

Abstract
Fungi and bacteria secrete glycoprotein cocktails to deconstruct cellulose. Cellulose-degrading enzymes (cellulases) are often modular, with catalytic domains for cellulose hydrolysis and carbohydrate-binding modules connected by linkers rich in serine and threonine with O-glycosylation. Few studies have probed the role that the linker and O-glycans play in catalysis. Since different expression and growth conditions produce different glycosylation patterns that affect enzyme activity, the structure-function relationships that glycosylation imparts to linkers are relevant for understanding cellulase mechanisms. Here, the linker of the Trichoderma reesei Family 7 cellobiohydrolase (Cel7A) is examined by simulation. Our results suggest that the Cel7A linker is an intrinsically disordered protein with and without glycosylation. Contrary to the predominant view, the O-glycosylation does not change the stiffness of the linker, as measured by the relative fluctuations in the end-to-end distance; rather, it provides a 16 Å extension, thus expanding the operating range of Cel7A. We explain observations from previous biochemical experiments in the light of results obtained here, and compare the Cel7A linker with linkers from other cellulases with sequence-based tools to predict disorder. This preliminary screen indicates that linkers from Family 7 enzymes from other genera and other cellulases within T. reesei may not be as disordered, warranting further study.
Introduction
Glycosylation is the covalent addition of polysaccharides to protein side chains, and is one of the most prevalent posttranslational modifications of proteins. Glycosylation serves many roles, such as facilitating recognition, imparting protease resistance, providing thermal stability, improving solubility, and modulating the rates of biological processes (1–3). Although the complex, heterogeneous nature of protein glycosylation makes it difficult to achieve a molecular-level understanding of the biochemical processes involved, this remains an area of active research (3–5).
Fungi and bacteria degrade cellulose and chitin with cocktails of synergistic enzymes, many of which contain N- and O-linked glycans (6,7). Because biomass is a vast source of fuels and products, elucidating the natural paradigms of carbohydrate conversion will enable the design of improved enzymes and enhance our understanding of how bacteria and fungi evolved with plants. The fungus Trichoderma reesei (Hypocrea jecorina) is one of the most thoroughly studied organisms that degrade plant cell walls (8). T. reesei secretes a cocktail of enzymes for cell wall deconstruction and is a common platform for enzyme production in biofuel processes. Studies in T. reesei have shown that the expression host and growth conditions change glycosylation patterns, thereby affecting enzyme activity (9–15). Gaining a molecular-level understanding of the means by which cellulases degrade crystalline cellulose, including the role of glycosylation in enzyme activity, will lead to an improved understanding of enzymatic routes for biomass conversion and to rational strategies for activity improvements (16).
The most abundantly produced cellulase from T. reesei is the Family 7 cellobiohydrolase (Cel7A), which is a multidomain, processive enzyme (Fig. 1). Cel7A consists of a carbohydrate-binding module (CBM) and a catalytic domain (CD) decorated with N-glycosylation (blue) connected by a linker with substantial O-glycosylation (yellow) (17–19). Cel7A is hypothesized to extract single chains from cellulose, thread the chain into the CD tunnel, and employ hydrolysis to cleave the chain at every cellobiose unit (18).
Figure 1.
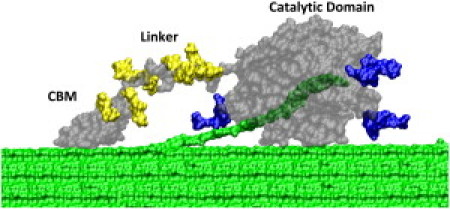
Cel7A from Trichoderma reesei on cellulose. The conformation is from Zhong et al. (63) with added N- and O-glycan. The enzyme is shown in gray, the O-glycosylation on the linker is in yellow, and the N-glycan on the CD is in blue. Cellulose is in green, with a single cellodextrin chain threaded into the CD tunnel. The pictures were prepared with VMD (64).
Because Cel7A provides most of the hydrolytic potential in T. reesei cocktails, many studies have aimed to elucidate the roles of each subdomain in Cel7A. The crystal structure of the CD has been solved (17,18), and mutagenesis has been conducted on the CD residues deemed relevant for catalysis and function (20,21). Various studies have demonstrated that expression host and growth conditions affect glycosylation (9–15). The CBM structure has been solved (19), and several studies have ascertained the role of specific residues (22,23). Additionally, on the basis of computational studies, our group predicted new functions of the Cel7A CBM (24–26) that are not readily accessible to experiment.
The Cel7A linker, however, has received less attention to date than the CBM and CD, and its role in catalysis is unknown. The Cel7A linker contains O-glycosylation, as shown in Figs. 1 and 2. The linker sequence and glycosylation pattern shown in Fig. 2 is one of the glycan patterns suggested in a study by Harrison et al. (11), in which they quantified the extent and heterogeneity of mannose residues. They reported that the S and T residues in the linker have O-glycosylation, with a range of mannoses between 1 and 3 on each site.
Figure 2.

Cel7A linker domain sequence, with the O-glycan shown in red. Adapted from Harrison et al. (11).
Cellulase linkers have been hypothesized to serve several functions, such as acting as a hinge between the CBM and the CD (27), as a torsional leash (11,27), as a spring (28), and as a driver of the relative orientations of the CBM and CD. Additionally, the glycosylation has been hypothesized to prevent proteolysis (29). Several studies have been conducted to characterize cellulase linkers from T. reesei (27,30–32) and other organisms (28,33–38). Early small-angle x-ray scattering (SAXS) studies on the Cel7A linker (30–32) suggested that the Cel7A and Cel6A enzymes adopt an extended conformation in solution.
