

Abstract
The routes water takes through membrane barriers is still a matter of debate. Although aquaporins only allow transmembrane water movement along an osmotic gradient, cotransporters are believed to be capable of water transport against the osmotic gradient. Here we show that the renal potassium-chloride-cotransporter (KCC1) does not pump a fixed amount of water molecules per movement of one K+ and one Cl−, as was reported for the analogous transporter in the choroid plexus. We monitored water and potassium fluxes through monolayers of primary cultured renal epithelial cells by detecting tiny solute concentration changes in the immediate vicinity of the monolayer. KCC1 extruded K+ ions in the presence of a transepithelial K+ gradient, but did not transport water. KCC1 inhibition reduced epithelial osmotic water permeability Pf by roughly one-third, i.e., the effect of inhibitors was small in resting cells and substantial in hormonal stimulated cells that contained high concentrations of aquaporin-2 in their apical membranes. The furosemide or DIOA (dihydroindenyl-oxy-alkanoic acid)-sensitive water flux was much larger than expected when water passively followed the KCC1-mediated ion flow. The inhibitory effect of these drugs on water flux was reversed by the K+–H+ exchanger nigericin, indicating that KCC1 affects water transport solely by K+ extrusion. Intracellular K+ retention conceivably leads to cell swelling, followed by an increased rate of endocytic AQP2 retrieval from the apical membrane.
Introduction
Water transport is essential to all life forms. However, the routes water takes through membrane barriers is still not entirely understood (compare also (1)). Although it is widely accepted that aquaporins allow transmembrane water movement along an osmotic gradient (2), the role of cotransporters is still a matter of debate (3). Uphill movement of water, against the osmotic gradient, is believed to occur by coupling to downhill solute transport (Fig. 1) along the chemical gradient (4,5). Although a number of water cotransporters have been identified, like the K+–Cl–-cotransporter (6), the H+–lactate-cotransporter (7), and the Na+–glucose-cotransporter (8), it has been argued that the transport rate and membrane abundance of these cotransporters are too low to be of any physiological significance for water homeostasis (3).
Figure 1.
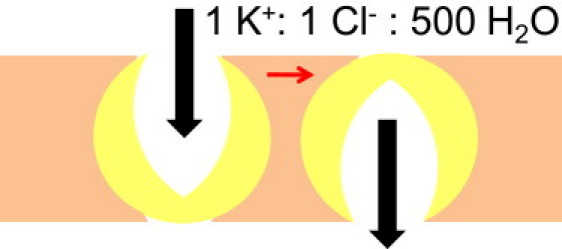
Model of stoichiometric solvent to solute coupling by potassium chloride cotransporters.
Moreover, some of the evidence presented in favor of secondary active water transport by the Na+–glucose-cotransporter seems to be hampered by unstirred layer (USL) effects, i.e., instead of being coupled to solute transport by a molecular device, water was reported to passively follow the solute that accumulates in the immediate membrane vicinity (9–11). At present, it is also possible that none of the above mechanisms work, and that coupling is rather indirect. In this case, the cotransporter would act to set the proper conditions for water channel function.
For the water pumping model to be valid, secondary active transport must occur down the electrochemical gradient. The energetic balance of the K+–Cl– cotransporter (KCC) was reported to be in accord with the 500 molecules of water which it pumps per chloride and potassium ion in the choroid plexus (12). The hypotheses of Loo et al. (5) and Zeuthen (13) about stoichiometrically linked water cotransport by homologous epithelial cotransporters, still awaits confirmation,
| (1) |
where J and Δμ are the fluxes and transmembrane differences in the electrochemical potentials of the species indicated in the subscripts. For
| (2) |
from Eq. 1 follows
| (3) |
Four members of the KCC cotransporter family have been identified. Three of them, KCC1, KCC3, and KCC4 are expressed both in the kidney and in the choroid plexus (14). The intracellular and the lateral K+ and Cl− concentrations for a typical kidney epithelial cell in the inner medulla of the collecting duct (IMCD) are Ki = 155 mM, Kl = 11 mM, Cli = 58 mM, and Cll = 250 mM. Because the overall transport is electroneutral, the magnitude of the electrical membrane potential is of minor importance. The difference between cellular and lateral water activities may be expressed via the osmotic pressure difference across the lateral membrane ΔcL and the molecular volume of water VW:
| (4) |
We arrive at
| (5) |
That is, dissipation of the K+ concentration gradient may drive water transport, as long as the local osmolyte concentration difference between transporter entrance and exit does not exceed ∼130 mOsm l−1.
