residues in S4-N. A loss of charge at R1 or R2 results in constitutively open, voltage-independent channels (Figs. 1 and 5). Compensating for the loss of charge at R1 or R2 with the addition of charge at Q3 conserves the total charge in S4 and may allow channels to regain voltage sensitivity. Indeed, mutating Q3R on the background of R1E restores voltage-dependent activation in both the absence and presence of KCNE1 (Fig. 5 A). However, R2Q/Q3R and R2E/Q3R (data not shown; similar phenotype as R2Q/Q3R) remain voltage-insensitive in Kv7.1 channels alone (Fig. 5 B). Of interest, when R2Q/Q3R is coexpressed with KCNE1, it regains voltage sensitivity (Fig. 5 B). R2E/Q3R+KCNE1 also has a phenotype similar to that of R2Q/Q3R+KCNE1 (data not shown). Thus, KCNE1 changes the ability of charge addition at Q3 to substitute for charge loss at R2.
Figure 5.
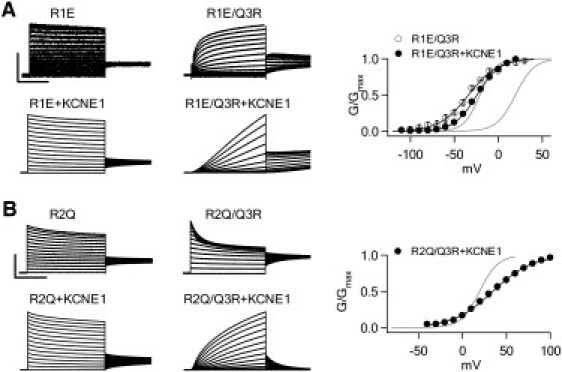
Charge addition to Q3 rescues voltage dependence in charge reduction mutations to R1 and R2. (A) Currents and G-V relationship from R1E and R1E/Q3R. Q3R restores voltage dependence to R1E in both the absence and presence of KCNE1. Voltage pulses of 5 s duration ranged from −80 to 60 mV in 10 mV increments for activating R1E-/+KCNE1 currents, and from −120 to 20 mV for R1E/Q3R-/+KCNE1 currents. The holding and repolarizing potentials were −80 mV for 32 s and −40 mV for 3 s, respectively. Scale: R1E 1 μA, R1C/Q3R 1.5 μA, R1E+KCNE1 25 μA, R1E/Q3R+KCNE1 10 μA; 2 s. (B) Currents and G-V relationship from R2Q and R2Q/Q3R. Q3R only restores voltage dependence to R2Q in the presence of KCNE1. Voltages ranged from −80 to 60 mV for activating R2Q-/+KCNE1 and R2Q/Q3R currents, and from −40 to 100 mV for R2Q/Q3R+KCNE1 currents. Scale: R2Q 2 μA, R2Q/Q3R 5 μA, R2Q+KCNE1 40 μA, R2Q/Q3R+KCNE1 15 μA; 2 s. In the G-V plots of both A and B, the black curves are fittings of the Boltzmann equation to the mutants, and the gray curves are the G-V relations of WT Kv7.1 (left) and Kv7.1+KCNE1 (right).