In another study concerning the T. reesei Cel7A linker, Srisodsuk et al. (27) constructed two Cel7A mutants: one with a putative hinge (G1–G10) removed, and one with the entire linker (G1–P27) removed. The authors hypothesized that the putative hinge provides flexibility (from G) and length (from P), and that the glycosylated, stiff region provides the linker with rigidity to maintain sufficient extension between the CBM and CD. They also hypothesized that if the flexible portion of the linker is removed, the CBM and CD must bind simultaneously to hydrolyze cellulose. However, for the hinge (G1–G10) mutant, activity on crystalline cellulose was not affected, whereas the binding capacity at high cellulase loading was reduced. This result is incompatible with the loss of flexibility as hypothesized. Thus, the experimental results were not explained (we address this issue further below). In the mutant in which the entire linker was removed, the enzyme did bind to cellulose, but the activity was significantly reduced (27).
Additional studies examined glycosylation from other cellulases from bacteria and fungi (33–38). Boisset et al. (37) examined the endoglucanase V from Humicola insolens with light scattering and determined that the linker adopts a conformation with an average of 2 Å per residue. Receveur et al. (36) examined the Family 45 cellulase from H. insolens, which has a 36-residue linker with ∼1.7 sugars per S or T residue, and used SAXS to measure the shape of the protein and several mutants. Their results indicate that the linker is extended and does not wrap around the CD. By examining an enzyme sans CBM, they found that the CBM does not alter the linker conformation. These results are important because they suggest that if the results are extendable to T. reesei Cel7A, studies of the conformational states of the linker alone will be relevant in terms of the conformational states of the enzyme with the CBM and CD.
Subsequently, von Ossowski et al. (28) constructed a double-headed cellulase from the two GH Family 6 cellulases from H. insolens connected by an 88-residue linker. On the basis of the SAXS results, the authors suggested that the linkers from these cellulases adopt random conformations. Most importantly, they noted that the linker can unwind to extended conformations with a low energetic cost. Additionally, the authors hypothesized that O-glycans drive the equilibrium distances between subdomains toward greater extension—a hypothesis that is addressed directly here.
Poon et al. (33) used NMR spectroscopy to examine the 20-residue linker from the Family 10 xylanase from Cellulomonas fimi. The C. fimi xylanase linker is composed of only two types of residues: P and T. Their results demonstrated that the linker samples different conformational states on picosecond-to-nanosecond timescales, which aligns with previous findings that linkers can unwind with relative ease (28). The authors also noted that glycosylation dampens the linker mobility in solution. In an additional study on linkers as they relate to cellulose deconstruction, Noach et al. (38) crystallized four cohesins from Acetivibrio celluloyticus with three short linkers of 5–6 residues and one linker with 45 residues. In all cases, they found significant conformational diversity of the linkers in the crystal structures, which suggests that the linkers adopt varied conformations in solution.
The results from two simulation studies that examined the Cel7A linker (39,40) have been interpreted as contradictory (41). Zhao et al. (39) used constrained molecular dynamics (MD) to study the free energy along a reaction coordinate (RC) described by the end-to-end distance of the linker above a cellulose slab. The RC was chosen to test the inchworm mechanism hypothesis (36). With 160 windows of 2 ns each, it was observed that the linker exhibits a significant free-energy barrier of ∼35 kcal/mol around 3.5 nm. However, Zhao et al. (39) reported that convergence along the RC is difficult to measure because the convergence of the simulations with a low-dimensional RC is a potential source of error in the free-energy profile. In another study, Zhong et al. (40) ran a 1.5 ns MD simulation of Cel7A on cellulose and stated that in this case the linker was quite flexible; however, the short simulation time used in that study is not adequate to make such a claim.
Ting et al. (41) constructed a kinetic model for the action of a processive cellulase, in which they modeled the CBM and CD as random walkers coupled via a spring for the linker. Their results indicate that the stiffness (as previously suggested by von Ossowski et al. (28)) and linker length are key to the hydrolysis rate and the intrinsic rate of hydrolysis of the CD. Although these results were obtained from a model with no atomistic details, they suggest that a molecular-level understanding of both the linker and the CD are important for improving cellulase activity. Additionally, Ting et al. (41) noted the lack of a reliable end-to-end distance distribution of a cellulase linker, which is provided here.
In this work, we studied the impact of O-glycosylation on the T. reesei Cel7A linker. The hypothesis examined here is that the glycosylated linker will adopt more-extended conformations because of excluded volume arising from glycosylation. To test this hypothesis, we conducted extensive simulations of the Cel7A linker with and without glycosylation. An implicit solvent model was used for enhanced sampling (42–44), and two methods (MD simulation and replica exchange MD (REMD)) were used to sample the linker conformations. We show that O-glycosylation affects the equilibrium distance of the linker but has no effect on the flexibility measured as relative fluctuations in the end-to-end distance distribution. Additionally, we show that both the putative hinge and stiff regions identified by Srisodsuk et al. (27) are flexible with and without glycosylation. We explain the results of Srisodsuk et al. (27) regarding the stiffness of the glycosylated region; specifically, we explain the reduction in binding affinity at high enzyme loading while maintaining wild-type activity by quantifying the flexibility of the stiff region. We provide quantitative measures of flexibility and equilibrium length for the linker, which are key parameters for coarse-grained models. Several parameters are given for comparisons with experimental measurements. On the basis of our results, the Cel7A linker is designated as an intrinsically disordered protein (IDP) (45–49). Sequence-based tools are applied to linkers from other cellulases from T. reesei (8) and to other Family 7 cellobiohydrolases to determine the likelihood of disorder among cellulase linkers. The sequence-based calculations warrant further detailed study; however, they suggest that linkers from other T. reesei cellulases may be more ordered than the Cel7A linker, which may have ramifications for catalysis.