If we now assume an isoosmotic KCC transport cycle, we find that each pair of K and Cl should be accompanied by
| (6) |
Here, x = 137 water molecules, where cW and co are the concentrations of water and of osmolyte in the lateral intercellular space (LIS), respectively. Increasing x to 500 would mean that co drops from normally 800 mOsm l−1 to
| (7) |
i.e., ∼220 mOsm l−1. In that case, transport would have to occur against the local concentration gradient of ∼580 mOsm l−1. Because dissipation of the potassium gradient cannot drive such transport (see Eq. 6), the role of renal KCC as secondary active water transporter seems questionable.
Of course, these very simple calculations require experimental verification, as fast diffusion away from the mouth of the transporter or K+ uptake by adjacent Na-K-ATPases may respectively lower or increase Δco considerably. For our study, we chose primary cultured IMCD cells. These cells express the KCC1 transporter (15) as well as aquaporin-2 (AQP2) (16), thereby offering the opportunity to dissect the contributions of the passive and the secondary active transepithelial water routes: the putative water pump KCC1 can be inhibited by furosemide or dihydroindenyl-oxy-alkanoic acid (DIOA); and the number of AQP2 copies in the apical membrane may be hormonally adjusted (17,18). AQP2 trafficking from intracellular vesicles to the plasma membrane allows for regulation of water balance in mammals. The AQP2-free apical membrane represents the main barrier to water movement in IMCD cells; i.e., transepithelial water flow is limited by the low permeability of this apical membrane. In contrast, the basolateral membrane always displays high water permeability because it constitutively harbors aquaporin-3 and aquaporin-4.
We observed that the inhibition of renal KCC1 led to a decrease of osmotic transepithelial water flux. This observation is consistent with earlier experimental results obtained for the choroid plexus epithelium (12). However, we also observed that the renal KCC1 did not transport water. It affects water transport exclusively via ion extrusion. Substitution of KCC1 with the potassium proton exchanger nigericin rescues the water transporting function of the epithelium by offering an alternative exit route for intracellular K+.
Materials and Methods
Cell culture
The primary cultured IMCD cells used in this study show a subcellular distribution of aquaporins (AQP2, AQP3, and AQP4), which is reminiscent of that in principal cells in situ, i.e., in the kidney inner medulla (18,19). Immunofluorescence microscopy reveals a rather homogenous intracellular staining of nearly all principal cells for AQP2, which switches to plasma membrane staining after stimulation with vasopressin, forskolin, or membrane-permeable cAMP analogs (17,20–22).
As previously described (17,18), Wistar rats were killed by decapitation, and kidney inner medullae (including papillae) were removed and cut into small pieces. Tissue was digested and cells were subsequently centrifuged (5 min at 300 × g), washed three times, and seeded at a density of ∼7 × 104 cells/cm2 on Transwell-polycarbonate filters (Costar, Bodenheim, Germany). These filters were 6.5 mm in diameter, had a thickness of 10 μm, and contained 108 pores/cm2. Because IMCD cells are accustomed to high osmotic challenges within the kidney medulla, Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, St. Louis, MO) was adjusted to 600 mOsm/kg by addition of 100 mM NaCl and 100 mM urea. pH of the medium was 7.4. It contained DBcAMP (dibutyryl cyclic AMP; 500 μM). In some experiments, the medium was replaced by a cell culture medium without DBcAMP for ∼18 h before the experiments, to render the cells susceptible to cAMP-elevating agents. Just before experiments, the cells were placed in a culture medium with osmolarity of 300 mOsm/kg (37°C). IMCD primary cultures were used from four to eight days after seeding (19).
Immunofluorescence microscopy
Analyses of primary cultured IMCD cells which were grown on Transwell filters (Costar) was performed as previously described (18,19). In brief, the cells were fixed with 2.5% paraformaldehyde in sodium cacodylate buffer (100 mM sodium cacodylate and 100 mM sucrose, pH 7.4) for 30 min. The cells were permeabilized by 0.1% Triton X-100. A quantity of 0.45% fish skin gelatin minimized nonspecific binding. AQP2 was detected using rabbit antisera. Goat antisera (Santa Cruz Biotechnology, Santa Cruz, CA) were used in a 1:50 ratio to detect AQP3 and AQP4. Secondary anti-rabbit IgG and anti-goat antibodies were coupled to Cy3 and Cy2 dyes, respectively. Samples were visualized by confocal microscopy on an Axiovert 730 microscope (Carl Zeiss, Jena, Germany).