Discussion
The effect of KCNE1 on the voltage sensor of Kv7.1 has been a controversial subject. Nakajo and Kubo (22) along with Rocheleau and Kobertz (21) examined the kinetics of S4 movement in Kv7.1 with and without KCNE1 using MTS modification of cysteines in S4. The debate as to whether KCNE1 slows S4 movement remains ongoing. In this study we focus on the thermodynamic changes induced by KCNE1 in the VSD, rather than the kinetics of S4 movement. Previous studies probed S4 via mutagenesis to determine how perturbations to S4 affect the voltage dependence of channel activation, as we also did in this study. However, we offer new interpretations of the data. Panaghie and Abbott (53) made Ala mutations of charged residues in S4 and concluded that the relative dearth of charge in S4 of Kv7.1 imparts a tendency for channels to become constitutively activated upon further charge reduction—an effect that is mimicked by Kv7.1 coexpression with KCNE3. From our neutralization and charge reversal mutagenesis of S4, we find that mutations of arginines in S4-N are susceptible to becoming constitutively open. Basic residues in S4-N stabilize the resting state, whereas basic residues in S4-C stabilize the activated state. KCNE1 coexpression perturbs the behavior of basic residues in S4-C (Fig. 1), suggesting that KCNE1 may disrupt electrostatic interactions in this region. Intragenic suppression of the nonfunctional E2R mutation consistently shows that S4 mutations can restore E2R current in the absence of KCNE1 but not with KCNE1 coexpression (Fig. 2), suggesting that KCNE1 disrupts E2-S4 electrostatic interactions in Kv7.1. Shamgar and colleagues (52) conducted a Trp scan of S4 and found that certain Trp mutations mimicked KCNE1 modulation. Trp mutation of certain residues also affected the ability of KCNE1 to modulate channel function (e.g., to prevent a positive shift in G-V). We also conducted a Trp scan of S4, but our analysis and interpretation are based on a side-by-side comparison of how each mutation affects channel activation relative to WT channels with and without KCNE1. This direct comparison reveals a cluster of residues in S4-C that is strongly perturbed by Trp substitution (Fig. 3). This same cluster also contains the basic residues that were perturbed by KCNE1. Thus, the results of these experiments all show that the association of KCNE1 disrupts the interactions of S4-C with other parts of the channel protein. In addition, KCNE1 also affects several Trp mutations in S4-N, albeit less severely. MTS probing shows that residues in the extracellular side of the VSD, E1C (8) and R2C (Fig. 4), have greater accessibility upon KCNE1 coexpression.
Crystal structures (49,54) and biophysical experiments (5,8,44,55–57) have shown that in Kv channels, S4 intimately interacts with other regions of the VSD and also forms contacts with the pore. These interactions include electrostatic interactions among charged residues (5,8,44,55) and steric interactions among uncharged residues (56,57). Our data show that KCNE1 disrupts electrostatic interactions involving S4, specifically perturbing interactions between E2 and S4 within the VSD. In addition, S4-C is more severely perturbed by Trp mutations in the presence of KCNE1, suggesting that KCNE1 also changes the steric interactions of S4.
Recent studies have increasingly focused on possible interactions between KCNE1 with the VSD of Kv7.1. Cross-linking experiments have established that E43C at the extracellular side of KCNE1 can form disulfide bonds with A226C in S4 (22), and disulfide linkages can also be made between the extracellular side of S1 and S5 with KCNE1 (41,42). On the intracellular side, cross-linking experiments have identified that the S4-S5 linker and the intracellular part of S6 can form disulfide bonds with the KCNE1 C-terminus (58). These constraints place KCNE1 in a cleft between adjacent Kv7.1 subunits.
Crystal structures capturing a KCNE1-bound Kv7.1 channel would provide the ultimate view of the interactions between the two proteins. Unfortunately, such a structure does not yet exist. However, the solution NMR structure of KCNE1 has been determined, and Kv7.1 models based on Kv crystal structures are available (43). Simulations docking KCNE1 to models of Kv7.1 in the open and closed states position KCNE1 in the aforementioned cleft between Kv7.1 subunits (43), which is consistent with disulfide cross-linking data. The strategic placement of KCNE1 in this cleft allows it to interact with both the VSD and the pore of Kv7.1. We show these models in Fig. 6, highlighting the high-impact residues in S4-C identified in this study (colored as in Fig. 3). The entire transmembrane region of KCNE1 (yellow) is shown as space-filling spheres (43). In the model, residues in S4-C are largely surrounded by protein, and interact with the VSD and pore in either the closed or open state. The docking simulations position KCNE1 proximal to S4. From our experiments, we cannot assert whether or not KCNE1 directly interacts with S4. In either case, the positioning of KCNE1 shown by the models suggests that KCNE1 may influence interactions involving S4. We hypothesize that KCNE1 repacks the protein environment around S4, which results in severe functional consequences in regions like S4-C that form significant protein-protein interactions. Changes in the packing of S4-C by KCNE1 disrupt electrostatic interactions with E2 and introduce additional steric constraints that make the region less tolerant of Trp mutations.
Figure 6.