Methods
The simulations described here were conducted with CHARMM (50). The CHARMM27 force field (50) with CMAP (51) was used for the peptide, and the C35 force field was used for the glycosylation (52). The O-glycosylation pattern used here was suggested in a previous study (11) and is shown in Fig. 2. The carbohydrates are linked by α1,2 bonds, as suggested by Deshpande et al. (5). The protein-carbohydrate bond model was described previously (40). The generalized Born model with molecular volume (GBMV2) was used as the implicit solvent model (42,53). The GBMV2 model circumvents the refitting of GB parameters for the C35 force field. The parameters for the GBMV2 model were as described previously (42). The linker was simulated with capped termini. All MD simulations were conducted with a 1.5 fs timestep, SHAKE for the covalent bonds to hydrogen (54), a cutoff for nonbonded interactions of 18 Å, and Langevin dynamics at 300 K. For the nonglycosylated and glycosylated linkers, 360 ns of MD simulation were collected. For the REMD simulations, the temperatures were distributed exponentially between 300 and 550 K for both cases. Twelve replicas were used for the nonglycosylated linker, and 16 replicas were used for the glycosylated linker. The number of replicas was tuned to achieve acceptance rates of 40–50%. For both linkers, the replicas were equilibrated for 150 ps before swapping. Swaps were attempted every 3 ps as described previously (55); 40,000 swaps were attempted for 120 ns for each linker. Clustering algorithms were used to determine whether stable structures were present (56). We varied the cluster counts from 2 to 30 using previously described metrics (56). The trajectories were oriented based on the root mean-square deviation for the protein backbone.
We also characterized the end-to-end distance (R), the end-to-end distance of the putative hinge (Rhinge), the end-to-end distance of the putative stiff region (Rstiff), and the mass-weighted radius of gyration (Rg). The end-to-end distance is from the α carbon on residue 1 (G) to the α carbon on residue 27 (P). The putative hinge region identified in Srisodsuk et al. (27) is from the α carbon on residue 1 (G) to residue 10 (G), and the stiff region is from residue 11 (T) to residue 27 (P). The potential of mean force (PMF) is calculated for each of these order parameters by binning the simulation results in 1 Å bins and converting the resulting histograms to relative free energies by computing F/kT = −ln[r] + C, where F = PMF, k = Boltzmann's constant, T = temperature, r = bin value, and C is a constant. See the Supporting Material for an additional discussion of convergence metrics (57) and a bootstrapping error analysis for the free-energy curves.
We employed several algorithms contained in the Prediction of Naturally Disordered Regions (PONDR) suite (Molecular Kinetics, Indianapolis, IN) (45–48) for predicting protein disorder, including the VL3 algorithm, which is a feed-forward neural network trained on 152 disordered proteins characterized by various methods (46). Also, the charge-hydrophobicity relationship previously described by Uversky et al. (58) was used to predict relative disorder in linker regions from other cellulases.
Results
The autocorrelation time of the end-to-end distances of the glycosylated and nonglycosylated linkers is 2–3 ns in implicit solvent (Fig. S1). For an MD simulation in explicit water, the autocorrelation time is >5 ns. Because simulations should be conducted to collect multiple autocorrelation times of variables of interest, this implies that the results of Zhao et al. (39) and Zhong et al. (40) are not converged. Before we performed the implicit solvent simulations in this study, we conducted MD umbrella sampling simulations in explicit solvent. Even though the total simulation time was >1 μs, the results did not converge. This suggests that using low-dimensional RCs to sample high-dimensional space for this particular problem is not computationally tractable with explicit solvent, unless one can conduct significantly longer simulations than those used previously (39,40).
Fig. 3 shows the relative free energy as a function of the end-to-end distance (R) from the REMD simulations. We report the free energy in kT units. The free energy as a function of R for the nonglycosylated linker exhibits a minimum at 37 Å. The glycosylated linker exhibits a minimum at 53 Å. Fig. 3 indicates that the most likely configuration for the glycosylated linker is at a 16 Å extension over the nonglycosylated linker from the excluded volume imparted by the O-glycan, as hypothesized. However, the linker is quite flexible in both cases measured by the stiffness of the linker defined by the shape of the distribution fitted to Hooke's law (i.e., a spring constant). With this definition of linker stiffness, the linker flexibility is not changed by O-glycosylation because the spring constants for the nonglycosylated and glycosylated linkers are equivalent (Table 1). The results shown in Fig. 3 are provided in Table S1 and Table S2. Table 1 provides the fitted parameters, namely, R0 and the spring constant k for both linkers, which is fit by minimizing the sum of square error between Hooke's law and the results in Fig. 3. At the extremes of the end-to-end distances sampled, the free energy to extension is ∼8–10 kT, which is readily accessible on the nanosecond timescale. Experimental methods can be applied to validate the results shown in Fig. 3, including SAXS on the whole Cel7A enzyme and Förster resonance energy transfer on the linker alone.
Figure 3.
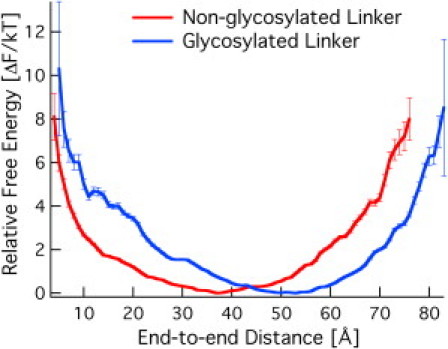
Relative free energy as a function of the end-to-end distance (R) from the REMD simulations for both glycosylated and nonglycosylated linkers from T. reesei Cel7A. R0 for the nonglycosylated linker is 37 Å, and R0 for the glycosylated linker is 53 Å.
Table 1.
Equilibrium end-to-end distance (R0) and spring constant (k) for the Cel7A linkers
| Linker | k (kT/ Å2) | R0 [Å] |
|---|---|---|
| Nonglycosylated | 5 × 10−2 | 37 |
| Glycosylated | 4 × 10−2 | 53 |
Spring constants are nearly equal, but the equilibrium length is 16 Å more extended with glycosylation.
We hypothesized that glycosylation would induce an excluded volume effect and provide extension. Another measure of the extension is the free energy as a function of protein side-chain contacts. In the Supporting Material, we provide the two-dimensional free-energy surfaces for the Cel7A linkers as a function of protein side-chain contacts and end-to-end distance. The glycosylated linker forms fewer side-chain contacts, and the free-energy minimum is located at a more extended set of conformations with fewer contacts (Fig. S2).
