

Abstract
The induced fit model has traditionally been invoked to describe the activating conformational change of the monomeric G-proteins, such as Ras and Rho. With this scheme, the presence or absence of the γ-phosphate of GTP leads to an instantaneous switch in conformation. Here we describe atomistic molecular simulations that demonstrate that both Ras and Rho superfamily members harbor an intrinsic susceptibility to sample multiple conformational states in the absence of nucleotide ligand. By comparing the distribution of conformers in the presence and absence of nucleotide, we show that conformational selection is the dominant mechanism by which Ras and Rho undergo nucleotide-dependent conformational changes. Furthermore, the pattern of correlated motions revealed by these simulations predicts a preserved allosteric coupling of the nucleotide-binding site with the membrane interacting C-terminus in both Rho and Ras.
Our understanding of biomolecular recognition has evolved with our knowledge of the growing repertoire of interactions that underpin life's processes. Early models that envisioned a rigid lock-and-key-like fit of interacting molecules with a single unique structure have given way to dynamic views of interacting molecules existing as an ensemble of interconverting conformers (1–8). The ability to visualize complex three-dimensional structures made it apparent that interacting biomolecules do not necessarily have a complementary shape before binding. This led to the induced fit model of receptor-ligand interaction (4). Induced fit relies on the formation of a loose initial complex followed by binding-induced conformational changes that lead to tighter binding. This model remains enormously influential despite limitations highlighted by early kinetics studies (1). For example, in the absence of some prior molecular match to provide sufficient affinity before conformational adaptation, a kinetic bottleneck would make a thermodynamically sound reaction nonviable (1). Such cases may be better described by a conformational selection and population shift mechanism, where the ligand acts to selectively stabilize and promote certain preexisting receptor conformations (7–10). This alternate view can account for cooperative changes including allosteric effects where binding to one site is coupled to a conformational change at a distant site (3,8,9). However, it is important to note that these models are not mutually exclusive. Indeed a combination of conformational selection and induced fit would seem to be the best description of the interaction between molecules that do not optimally fit a priori.
Conformational selection and induced fit can be considered two extremes of possible mechanisms underlying biomolecular recognition (11). Clearly, the intrinsic dynamics of binding partners may strongly affect their interactions. However, it is unclear whether induced dynamics or such intrinsic dynamics play a dominant role in ligand binding that often entails highly specific localized interactions. Using advanced methods for probing protein dynamics, such as multidimensional nuclear magnetic resonance, a number of recent reports demonstrated the dominant role of conformational selection in a variety of proteins (e.g., (8)). From a kinetic standpoint, one can demonstrate conformational selection by showing that the rate of formation of the ligand-receptor complex is linearly proportional to the concentration of a conformational species fit for binding and nonlinearly proportional to the total concentration (see Fig. S1 in the Supporting Material). An equivalent way of demonstrating conformational selection through molecular simulation is by assessing the equilibrium distribution of conformations in the absence of ligand compared to their relative distribution in the presence of ligand. Employing this latter approach, we demonstrate that conformational selection is the dominant mechanism underlying the function of the monomeric Ras and Rho G-proteins.
Monomeric G-proteins cycle between GTP-bound active and GDP-bound inactive conformations to regulate diverse cellular processes from signal transduction to cytoskeletal remodeling. The critical activating conformational change common to all G-protein superfamily members has traditionally been described by the induced fit model, where the presence or absence of the γ-phosphate of GTP leads to an instantaneous switch in conformation (12). This view has been shaped by the extensive use of nucleotide analogs in crystallographic studies (12,13). Examination of such structures indicates that differences indicative of a particular conformational state are largely concentrated in local regions responsible for effector and modulator binding, namely, switch 1 (residues 25–40) and switch 2 (residues 57–75) (12–14). The apparent lack of structural changes in the C-terminal portion of the structure that, in the context of the biologically active full-length protein, leads to the membrane interacting C-terminus is intriguing because membrane binding has been shown to be modulated by the nature of the bound nucleotide (15). The structural and thermodynamic basis for the coupling between these distinct functional surfaces remains undetermined, and cannot be easily explained by the local induced fit mechanism.
Principal component analysis of available crystal structures of Ras and Rho, shown in previous work (14,16,17) to be successful in succinctly describing important conformational phenomena (see Fig. 1 and Movie S1 in the Supporting Material), support the conclusion that distinct chemical species in the active site are correlated with distinct global conformations (Fig. 1). However, there are notable exceptions to this general trend. For example, the Rho-GTP bound structure 2GCO most closely resembles other Rho GDP-bound structures (rather than any other GTP bound structures). These exceptions to the induced fit model become more striking when one considers the related kinesin and myosin protein families. Here crystal structures with similar global conformations have little correlation to the nucleotide that is bound in the active site of the respective structures (Fig. S2).
Figure 1.