Water permeability, Pf of a confluent epithelial monolayer
The cell monolayer with its basal membrane attached to a Transwell-polycarbonate membrane (Costar) was vertically mounted in a Teflon chamber, which was therefore divided into two compartments. The solutions in both compartments were stirred continuously to guarantee an invariant size of unstirred layers (USL) in the immediate epithelial vicinity. The USL thickness, δ, is defined in terms of the Mg2+ concentration gradient at the membrane water interface,
| (8) |
where x is the distance from the membrane. The values cb and cs denote the Mg2+ concentrations in the bulk and at the interface, respectively. Osmotic water flux was induced by additions of polyethylene glycol 600 (PEG) (Sigma-Aldrich) to the basolateral compartment. The resulting steady-state Mg2+ dilution in the hypertonic USL was used to derive the linear velocity of the osmotic volume flow, v (23),
| (9) |
where DMg and a are the Mg2+ diffusion coefficient and the stirring parameter, respectively. The value v allows calculation of Pf,
| (10) |
where Vw is the partial molar volume of water, and cosm is the osmolyte concentration at the surface of the filter. PEG is diluted by the osmotic water flow it induces, so that the actual osmotic pressure cosm is smaller than would have been expected from PEG bulk concentration cosm,b. Calculation of cosm according to (24)
| (11) |
requires the PEG diffusion coefficient, DP to be known. Because the increased viscosity should have a larger effect on DP than on DMg, we exploited fluorescence correlation spectroscopy to estimate DP and conductivity measurements to derive DMg (compare the Supporting Material).
K+ flux measurements
The K+ flux was measured by first establishing a transepithelial K+ ion gradient. To ensure isoosmotic conditions, proper amounts of sorbitol were added to the aqueous solutions in the basolateral compartment while KCl was added to the apical compartment. The resulting tiny changes of the K+-concentration, cK, were detected in the immediate vicinity of the basolateral membrane by a potassium-sensitive microelectrode, which was moved perpendicular to the epithelium. The transepithelial flux of K+-ions, JK, was calculated as
| (12) |
where x is the distance to the basolateral membrane. The value dck/dx was determined from a linear fit to the concentration profiles in the interval 0 < x < 50 μm.
Microelectrodes
The Mg2+- and K+-sensitive microelectrodes were made of glass capillaries. Their tips (1–2 μm in diameter) were filled with cocktail A of Magnesium Ionophore II or cocktail B of Potassium Ionophore I (both Fluka, Dreisenhofen, Germany). Movement of the electrodes relative to the epithelial monolayer was realized by a hydraulic stepdrive (Narishige Scientific, Tokyo, Japan) with a velocity of 2–5 μm/s.
Measurements of cell monolayer resistance
Monolayer resistance was monitored just before and after volume flux measurements. Two pairs of electrodes were exploited. The first pair of Ag/AgCl pellets was used to transfer a 1-kHz square-wave input voltage (source: Model 33120A; Hewlett-Packard, Loveland, CO) to the bathing solution in the first compartment and to record the output signal from the second compartment. First it was amplified by a picoamperemeter (Model 428; Keithley Instruments, Cleveland, OH), and then transferred to an oscilloscope (Model TDS 220; Tektronix, Wilsonville, OR). The resulting potential difference was recorded by an operational amplifier (AD 549; Analog Devices, Norwood, MA) via the second pair of pellets and displayed on the second channel of the oscilloscope.
Results
Fluorescence correlation spectroscopy measurements revealed that PEG600 significantly decreases mobility of the dye rhodamine 6G in solution. Fig. S1 in the Supporting Material shows the shift in autocorrelation functions to longer times. The corresponding relative decrease of the diffusion coefficient is also shown. In all subsequent calculations, we assume that the ratio of the diffusion coefficients in the presence and absence of PEG is similar for rhodamine 6G and PEG, because there is only a minor difference between their molecular masses. In contrast, conductivity measurements showed that the effect of increasing viscosity on Mg2+ mobility was less pronounced (Fig. S1, inset).