The relative free energy as a function of the mass-weighted Rg of both linkers is shown in Fig. 4. The Rg for the glycosylated Cel7A linker is significantly narrower than the nonglycosylated linker. The narrower distribution in Rg results from the fact that the glycans are distributed around the center of the sequence (Fig. 2).
Figure 4.
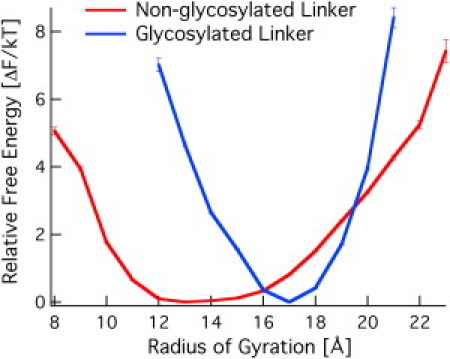
Relative free energy as a function of the mass-weighted Rg for the glycosylated and nonglycosylated T. reesei Cel7A linkers. Rg for the glycosylated linker includes the glycans, and thus the distribution is narrower because the sugars are close to the center of the peptide.
As noted previously by Srisodsuk et al. (27), the Cel7A linker exhibits a region from residues G1–G10 with four G residues and four P residues. The authors identified this as a putative hinge because of the inherent flexibility of G for rotation and the stiffness of the P residues. They hypothesized that the region from T11 to P27 is stiff because of glycosylation. Thus, they posited that removal of the hinge will reduce the conformational freedom of the CBM and CD, and thus influence the activity. However, they demonstrated that upon removal of this region, activity on crystalline cellulose was not affected, and the binding capacity at high enzyme loadings was reduced (27). Upon removal of the entire linker, activity on crystalline cellulose was significantly reduced. To explain their observations, we examined the end-to-end distance distributions of the putative hinge and stiff regions (Rhinge and Rstiff,, respectively). Fig. 5 shows the free energy for both linkers for both sections of the linker. As shown in Fig. 5 a, the presence of glycan has no effect on the hinge, which is stabilized at 16 Å. The putative stiff region, however, is significantly impacted in terms of the equilibrium distance, and it is this region that primarily gives rise to the differences seen in Fig. 3.
Figure 5.
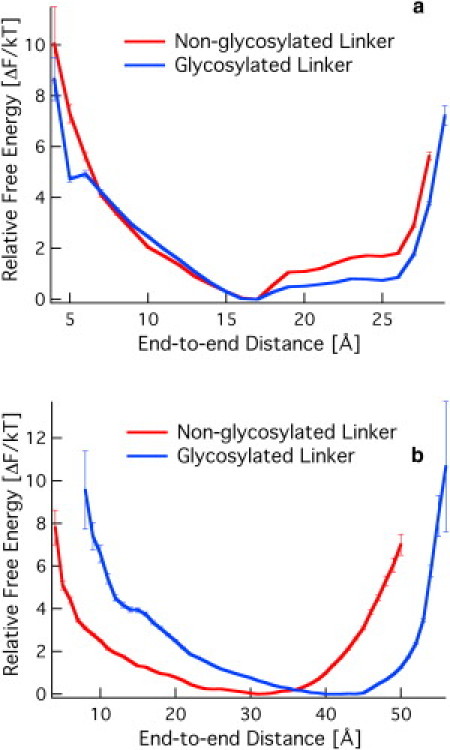
Relative free energy as a function of the end-to-end distance for (a) the putative hinge (Rhinge) and (b) the glycosylated stiff region (Rstiff).
The REMD and MD simulations were analyzed for structural states and to examine the local effects of glycosylation on the intramolecular flexibility of each residue. Ramachandran maps were assembled for each residue in the linker (Fig. S3). The differences in the Ramachandran angles are seen near the glycosylation sites; specifically, T13, T14, and R15 exhibit helical content in the nonglycosylated linker, whereas there is no helical content in the glycosylated linker. It was not known a priori whether secondary structure is present in the Cel7A linker, but previous studies on other linkers suggest that cellulase linkers have no secondary structure (28,33,36,37). Additionally, from the results presented in Figs. 3–5, we expected that no long-lived secondary structure would be formed. The only secondary structure found from the REMD simulations is a small α-helix from residues 9–15 (Fig. S4). However, this helix readily exchanges with other conformations, suggesting that it is not thermodynamically more favorable than an extended conformation. In the glycosylated REMD simulations, no secondary structure is observed. Additionally, a clustering analysis was conducted with multiple algorithms. Based on three clustering metrics, the Davies-Bouldin index, the pseudo-F statistic, and the ratio of the sum-of-squares regression to the total sum of squares, we conclude that there is no optimal cluster size (Fig. S7). On the basis of the simulations, this is expected because of the conformational flexibility observed in the linker. Fig. S8 shows the conformations of both linkers.
Because of the absence of secondary structure for the T. reesei Cel7A linker tested via exhaustive simulations, we designate it as an IDP (45–49). Over the last decade, there has been significant interest in describing IDPs, and tools have been developed to predict intrinsic disorder based on sequence. From the predictions obtained here for the Cel7A linker, we conducted a sequence-based analysis of the entire Cel7A cellulase to determine whether available algorithms would select the linker region as the most disordered portion of the enzyme, and to ascertain the applicability of these tools for distinguishing classes of cellulases based on their linker flexibilities. The VL3 algorithm within the PONDR software (45–48) was applied to T. reesei Cel7A. The VL3 algorithm predicts disordered regions with a VL3 PONDR score > 0.5. The linker from T. reesei Cel7A is predicted to be a disordered region (Fig. S9 a).