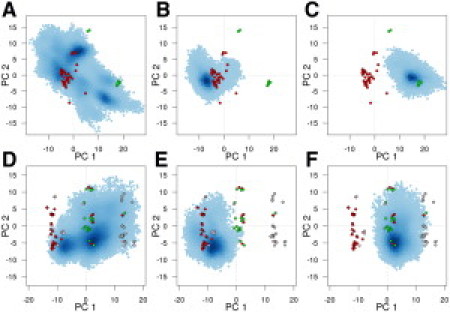
Ras (A–C) and Rho (D–F) conformations from crystallography and simulation. Crystallographic GTP conformers are colored red, GDP green, and nucleotide free conformers gray. The distribution of MD conformers is depicted with density-shaded blue points from (A) Ras nucleotide free, (B) Ras GTP-bound, (C) Ras GDP-bound, (D) Rho nucleotide free, (E) Rho GTP-bound, and (F) Rho GDP-bound simulations.
Also shown in Fig. 1 are the results of novel accelerated molecular dynamics (aMD, see the Supporting Material) simulations of nucleotide free Rho and Ras indicating the conformational subspace accessible to each family. Note the intrinsic susceptibility of nucleotide free simulations to sample multiple conformations (Fig. 1, A and D, and see Table S1 in the Supporting Material). In particular, these simulations sampled regions populated by the major GTP and GDP crystal structures highlighting the ability to interconvert between conformations in the absence of nucleotide (preexisting equilibrium). In the presence of GDP or GTP, aMD simulations under the same conditions resulted in restricted sampling in regions around the corresponding cluster of crystallographic structures (conformational selection) (Fig. 1, B, C, E, and F). This finding that nucleotide free simulations of Ras and Rho sample multiple states whereas nucleotide bound simulations sample a more restricted range of conformers is consistent with our previous studies that have demonstrated that swapping GTP for GDP (and vice versa) in simulations led to transitions between nucleotide states (16). Furthermore, point mutations in Ras such as G12V and A59G that populate intermediate regions of the conformational landscape are particularly susceptible to transitions (14,18,19). Together these results indicate that these families operate predominantly via a nucleotide-induced conformational shift mechanism rather than a conventional induced fit mechanism.
The pattern of correlated motions revealed by aMD simulations of Rho and Ras representatives predict coupling of the nucleotide-binding site with the membrane interacting C-terminus. These correlated motions link the active site to the C-terminus via loop 3 (Fig. 2 A). Loop 3 and the C-terminus of α5 were recently proposed to constitute a third switch responsible for a nucleotide-dependent membrane orientation of full-length Ras (20). To further probe this conserved coupling we carried out unaccelerated MD simulations on a D47A/E49A double mutant of Ras (Fig. 2 B). These loop 3 mutant simulations display a greater flexibility in the switch 1 and switch 2 regions. The results provide further evidence that Loop 3 may represent an important allosteric site that can be disrupted via mutations, which in turn affect the flexibility of the nucleotide-binding site (Fig. 2 B). This is in line with several biophysical and crystallographic studies that provided strong evidence for the allosteric modulation of Ras function (20,21). For example, the D47A/E49A mutation was found to enhance MAP kinase activation by stabilizing active H-Ras in a more productive membrane orientation (20), whereas binding of calcium acetate at a remote site resulted in a shift in helix 3/loop 7 and a network of H-bonding interactions that led to the ordering of switch 2 (21).
Figure 2.
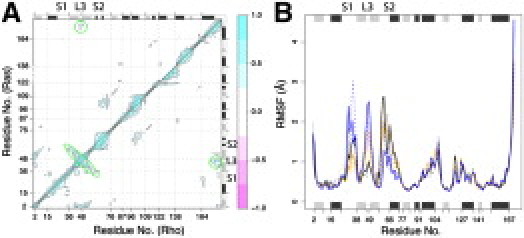
Wild-type and mutant simulations suggest that loop 3 (L3) is a potential allosteric site. (A) Correlated motions during wild-type Ras (upper triangle) and Rho (lower triangle) simulations. Correlations with L3-S1-S2 are highlighted with dashed green ovals. (B) Simulations with L3 D47A/E49A mutants (blue lines) display increased flexibility of SI and L3 over wild-type simulations (black lines). α5 R161A/R164A mutants (orange lines) are also shown. Dashed lines represent replicate simulations, two for each system (see the Supporting Material for details).
Taken together the accumulated computational and experimental evidence on the allosteric regulation of Ras function is consistent with our conclusion that conformational selection is the dominant mechanism by which small G-proteins accomplish their switching function. This finding is also consistent with the coupled motion of physically separated regions previously found in Ras and now found to be present in Rho. That coupled motions exist in identical regions in Ras and Rho is not necessarily expected, because residue identities are not conserved within the Ras superfamily. Furthermore, in this work we confirmed the importance of the previously highlighted correlated motions in H-Ras by mutant simulation results that clearly demonstrated the link between the semiconserved Loop3 amino acids with the fully conserved amino acids at the canonical switches.