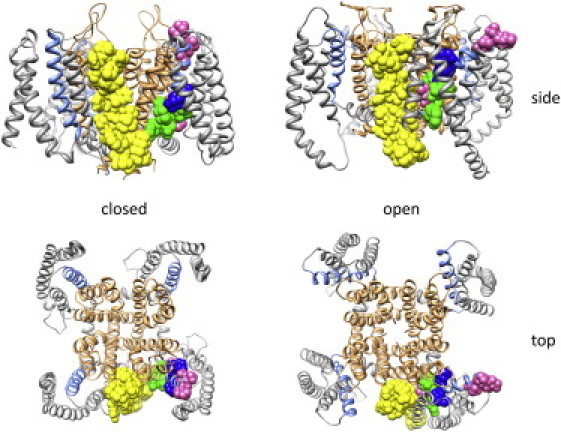
KCNE1 docked to structural models of Kv7.1 in the closed and open state from Kang et al. (43). The pore is displayed as gold ribbons and the voltage-sensing domains are displayed as gray (S1–S3) or blue (S4) ribbons. The transmembrane segment of KCNE1 and the high impact mutants in S4 of Kv7.1 that show KCNE1 dependent changes in channel function are highlighted: KCNE1 is depicted with yellow space-filling spheres and the high impact residues in S4 are depicted as space-filling spheres with the same colors shown in Fig. 3.
The repacking of S4 may also have consequences in S4-N, albeit to a lesser extent than in S4-C. As shown in Figs. 3 and 4, KCNE1 coexpression affects Trp mutants in S4-N and increases the extracellular accessibility of R2C located in this region. An interesting hypothesis is that the repacking of S4 by KCNE1 changes the solvent exposure of S4 and thus reshapes the electric field acting on charges in S4. Increased solvent exposure to R2 may reduce the strength of the electric field that R2 traverses relative to Q3. Of course, Q3 would not sense the electric field in its native state, but adding a charge via mutation (Q3R) would subject it to the electric field. We found that Q3R can compensate for loss of charge at R2 in the presence of KCNE1 but not in its absence (Fig. 5 B), which is consistent with a change in the electric field such that Q3R may sense a stronger electric field in the presence of KCNE1. Thus, the association of KCNE1 may not only remodel the interaction of S4 with its environment but also alter the electric field to extend the effect beyond the high-impact cluster in S4-C. Gating currents or fluorometry measurements of S4 movement would more directly indicate whether KCNE1 affects the gating charge of Kv7.1. Unfortunately, gating currents have not been reported for Kv7.1, possibly due to a combination of low channel expression, slow gating charge movements, and relatively few S4 charges compared to other channels. Likewise, fluorometry measurements have not been reported for Kv7.1, possibly due to low channel expression or the lack of a suitable reporter residue.
In summary, in this study we focused on the behavior of S4 of Kv7.1 in the absence and presence of KCNE1. Our experimental data and modeling from other studies suggest that KCNE1 causes a repacking of the protein environment around S4. This repacking affects electrostatic interactions that involve S4. There are three conserved countercharges in the VSD of Kv channels. We showed in a previous study that E1-S4 interactions are mildly altered by KCNE1. Here, we show that E2-S4 interactions are more severely perturbed by KCNE1. We also attempted to study the effect of KCNE1 on D3-S4 interactions, but were limited by experimental difficulties as previously indicated in the Results. The repacking of S4 also affects steric interactions of S4 with its protein environment. In this study, we did not identify the specific residues that interact sterically with S4. Such an effort would involve more extensive mutational scanning of transmembrane segments in the VSD and the pore, which should be the subject of future studies. Efforts to map out interacting partners with S4 in both the absence and presence of KCNE1 may provide additional details with regard to how KCNE1 remodels the environment around S4.
Acknowledgments
D.W. and J.C. designed the research shown in Figs. 1, 2, 4, and 5. H.P. and J.C. designed the research shown in Fig. 3. D.W. performed the experiments shown in Figs. 1–5. H.P. performed the experiments shown in Fig. 3. K.D. performed the mutagenesis. D.W. created Fig. 6. D.W. and J.C. wrote the manuscript.
This study was funded by grants from the American Heart Association (Scientist Development Grant 9930025N and Established Investigator Award 0440066N), the United States-Israel Binational Science Foundation (research grant 2001229), and the National Science Foundation of China (grant 30528011 to J.C.). D.W. received a predoctoral fellowship (No. 0910020G) from the American Heart Association. J.C. is the Spencer T. Olin Professor of Biomedical Engineering at Washington University.