Because the PONDR algorithm identifies the Cel7A linker as disordered, we conducted a similar analysis for the Family 6 cellulase (Cel6A) and the four endoglucanases from T. reesei that have linkers, i.e., Family 7 endoglucanase I (Cel7B), Family 5 endoglucanase II (Cel5A), Family 61 endoglucanase IV (Cel61A), and Family 45 endoglucanase V (Cel45A). The sequences were obtained from the CAZy database (59). The results are provided in the Supporting Material and illustrate that the PONDR algorithm predicts the linkers of the other cellulases from T. reesei to be disordered regions. Additionally, we screened a library of 11 other processive Family 7 cellobiohydrolases with the VL3 algorithm, and found that the PONDR algorithm identifies the linker region in each one as an intrinsically disordered region. These results are shown in the Supporting Material. An additional metric of protein disorder is the charge-hydropathy scale (58). The basis of this algorithm is that at high absolute net charge and low hydropathy (or hydrophobicity), a protein is likely to be disordered, and vice versa at low absolute net charge and high hydropathy. The hydropathy is measured by means of the Kyte-Doolitle scale (60). Fig. S14 shows the absolute mean net charge as a function of mean scaled hydropathy for Cel7A, Cel6A, and the four endoglucanases from T. reesei. The Cel7A linker is predicted to be more disordered than the other linkers in T. reesei cellulases. It should be noted that the charge-hydropathy relationship is calculated only for the linker section. The linker regions were chosen from the known sequences and are provided in the Supporting Material.
Fig. S14 shows the charge/hydropathy relationship for additional processive Family 7 cellulases from other organisms. These results show two primary clusters, and the linkers from the Trichoderma genera (Hypocrea koningii, T. viride, and T. reesei) are predicted to be more disordered than linkers from other cellulases. The entire sequences of the Cel7A enzymes shown in Fig. S14 are listed in the Supporting Material. The primary sequence differences in the Family 7 cellulase linkers from the Trichoderma genera relative to the other linkers is the presence of one (T. viride), two (T. reesei), or three arginines (H. koningii) with few to no hydrophobic residues.
Discussion
Experimental implications
The results presented here demonstrate that the Cel7A linker is highly flexible, making it an IDP, both with and without glycosylation (45,46,49). Glycosylation provides excluded volume for extension and prevents the formation of secondary structure. The end-to-end distance distribution shown in Fig. 3 indicates that the Cel7A linker can change conformation rapidly. Few long-lived hydrogen bonds exist in either case. Overall, the results presented here demonstrate that the CBM and CD of Cel7A likely have significant conformational freedom relative to one another, and a relative operating range of ∼10–80 Å on the cellulose surface. Coupled with previous results, this suggests that the CD has significant freedom to search for an available reducing end of cellulose, whereas the CBM anchors Cel7A in discrete 1 nm wells on the hydrophobic face of cellulose (25,26,61).
An important aspect of the data presented here is that the results do not support a two-state model of the Cel7A linker, which is related to the inchworm mechanism hypothesis (36). Although our results do not directly probe the behavior of the linker on cellulose, given the flexibility of the linker and the size of the CBM and CD, it is likely that the linker remains ∼≥10 Å above the cellulose surface while Cel7A is conducting catalysis. We hypothesize that, geometrically, the linker will have few interactions with cellulose, and thus the presence of a cellulose surface will likely not impact the linker flexibility in such a way as to give rise to two metastable states.
Srisodsuk et al. (27) described the activity observed after two changes to the Cel7A linker. The authors hypothesized that the portion of the Cel7A linker closest to the CD is a hinge, and that the glycosylated portion is stiff. Removal of the putative hinge, however, did not affect enzyme activity and reduced the binding affinity at high enzyme loadings. This result is incompatible with the hypothesis of Srisodsuk et al. that removal of the hinge would affect flexibility and thus impact activity. The results presented here indicate that the putative stiff region is actually quite flexible. Fig. 5 demonstrates that removal of the putative hinge region does not reduce the flexibility of the remaining linker region, and only reduces the maximum operating range from 7–8 cellobiose units to 5–6 cellobiose units. This in turn explains the observation of reduced binding affinity at high enzyme loading. Because the hinge linker is shorter, the distance between the CBM and CD is shorter. At high enzyme loadings, many of the enzymes will be bound to cellulose via CBM-cellulose interactions and will crowd the surface. With many enzymes bound to cellulose via their CBMs, and shorter (yet still flexible) linkers in the hinge mutant, the available volume to pack the same molar amount of a less extended enzyme is reduced because of the shortened tether from the CBM to the CD. This result suggests a further experiment: If the nonglycosylated Cel7A linker is indeed less extended than the glycosylated linker, as shown in Fig. 3, we predict that removal of the O-linked glycans on the Cel7A linker (perhaps enzymatically) will not significantly impact activity, but the binding affinity may be reduced at high enzyme loadings, as previously observed by Srisodsuk et al. (27).
To the extent of the glycosylation studied here on the Cel7A linker, the stiffness is not altered, as measured by the relative fluctuations in R. We note that more glycosylation could change the linker stiffness, which is now being probed computationally. Higher extents of glycosylation or glycans bonded via different types of linkages could be obtained experimentally by using a different expression host (9,10), different growth conditions, or different T. reesei strains (13,14). Further work is needed to measure the glycosylation extents and linkage types imparted to the Cel7A linker by these variables.
Ting et al. (41) suggested that changes in the Cel7A linker stiffness can affect enzyme activity, and demonstrated that changes in the linker stiffness impact the hydrolysis rate. They noted that there is no reliable measure of the stiffness and equilibrium distance for a cellulase linker, which we have provided here for one of the most relevant cellulases. This will allow direct modeling of the Cel7A cellulase with their model (41). The results of our study will also enable investigators to make in silico mutations on the Cel7A linker to ascertain differences in protein flexibility, and to determine whether such differences or the addition of more glycans changes the inherent flexibility of the linker or the maximum operating range.
The results presented in the Supporting Material demonstrate that there may be a difference in the Cel7A linker relative to the endoglucanases and the Cel6A linker from T. reesei and the linkers from other organisms. In particular, the presence of arginine in the Cel7A linkers from Trichoderma species and the lack of hydrophobic residues lead to predictions of greater disorder. These observations warrant further study, both to validate the accuracy of the sequence-based charge-hydropathy measure of protein disorder and to probe the relative structure-function differences in fungal cellulase linkers.