Osmotic water flow through IMCD monolayers resulted in solute dilution in the hyperosmotic compartment. We monitored Mg2+ dilution using ion-sensitive scanning microelectrodes (Fig. 2). The spatial distribution of Mg2+ indicated that the apparent osmotic water permeability, Pf, of resting cells was equal to (22 ± 4) μm s−1. This permeability is close to that of bare lipid bilayers which mimic lipid composition and leaflet asymmetry of epithelial cells (25). Consequently, there is little contribution of proteinaceous pathways or tight junctions to water flow under these conditions. Conventional immunofluorescence microscopy with anti-AQP2, anti-AQP3, and anti-AQP4 antibodies confirmed the conclusion revealing AQP-free apical membranes. AQP2 exhibited an intracellular location, whereas both AQP3 and AQP4 resided in the basolateral membrane (Fig. 3).
Figure 2.
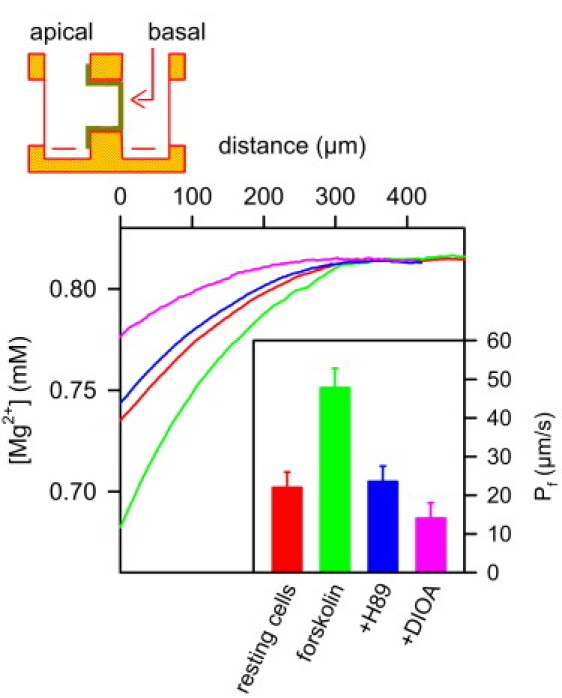
Inhibition of KCC1 has a minor effect on osmotic water flux through IMCD cells in the absence of apical AQP2. A scheme of the experimental setup is shown at the top. The confluent cell monolayer on a Transwell filter is mounted vertically in a Teflon chamber. The Mg2+-sensitive microelectrode and a reference electrode are placed in the hyperosmotic (basolateral) compartment. The osmotic pressure was induced by 450 mM PEG600. The resulting water flux led to Mg2+ dilution, which allowed calculation of the apparent water permeability Pf in the steady state (inset). Therefore, we took both the near-membrane dilution of PEG600 and the decrease of the PEG and Mg2+ diffusion coefficients (compare Fig. S1) into account. First we measured the Mg2+ dilution in the immediate membrane vicinity of resting cells. To show that the cells are functionally active, we then added forskolin, which induced exocytic insertion of aquaporin-2 (AQP2) water channels into the apical membrane. As a result Pf, and thus, the Mg2+ dilution, increased. Subsequent application of the exocytosis inhibitor H89 (50 μM) resulted in endocytic retrieval of AQP2, and thus, reestablished Pf of resting cells. The subsequent addition of DIOA (15 μM) induced a very modest decrease of Pf due to KCC1 inhibition. (Inset) Average Pf values for six runs of the above-described sequence of additions. The water fluxes values for the representative records shown were 0.9, 1.9, 0.9, and 0.6 μmol cm2 s−1 for, respectively, the control monolayer and the monolayers subsequently treated by forskolin, H89, and DIOA.
Figure 3.