Supporting Material
References
- 1.Tombola F., Pathak M.M., Isacoff E.Y. How does voltage open an ion channel? Annu. Rev. Cell Dev. Biol. 2006;22:23–52. doi: 10.1146/annurev.cellbio.21.020404.145837. [DOI] [PubMed] [Google Scholar]
- 2.Aggarwal S.K., MacKinnon R. Contribution of the S4 segment to gating charge in the Shaker K+ channel. Neuron. 1996;16:1169–1177. doi: 10.1016/s0896-6273(00)80143-9. [DOI] [PubMed] [Google Scholar]
- 3.Seoh S.A., Sigg D., Bezanilla F. Voltage-sensing residues in the S2 and S4 segments of the Shaker K+ channel. Neuron. 1996;16:1159–1167. doi: 10.1016/s0896-6273(00)80142-7. [DOI] [PubMed] [Google Scholar]
- 4.Yang N., George A.L., Jr., Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- 5.Tiwari-Woodruff S.K., Schulteis C.T., Papazian D.M. Electrostatic interactions between transmembrane segments mediate folding of Shaker K+ channel subunits. Biophys. J. 1997;72:1489–1500. doi: 10.1016/S0006-3495(97)78797-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L., Sato Y., Uozumi N. Contribution of hydrophobic and electrostatic interactions to the membrane integration of the Shaker K+ channel voltage sensor domain. Proc. Natl. Acad. Sci. USA. 2007;104:8263–8268. doi: 10.1073/pnas.0611007104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silva J.R., Pan H., Rudy Y. A multiscale model linking ion-channel molecular dynamics and electrostatics to the cardiac action potential. Proc. Natl. Acad. Sci. USA. 2009;106:11102–11106. doi: 10.1073/pnas.0904505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu D., Delaloye K., Cui J. State-dependent electrostatic interactions of S4 arginines with E1 in S2 during Kv7.1 activation. J. Gen. Physiol. 2010;135:595–606. doi: 10.1085/jgp.201010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decaen P.G., Yarov-Yarovoy V., Catterall W.A. Sequential formation of ion pairs during activation of a sodium channel voltage sensor. Proc. Natl. Acad. Sci. USA. 2009;106:22498–22503. doi: 10.1073/pnas.0912307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sigg D., Bezanilla F. A physical model of potassium channel activation: from energy landscape to gating kinetics. Biophys. J. 2003;84:3703–3716. doi: 10.1016/S0006-3495(03)75099-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pongs O., Schwarz J.R. Ancillary subunits associated with voltage-dependent K+ channels. Physiol. Rev. 2010;90:755–796. doi: 10.1152/physrev.00020.2009. [DOI] [PubMed] [Google Scholar]
- 12.Liu G., Zakharov S.I., Marx S.O. Locations of the β1 transmembrane helices in the BK potassium channel. Proc. Natl. Acad. Sci. USA. 2008;105:10727–10732. doi: 10.1073/pnas.0805212105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu G., Niu X., Karlin A. Location of modulatory β subunits in BK potassium channels. J. Gen. Physiol. 2010;135:449–459. doi: 10.1085/jgp.201010417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrow J.P., Zakharov S.I., Marx S.O. Defining the BK channel domains required for β1-subunit modulation. Proc. Natl. Acad. Sci. USA. 2006;103:5096–5101. doi: 10.1073/pnas.0600907103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orio P., Latorre R. Differential effects of β1 and β2 subunits on BK channel activity. J. Gen. Physiol. 2005;125:395–411. doi: 10.1085/jgp.200409236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bao L., Cox D.H. Gating and ionic currents reveal how the BKCa channel's Ca2+ sensitivity is enhanced by its β1 subunit. J. Gen. Physiol. 2005;126:393–412. doi: 10.1085/jgp.200509346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Savalli N., Kondratiev A., Olcese R. Modes of operation of the BKCa channel β2 subunit. J. Gen. Physiol. 2007;130:117–131. doi: 10.1085/jgp.200709803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang H., Zhang G., Cui J. Subunit-specific effect of the voltage sensor domain on Ca2+ sensitivity of BK channels. Biophys. J. 2008;94:4678–4687. doi: 10.1529/biophysj.107.121590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dougherty K., Tu L., Covarrubias M. The dipeptidyl-aminopeptidase-like protein 6 is an integral voltage sensor-interacting β-subunit of neuronal K(V)4.2 channels. Channels (Austin) 2009;3:122–128. doi: 10.4161/chan.3.2.8333. [DOI] [PubMed] [Google Scholar]