Computational implications
Computationally, our approach provides a general means of examining small IDPs and glycoproteins. The limitations of this study primarily center on the use of an implicit solvent model. It is not possible to determine whether atomistic water structuring plays a role in stabilizing a given conformation. However, we note that the GBMV2 method is the most accurate implicit solvent method available for predicting the solvation-free energies of proteins and obtaining thermodynamically accurate descriptions of energy landscapes relative to explicit solvent and experiment (43,44,62). As demonstrated previously by some of the authors of this study (39,40,63), and as shown in Fig. 3, it is difficult to adequately sample the conformational space in explicit solvent, and such an approach may take many tens of microseconds in explicit water. The use of implicit solvent, however, makes it possible to achieve enhanced conformational sampling at a fraction of the cost for explicit solvent.
Conclusions
In this study, we conducted thorough MD simulations to probe the molecular-level characteristics of the Cel7A linker. The salient points of this study are as follows:
-
1.
The T. reesei Cel7A linker is an IDP with significant flexibility with or without O-glycans.
-
2.
If the Cel7A CBM anchors the entire enzyme on the hydrophobic cellulose surface in 1 nm increments, as predicted in previous studies (25,26), the glycosylated linker will enable the CD to search for reducing ends on the cellulose surface with a maximum operating range of 7–8 cellobiose units with the native glycosylation pattern.
-
3.
The biochemical results from the study by Srisodsuk et al. (27), in which the putative hinge region was removed, can be explained in terms of a reduction in operating range in the flexible linker peptide, not by changes in flexibility as originally hypothesized.
-
4.
Two parameters for coarse-grained modeling of Cel7A—the spring constant (k) and equilibrium distance (R0) for the linker—are provided for both linkers.
-
5.
Multiple measures for comparison with experiments, including the Rg (SAXS), end-to-end distance distributions (Förster resonance energy transfer), and hydrogen-bonding patterns and transient secondary structure prediction (NMR), are provided.
-
6.
The T. reesei Cel7A linker may be more disordered than other cellulases from the same organism, as well as other processive Family 7 cellobiohydrolases from other genera.
Acknowledgments
We thank Scott Shell, Michael Shirts, and Vladimir Uversky for helpful discussions, and Alan Grossfield for the use of his bootstrapping code for error analysis.
This study was supported by the U.S. Department of Energy Office of the Biomass Program and the Sweden-America Foundation (J.W. and M.B.). Computational time for this research was provided in part by the Texas Advanced Computing Center Ranger cluster under National Science Foundation Teragrid grants MCB090159 and MCB080117N, from resources provided by the National Institute of Computational Sciences. Resources were also provided by the National Energy Research Scientific Computing Center, supported by the Office of Science of the Department of Energy under Contract No. DE-AC02-05CH11231.
Contributor Information
Gregg T. Beckham, Email: Gregg.Beckham@nrel.gov.
Michael F. Crowley, Email: Michael.Crowley@nrel.gov.
Supporting Material
References
- 1.Varki A. Biological roles of oligosaccharides: all of the theories are correct. Glycobiology. 1993;3:97–130. doi: 10.1093/glycob/3.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spiro R.G. Protein glycosylation: nature, distribution, enzymatic formation, and disease implications of glycopeptide bonds. Glycobiology. 2002;12:43R–56R. doi: 10.1093/glycob/12.4.43r. [DOI] [PubMed] [Google Scholar]
- 3.Goto M. Protein O-glycosylation in fungi: diverse structures and multiple functions. Biosci. Biotechnol. Biochem. 2007;71:1415–1427. doi: 10.1271/bbb.70080. [DOI] [PubMed] [Google Scholar]
- 4.Wormald M.R., Petrescu A.J., Dwek R.A. Conformational studies of oligosaccharides and glycopeptides: complementarity of NMR, X-ray crystallography, and molecular modelling. Chem. Rev. 2002;102:371–386. doi: 10.1021/cr990368i. [DOI] [PubMed] [Google Scholar]
- 5.Deshpande N., Wilkins M.R., Nevalainen H. Protein glycosylation pathways in filamentous fungi. Glycobiology. 2008;18:626–637. doi: 10.1093/glycob/cwn044. [DOI] [PubMed] [Google Scholar]
- 6.Himmel M.E., Ding S.Y., Foust T.D. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science. 2007;315:804–807. doi: 10.1126/science.1137016. [DOI] [PubMed] [Google Scholar]
- 7.Eijsink V.G.H., Vaaje-Kolstad G., Horn S.J. Towards new enzymes for biofuels: lessons from chitinase research. Trends Biotechnol. 2008;26:228–235. doi: 10.1016/j.tibtech.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 8.Martinez D., Berka R.M., Brettin T.S. Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina) Nat. Biotechnol. 2008;26:553–560. doi: 10.1038/nbt1403. [DOI] [PubMed] [Google Scholar]
- 9.Adney W.S., Jeoh T., Himmel M.E. Probing the role of N-linked glycans in the stability and activity of fungal cellobiohydrolases by mutational analysis. Cellulose. 2009;16:699–709. [Google Scholar]
- 10.Jeoh T., Michener W., Adney W.S. Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnol. Biofuels. 2008;1:10. doi: 10.1186/1754-6834-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison M.J., Nouwens A.S., Packer N.H. Modified glycosylation of cellobiohydrolase I from a high cellulase-producing mutant strain of Trichoderma reesei. Eur. J. Biochem. 1998;256:119–127. doi: 10.1046/j.1432-1327.1998.2560119.x. [DOI] [PubMed] [Google Scholar]