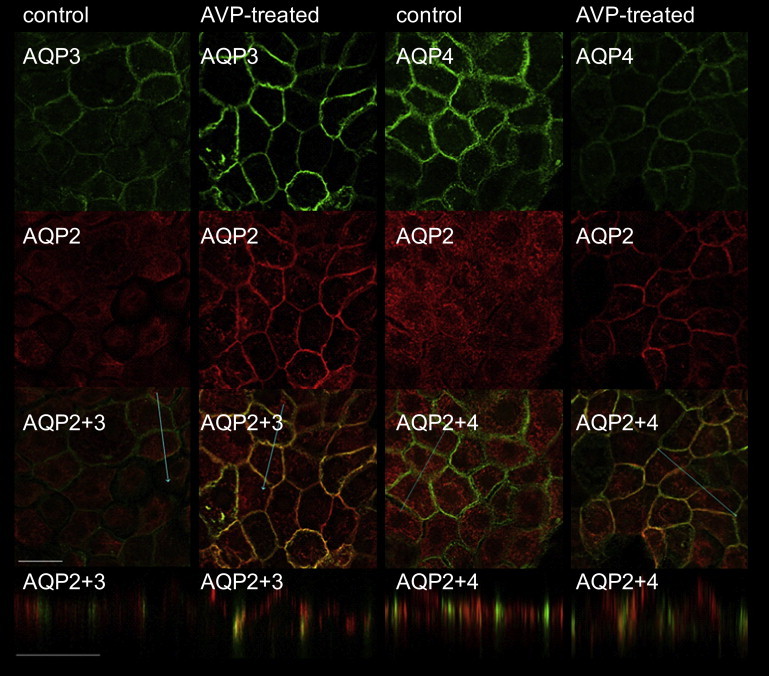
Visualization of AQP2, AQP3, and AQP4 localization by immunofluorescence. Control and AVP-treated (100 nM, 15 min) IMCD cells were incubated with anti-AQP2 and AQP3 or anti-AQP2 and AQP4 antibodies. (Upper two lines) xy scans with Cy2 and Cy3 staining for single AQP species. The blue lines in the overlay pictures (third line) indicate where the Z-scan (lower line) has been performed. AQP3 and AQP4 are exclusively located in the basolateral membrane at all times. Control cells show intracellular AQP2 staining. Upon AVP stimulation, exocytosis of AQP2-bearing vesicles occurs. To show apical AQP2 localization in the very flat IMCD cells (∼8 μm in height), the z axis is shown at a fivefold higher magnification than the xy axes (scale bar, 20 μm). Some of the AQP2 also inserts into the lateral membrane (compare overlay).
The expression rates of all three aquaporins were rather homogenous for all cells of a Transwell filter, as would be expected of primary cultured cells. In line with previous results (17), we observed exocytic insertion of AQP2-bearing vesicles into the apical membrane after hormonal stimulation (Fig. 3). The accompanying increase in Mg2+ ion dilution adjacent to the epithelial monolayer indicated that Pf increased by a factor of 2.2 (Fig. 2). PEG was also diluted, i.e., its concentration dropped from 450 mM in the bulk to 400 mM on the surface (compare Eq. 11). Because Eqs. 8, 9, 11, and 12 are only valid in the immediate vicinity of the surface, and because DMg and DP changed by <5% for 0 ≤ x ≤ 50 μm (compare Fig. S1), we calculated DMg and DP for x ≈ 0 and assumed them to be spatially invariant. Application of H89 reversed the effect of forskolin by inhibiting AQP2 exocytosis while leaving the machinery for endocytic AQP2 retrieval intact. Pf returned to the value of resting cells. KCC1 inhibition by DIOA reduced Pf to roughly two-thirds of the resting value (Fig. 3).
For our calculations of Pf we assumed that the dilution of the osmolyte within the pores of the filter can be neglected. Simple extrapolation of the measured concentration profiles (Fig. 2) to −10 μm would reveal that cosm was overestimated by <1.5%. This number increased to <3.5% if the 10-fold increase in v was considered, which was caused by the decrease in cross-sectional flow area (the pores covered only ∼12.6% of the filter area). Osmolyte dilution in the LIS was also neglected because the cells were rather flat (∼7 μm in height). Even for swollen cells, the combined effects of pores and LIS resulted in an overestimation of cosm < 5%.
To test the possibility that the KCC1 mediated K+ net flux JK is stoichiometrically linked to the water flux JW, we measured both fluxes under isoosmotical conditions. In DBcAMP-activated cells, JK was undetectably small in the absence of transepithelial osmotic and K+ gradients, i.e., the amount of K+ pumped into the cells was roughly identical to the amount of K+ leaving the cells through KCC1 and K+ channels (Fig. 4 A). We raised the K+ apical concentration isoosmotically from 5.4 to 45 to 90 and 150 mM. No dilution of Mg2+ ions occurred, i.e., if there was a net water flow, it was undetectably small (not shown). Nevertheless, we observed JK increased roughly proportional to the transepithelial K+ gradient (Fig. 4 A). It is likely that both intracellular and paracellular routes contributed to JK. K+ may enter the cell via the H+-K+-ATPase at the apical membrane and leave the cell via KCC1 and Kir2.3, the dominant basolateral K+ channel in IMCD cells.
Figure 4.