- 20.Börjesson S.I., Elinder F. Structure, function, and modification of the voltage sensor in voltage-gated ion channels. Cell Biochem. Biophys. 2008;52:149–174. doi: 10.1007/s12013-008-9032-5. [DOI] [PubMed] [Google Scholar]
- 21.Rocheleau J.M., Kobertz W.R. KCNE peptides differently affect voltage sensor equilibrium and equilibration rates in KCNQ1 K+ channels. J. Gen. Physiol. 2008;131:59–68. doi: 10.1085/jgp.200709816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nakajo K., Kubo Y. KCNE1 and KCNE3 stabilize and/or slow voltage sensing S4 segment of KCNQ1 channel. J. Gen. Physiol. 2007;130:269–281. doi: 10.1085/jgp.200709805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi E., Abbott G.W. A shared mechanism for lipid- and β-subunit-coordinated stabilization of the activated K+ channel voltage sensor. FASEB J. 2010;24:1518–1524. doi: 10.1096/fj.09-145219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barhanin J., Lesage F., Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 25.Sanguinetti M.C., Curran M.E., Keating M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 26.Nerbonne J.M., Kass R.S. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005;85:1205–1253. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- 27.Splawski I., Shen J., Keating M.T. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 28.Keating M.T., Sanguinetti M.C. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104:569–580. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 29.Jespersen T., Grunnet M., Olesen S.P. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–416. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 30.Tapper A.R., George A.L., Jr. Location and orientation of minK within the I(Ks) potassium channel complex. J. Biol. Chem. 2001;276:38249–38254. doi: 10.1074/jbc.M103956200. [DOI] [PubMed] [Google Scholar]
- 31.Tai K.K., Goldstein S.A. The conduction pore of a cardiac potassium channel. Nature. 1998;391:605–608. doi: 10.1038/35416. [DOI] [PubMed] [Google Scholar]
- 32.Chen H., Sesti F., Goldstein S.A. Pore- and state-dependent cadmium block of I(Ks) channels formed with MinK-55C and wild-type KCNQ1 subunits. Biophys. J. 2003;84:3679–3689. doi: 10.1016/S0006-3495(03)75097-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kurokawa J., Motoike H.K., Kass R.S. TEA(+)-sensitive KCNQ1 constructs reveal pore-independent access to KCNE1 in assembled I(Ks) channels. J. Gen. Physiol. 2001;117:43–52. doi: 10.1085/jgp.117.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Melman Y.F., Um S.Y., McDonald T.V. KCNE1 binds to the KCNQ1 pore to regulate potassium channel activity. Neuron. 2004;42:927–937. doi: 10.1016/j.neuron.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 35.Chen J., Zheng R., McDonald T.V. Functional interactions between KCNE1 C-terminus and the KCNQ1 channel. PLoS ONE. 2009;4:e5143. doi: 10.1371/journal.pone.0005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haitin Y., Wiener R., Attali B. Intracellular domains interactions and gated motions of I(KS) potassium channel subunits. EMBO J. 2009;28:1994–2005. doi: 10.1038/emboj.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tapper A.R., George A.L., Jr. MinK subdomains that mediate modulation of and association with KvLQT1. J. Gen. Physiol. 2000;116:379–390. doi: 10.1085/jgp.116.3.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng R., Thompson K., McDonald T.V. Analysis of the interactions between the C-terminal cytoplasmic domains of KCNQ1 and KCNE1 channel subunits. Biochem. J. 2010;428:75–84. doi: 10.1042/BJ20090977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y.H., Xu S.J., Huang W. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- 40.Restier L., Cheng L., Sanguinetti M.C. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J. Physiol. 