- 12.Hui J.P.M., Lanthier P., Thibault P. Characterization of cellobiohydrolase I (Cel7A) glycoforms from extracts of Trichoderma reesei using capillary isoelectric focusing and electrospray mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2001;752:349–368. doi: 10.1016/s0378-4347(00)00373-x. [DOI] [PubMed] [Google Scholar]
- 13.Stals I., Sandra K., Claeyssens M. Factors influencing glycosylation of Trichoderma reesei cellulases. II: N-glycosylation of Cel7A core protein isolated from different strains. Glycobiology. 2004;14:725–737. doi: 10.1093/glycob/cwh081. [DOI] [PubMed] [Google Scholar]
- 14.Stals I., Sandra K., Claeyssens M. Factors influencing glycosylation of Trichoderma reesei cellulases. I: Postsecretorial changes of the O- and N-glycosylation pattern of Cel7A. Glycobiology. 2004;14:713–724. doi: 10.1093/glycob/cwh080. [DOI] [PubMed] [Google Scholar]
- 15.Godbole S., Decker S.R., Himmel M.E. Cloning and expression of Trichoderma reesei cellobiohydrolase I in Pichia pastoris. Biotechnol. Prog. 1999;15:828–833. doi: 10.1021/bp9901116. [DOI] [PubMed] [Google Scholar]
- 16.Wilson D.B. Cellulases and biofuels. Curr. Opin. Biotechnol. 2009;20:295–299. doi: 10.1016/j.copbio.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Divne C., Ståhlberg J., Jones T.A. The three-dimensional crystal structure of the catalytic core of cellobiohydrolase I from Trichoderma reesei. Science. 1994;265:524–528. doi: 10.1126/science.8036495. [DOI] [PubMed] [Google Scholar]
- 18.Divne C., Ståhlberg J., Jones T.A. High-resolution crystal structures reveal how a cellulose chain is bound in the 50 A long tunnel of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 1998;275:309–325. doi: 10.1006/jmbi.1997.1437. [DOI] [PubMed] [Google Scholar]
- 19.Kraulis J., Clore G.M., Gronenborn A.M. Determination of the three-dimensional solution structure of the C-terminal domain of cellobiohydrolase I from Trichoderma reesei. A study using nuclear magnetic resonance and hybrid distance geometry-dynamical simulated annealing. Biochemistry. 1989;28:7241–7257. doi: 10.1021/bi00444a016. [DOI] [PubMed] [Google Scholar]
- 20.Ståhlberg J., Divne C., Jones T.A. Activity studies and crystal structures of catalytically deficient mutants of cellobiohydrolase I from Trichoderma reesei. J. Mol. Biol. 1996;264:337–349. doi: 10.1006/jmbi.1996.0644. [DOI] [PubMed] [Google Scholar]
- 21.von Ossowski I., Ståhlberg J., Teeri T.T. Engineering the exo-loop of Trichoderma reesei cellobiohydrolase, Cel7A. A comparison with Phanerochaete chrysosporium Cel7D. J. Mol. Biol. 2003;333:817–829. doi: 10.1016/s0022-2836(03)00881-7. [DOI] [PubMed] [Google Scholar]
- 22.Reinikainen T., Ruohonen L., Teeri T.T. Investigation of the function of mutated cellulose-binding domains of Trichoderma reesei cellobiohydrolase I. Proteins. 1992;14:475–482. doi: 10.1002/prot.340140408. [DOI] [PubMed] [Google Scholar]
- 23.Srisodsuk M., Lehtiö J., Teeri T.T. Trichoderma reesei cellobiohydrolase I with an endoglucanase cellulose-binding domain: action on bacterial microcrystalline cellulose. J. Biotechnol. 1997;57:49–57. doi: 10.1016/s0168-1656(97)00088-6. [DOI] [PubMed] [Google Scholar]
- 24.Nimlos M.R., Matthews J.F., Himmel M.E. Molecular modeling suggests induced fit of Family I carbohydrate-binding modules with a broken-chain cellulose surface. Protein Eng. Des. Sel. 2007;20:179–187. doi: 10.1093/protein/gzm010. [DOI] [PubMed] [Google Scholar]
- 25.Bu L., Beckham G.T., Nimlos M.R. The energy landscape for the interaction of the family 1 carbohydrate-binding module and the cellulose surface is altered by hydrolyzed glycosidic bonds. J. Phys. Chem. B. 2009;113:10994–11002. doi: 10.1021/jp904003z. [DOI] [PubMed] [Google Scholar]
- 26.Beckham G.T., Matthews J.F., Crowley M.F. Identification of amino acids responsible for processivity in a Family 1 carbohydrate-binding module from a fungal cellulase. J. Phys. Chem. B. 2010;114:1447–1453. doi: 10.1021/jp908810a. [DOI] [PubMed] [Google Scholar]
- 27.Srisodsuk M., Reinikainen T., Teeri T.T. Role of the interdomain linker peptide of Trichoderma reesei cellobiohydrolase I in its interaction with crystalline cellulose. J. Biol. Chem. 1993;268:20756–20761. [PubMed] [Google Scholar]
- 28.von Ossowski I., Eaton J.T., Receveur-Bréchot V. Protein disorder: conformational distribution of the flexible linker in a chimeric double cellulase. Biophys. J. 2005;88:2823–2832. doi: 10.1529/biophysj.104.050146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langsford M.L., Gilkes N.R., Kilburn D.G. Glycosylation of bacterial cellulases prevents proteolytic cleavage between functional domains. FEBS Lett. 1987;225:163–167. doi: 10.1016/0014-5793(87)81150-x. [DOI] [PubMed] [Google Scholar]
- 30.Schmuck M., Pilz I., Esterbauer H. Investigation of cellobiohydrolase from Trichoderma reesei by small-angle X-ray scattering. Biotechnol. Lett. 1986;8:397–402. [Google Scholar]
- 31.Abuja P.M., Schmuck M., Esterbauer H. Structural and functional domains of cellobiohydrolase I from Trichoderma reesei. Eur. Biophys. J. 1988;15:339–342. [Google Scholar]
- 32.Abuja P.M., Pilz I., Tomme P. Domain structure of cellobiohydrolase II as studied by small angle X-ray scattering: close resemblance to cellobiohydrolase I. Biochem. Biophys. Res. Commun. 1988;156:180–185. doi: 10.1016/s0006-291x(88)80821-0. [DOI] [PubMed] [Google Scholar]
- 33.Poon D.K.Y., Withers S.G., McIntosh L.P. Direct demonstration of the flexibility of the glycosylated proline-threonine linker in the Cellulomonas fimi Xylanase Cex through NMR spectroscopic analysis. J. Biol. Chem. 2007;282:2091–2100. doi: 10.1074/jbc.M609670200. [DOI] [PubMed] [Google Scholar]