In summary, atomistic molecular simulations indicate that both Rho and Ras superfamily members harbor an intrinsic susceptibility to sample multiple conformational states regardless of the bound nucleotide. Furthermore, the distribution of conformers in the absence of the nucleotide ligand suggests conformational selection to be the mechanism by which Rho and Ras undergo nucleotide-dependent conformational changes. However, a closer inspection of the sampled conformers suggests a role, albeit secondary, for induced fit. This involves the ligand's ability to organize the side chains once an initial favorable interaction has been established. Furthermore, these simulations, together with in silico mutations, provide evidence for a common preserved dynamic linkage, between the nucleotide-binding site and the distal C-terminus critical for membrane interaction. The apparent allosteric coupling of these functional sites is likely to exist in all monomeric G-proteins.
Acknowledgments
We thank Dr. Ana Rodrigues for valuable discussions, and acknowledge the Center for Theoretical Biological Physics, the National Science Foundation, National Institutes of Health, Howard Hughes Medical Institute, and National Biomedical Computation Resource.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Supporting Material
References and Footnotes
- 1.Bosshard H.R. Molecular recognition by induced fit: how fit is the concept? News Physiol. Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 2.Fisher E. Influence of configuration on the action of enzymes [Einfluss der konfiguration auf die wirkung der enzyme] Ber. Dt. Chem. Ges. 1894;27:2985–2993. [Google Scholar]
- 3.James L.C., Tawfik D.S. Conformational diversity and protein evolution—a 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- 4.Koshland D.E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma B., Kumar S., Nussinov R. Folding funnels and binding mechanisms. Protein Eng. 1999;12:713–720. doi: 10.1093/protein/12.9.713. [DOI] [PubMed] [Google Scholar]
- 6.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 7.Tsai C.J., Ma B., Nussinov R. Folding and binding cascades: shifts in energy landscapes. Proc. Natl. Acad. Sci. USA. 1999;96:9970–9972. doi: 10.1073/pnas.96.18.9970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition (vol 5, pg 789, 2009) Nat. Chem. Biol. 2009;5:954. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gunasekaran K., Ma B.Y., Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 10.Tobi D., Bahar I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA. 2005;102:18908–18913. doi: 10.1073/pnas.0507603102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou H.X. From induced fit to conformational selection: a continuum of binding mechanism controlled by the timescale of conformational transitions. Biophys. J. 2010;98:L15–L17. doi: 10.1016/j.bpj.2009.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vetter I.R., Wittinghofer A. The guanine nucleotide-binding switch in three dimensions. Science. 2001;294:1299–1304. doi: 10.1126/science.1062023. [DOI] [PubMed] [Google Scholar]
- 13.Sprang S.R. G proteins, effectors and GAPs: structure and mechanism. Curr. Opin. Struct. Biol. 1997;7:849–856. doi: 10.1016/s0959-440x(97)80157-1. [DOI] [PubMed] [Google Scholar]
- 14.Gorfe A.A., Grant B.J., McCammon J.A. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885–896. doi: 10.1016/j.str.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rotblat B., Prior I.A., Hancock J.F. Three separable domains regulate GTP-dependent association of H-Ras with the plasma membrane. Mol. Cell. Biol. 2004;24:6799–6810. doi: 10.1128/MCB.24.15.6799-6810.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grant B.J., Gorfe A.A., McCammon J.A. Ras conformational switching: simulating nucleotide-dependent conformational transitions with accelerated molecular dynamics. PLOS Comput. Biol. 2009;5:e1000325. doi: 10.1371/journal.pcbi.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant B.J., McCammon J.A., Cross R.A. Multivariate analysis of conserved sequence-structure relationships in kinesins: coupling of the active site and a tubulin-binding sub-domain. J. Mol. Biol. 2007;368:1231–1248. doi: 10.1016/j.jmb.2007.02.049. [DOI] [PubMed] [Google Scholar]
- 18.Grant B.J., Gorfe A.A., McCammon J.A. Large conformational changes in proteins: signaling and other functions. Curr. Opin. Struct. Biol. 2010;20:142–147. doi: 10.1016/j.sbi.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lukman S., Grant B.J., McCammon J.A. The distinct conformational dynamics of K-Ras and H-Ras A59G. PLOS Comput. Biol. 2010;6:e1000922. doi: 10.1371/journal.pcbi.1000922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abankwa D., Hanzal-Bayer M., Hancock J.F. A novel switch region regulates H-Ras membrane orientation and signal output. EMBO J. 2008;27:727–735. doi: 10.1038/emboj.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buhrman G., Holzapfel G., Mattos C. Allosteric modulation of Ras positions Q61 for a direct role in catalysis. Proc. Natl. Acad. Sci. USA. 2010;107:4931–4936. doi: 10.1073/pnas.0912226107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.