2008;586:4179–4191. doi: 10.1113/jphysiol.2008.157511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chung D.Y., Chan P.J., Kass R.S. Location of KCNE1 relative to KCNQ1 in the I(KS) potassium channel by disulfide cross-linking of substituted cysteines. Proc. Natl. Acad. Sci. USA. 2009;106:743–748. doi: 10.1073/pnas.0811897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu X., Jiang M., Tseng G.N. KCNQ1 and KCNE1 in the IKs channel complex make state-dependent contacts in their extracellular domains. J. Gen. Physiol. 2008;131:589–603. doi: 10.1085/jgp.200809976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang C., Tian C., Sanders C.R. Structure of KCNE1 and implications for how it modulates the KCNQ1 potassium channel. Biochemistry. 2008;47:7999–8006. doi: 10.1021/bi800875q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Papazian D.M., Shao X.M., Wainstock D.H. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 1995;14:1293–1301. doi: 10.1016/0896-6273(95)90276-7. [DOI] [PubMed] [Google Scholar]
- 45.Franqueza L., Lin M., Sanguinetti M.C. Long QT syndrome-associated mutations in the S4-S5 linker of KvLQT1 potassium channels modify gating and interaction with minK subunits. J. Biol. Chem. 1999;274:21063–21070. doi: 10.1074/jbc.274.30.21063. [DOI] [PubMed] [Google Scholar]
- 46.Chouabe C., Neyroud N., Barhanin J. Novel mutations in KvLQT1 that affect Iks activation through interactions with Isk. Cardiovasc. Res. 2000;45:971–980. doi: 10.1016/s0008-6363(99)00411-3. [DOI] [PubMed] [Google Scholar]
- 47.Park K.H., Piron J., Loussouarn G. Impaired KCNQ1-KCNE1 and phosphatidylinositol-4,5-bisphosphate interaction underlies the long QT syndrome. Circ. Res. 2005;96:730–739. doi: 10.1161/01.RES.0000161451.04649.a8. [DOI] [PubMed] [Google Scholar]
- 48.Long S.B., Campbell E.B., Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- 49.Long S.B., Tao X., MacKinnon R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature. 2007;450:376–382. doi: 10.1038/nature06265. [DOI] [PubMed] [Google Scholar]
- 50.Monks S.A., Needleman D.J., Miller C. Helical structure and packing orientation of the S2 segment in the Shaker K+ channel. J. Gen. Physiol. 1999;113:415–423. doi: 10.1085/jgp.113.3.415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong K.H., Miller C. The lipid-protein interface of a Shaker K(+) channel. J. Gen. Physiol. 2000;115:51–58. doi: 10.1085/jgp.115.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shamgar L., Haitin Y., Attali B. KCNE1 constrains the voltage sensor of Kv7.1 K+ channels. PLoS One. 2008;3:e1943. doi: 10.1371/journal.pone.0001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Panaghie G., Abbott G.W. The role of S4 charges in voltage-dependent and voltage-independent KCNQ1 potassium channel complexes. J. Gen. Physiol. 2007;129:121–133. doi: 10.1085/jgp.200609612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Long S.B., Campbell E.B., Mackinnon R. Crystal structure of a mammalian voltage-dependent Shaker family K+ channel. Science. 2005;309:897–903. doi: 10.1126/science.1116269. [DOI] [PubMed] [Google Scholar]
- 55.Elinder F., Männikkö R., Larsson H.P. S4 charges move close to residues in the pore domain during activation in a K channel. J. Gen. Physiol. 2001;118:1–10. doi: 10.1085/jgp.118.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith-Maxwell C.J., Ledwell J.L., Aldrich R.W. Uncharged S4 residues and cooperativity in voltage-dependent potassium channel activation. J. Gen. Physiol. 1998;111:421–439. doi: 10.1085/jgp.111.3.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soler-Llavina G.J., Chang T.H., Swartz K.J. Functional interactions at the interface between voltage-sensing and pore domains in the Shaker K(v) channel. Neuron. 2006;52:623–634. doi: 10.1016/j.neuron.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 58.Lvov A., Gage S.D., Kobertz W.R. Identification of a protein-protein interaction between KCNE1 and the activation gate machinery of KCNQ1. J. Gen. Physiol. 2010;135:607–618. doi: 10.1085/jgp.200910386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.