- 34.Sonan G.K., Receveur-Brechot V., Gerday C. The linker region plays a key role in the adaptation to cold of the cellulase from an Antarctic bacterium. Biochem. J. 2007;407:293–302. doi: 10.1042/BJ20070640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen H., Schmuck M., Warren R.A.J. Deletion of the linker connecting the catalytic and CBD of endoglucanase-A (CenA) of Cellulomonas fimi alters its conformation and catalytic activity. J. Biol. Chem. 1991;266:11335–11340. [PubMed] [Google Scholar]
- 36.Receveur V., Czjzek M., Henrissat B. Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle X-ray scattering. J. Biol. Chem. 2002;277:40887–40892. doi: 10.1074/jbc.M205404200. [DOI] [PubMed] [Google Scholar]
- 37.Boisset C., Borsali R., Henrissat B. Dynamic light scattering study of the two-domain structure of Humicola insolens endoglucanase V. FEBS Lett. 1995;376:49–52. doi: 10.1016/0014-5793(95)01244-0. [DOI] [PubMed] [Google Scholar]
- 38.Noach I., Frolow F., Bayer E.A. Intermodular linker flexibility revealed from crystal structures of adjacent cellulosomal cohesins of Acetivibrio cellulolyticus. J. Mol. Biol. 2009;391:86–97. doi: 10.1016/j.jmb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 39.Zhao X., Rignall T.R., Himmel M.E. Molecular simulation evidence for processive motion of Trichoderma reesei Cel7A during cellulose depolymerization. Chem. Phys. Lett. 2008;460:284–288. [Google Scholar]
- 40.Zhong L., Matthews J.F., Brady J.W. Interactions of the complete cellobiohydrolase I from Trichoderma reesei with microcrystalline cellulose Iβ. Cellulose. 2008;15:261–273. [Google Scholar]
- 41.Ting C.L., Makarov D.E., Wang Z.G. A kinetic model for the enzymatic action of cellulase. J. Phys. Chem. B. 2009;113:4970–4977. doi: 10.1021/jp810625k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chocholousová J., Feig M. Balancing an accurate representation of the molecular surface in generalized born formalisms with integrator stability in molecular dynamics simulations. J. Comput. Chem. 2006;27:719–729. doi: 10.1002/jcc.20387. [DOI] [PubMed] [Google Scholar]
- 43.Feig M. Kinetics from implicit solvent simulations of biomolecules as a function of viscosity. J. Chem. Theory Comput. 2007;3:1734–1748. doi: 10.1021/ct7000705. [DOI] [PubMed] [Google Scholar]
- 44.Feig M., Onufriev A., Brooks C.L., 3rd Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J. Comput. Chem. 2004;25:265–284. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 45.Dunker A.K., Brown C.J., Obradović Z. Intrinsic disorder and protein function. Biochemistry. 2002;41:6573–6582. doi: 10.1021/bi012159+. [DOI] [PubMed] [Google Scholar]
- 46.Radivojac P., Iakoucheva L.M., Dunker A.K. Intrinsic disorder and functional proteomics. Biophys. J. 2007;92:1439–1456. doi: 10.1529/biophysj.106.094045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunker A.K., Lawson J.D., Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001;19:26–59. doi: 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- 48.Romero P., Obradovic Z., Dunker A.K. Sequence complexity of disordered protein. Proteins. 2001;42:38–48. doi: 10.1002/1097-0134(20010101)42:1<38::aid-prot50>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Dyson H.J., Wright P.E. Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 50.Brooks B.R., Brooks C.L., 3rd, Karplus M. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mackerell A.D., Jr., Feig M., Brooks C.L., 3rd Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 52.Guvench O., Hatcher E.R., Mackerell A.D. CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 2009;5:2353–2370. doi: 10.1021/ct900242e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee M.S., Salsbury F.R., Brooks C.L. Novel generalized Born methods. J. Chem. Phys. 2002;116:10606. [Google Scholar]
- 54.Ryckaert J., Ciccotti G., Berendsen H. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-Alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 55.Lin E., Shell M.S. Convergence and heterogeneity in peptide folding with replica exchange molecular dynamics. J. Chem. Theory Comput. 2009;5:2062–2073. doi: 10.1021/ct900119n. [DOI] [PubMed] [Google Scholar]
- 56.Shao J., Tanner S.W., Cheatham T.E. Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 57.Abraham M.J., Gready J.E. Ensuring mixing efficiency of replica-exchange molecular dynamics simulations. J. Chem. Theory Comput. 2008;4:1119–1128. doi: 10.1021/ct800016r. [DOI] [PubMed] [Google Scholar]
- 58.Uversky V.N., Gillespie J.R., Fink A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins. 2000;41:415–427. doi: 10.1002/1097-0134(20001115)41:3<415::aid-prot130>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 59.Cantarel B.L., Coutinho P.M., Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kyte J., Doolittle R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 61.Lehtiö J., Sugiyama J., Teeri T.T. The binding specificity and affinity determinants of family 1 and family 3 cellulose binding modules. Proc. Natl. Acad. Sci. USA. 2003;100:484–489. doi: 10.1073/pnas.212651999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeh I.C., Wallqvist A. Structure and dynamics of end-to-end loop formation of the penta-peptide Cys-Ala-Gly-Gln-Trp in implicit solvents. J. Phys. Chem. B. 2009;113:12382–12390. doi: 10.1021/jp904064z. [DOI] [PubMed] [Google Scholar]
- 63.Zhong L.H., Matthews J.F., Brady J.W. Computational simulations of the Trichoderma reesei cellobiohydrolase I acting on microcrystalline cellulose Iβ: the enzyme-substrate complex. Carbohydr. Res. 2009;344:1984–1992. doi: 10.1016/j.carres.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 64.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.