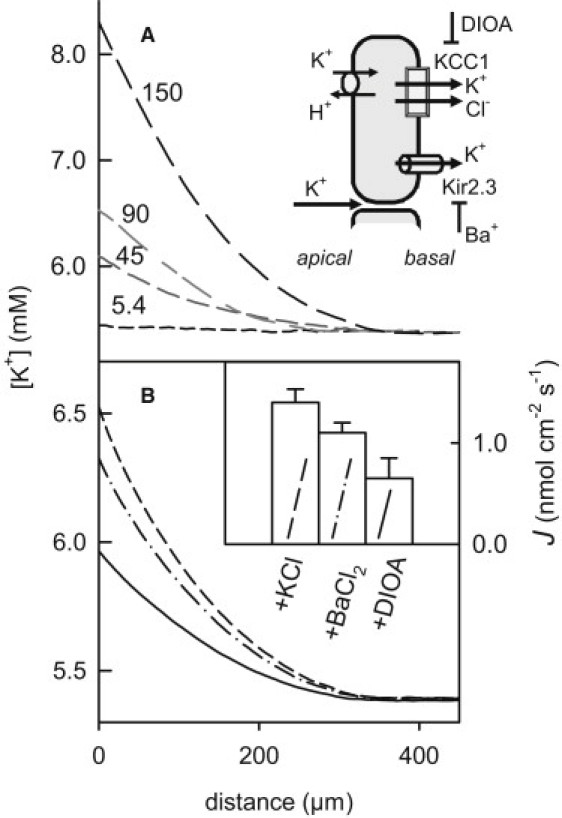
Visualization of K+ transport by KCC1. (A) While the basolateral K+ concentration was kept at 5.4 mM, the apical concentration was augmented to the indicated values (in mM). Isoosmotic conditions were maintained by adding sorbitol to the basolateral aqueous solutions. The resulting transepithelial ion flux was monitored via the accompanying increase in K+ concentration close to the basolateral membrane. The representative K+ concentration profiles allowed flux calculation (0, 900, 1300, and 3000 pmol cm−2 s−1). (B) Inhibition of transcellular potassium ion flow. Potassium ion flux was induced by adding 90 mM KCl to the apical aqueous solution. Isoosmotic conditions were maintained by adding sorbitol to the basolateral aqueous solutions. (Inset) Subsequent additions of 500 μM BaCl2 and 15 μM DIOA to both parts of the chamber decreased K+ flux density. Before each addition, the system was allowed to equilibrate for ∼30 min. Mean values and standard errors were calculated from seven independent runs of the experiments (inset).
Inhibition of Kir2.3 by Ba2+ (26) and KCC1 by DIOA allowed the dissection of paracellular and transcellular pathways (Fig. 4 B). In the presence of 90 mM K+ in the apical compartment, Ba2+ administration reduced JK from 1.4 nmol cm−2 s−1 to 1.1 nmol cm−2 s−1, whereas inhibition of KCC1 by DIOA dropped JK to 0.6 nmol cm−2 s−1 (Fig. 4 B). Thus, ∼50% of the net flux was transcellular. Provided that the major barrier to K+ transport is the apical membrane, which has a permeability of ∼10−5 cm/s (27), a transepithelial K+ concentration difference of 85 mM is expected to result in a net transcellular flux of ∼0.85 nmol cm−2 s−1. Thus, the measured DIOA-sensitive JK of 0.5 nmol cm−2 s−1 (Fig. 4 B) indicates that the major K+ transporters both in the apical and basolateral membranes are not downregulated, i.e., KCC1 is present, and functions in high-copy-numbers in our primary cultured cells. The lack of measureable Jw is at odds with the idea of secondary active water transport, because according to Zeuthen (12), Jw should be in the range of 500 times JK, i.e., ∼0.25 μmol cm−2 s−1.
There is little doubt that ion extrusion by cotransporters such as KCC1 provides part of the osmotic driving force for water transport across membranes as there is a continuous K+ flux directed from the apical solution (50 mM K+) to the basal solution (11 mM K+). Here we have tested whether KCC1 may affect water transport in the absence of a transepithelial K+ gradient. At a K+ concentration of 5.4 mM in both compartments, inhibition of K+ and Cl− efflux by DIOA decreased Pf from (51 ± 6) μm s−1 to (34 ± 5) μm s−1 (Fig. 5). If the accumulation of K+ inside the cell was responsible for the effect, rescuing the K+ extrusion pathway should restore Pf.
Figure 5.
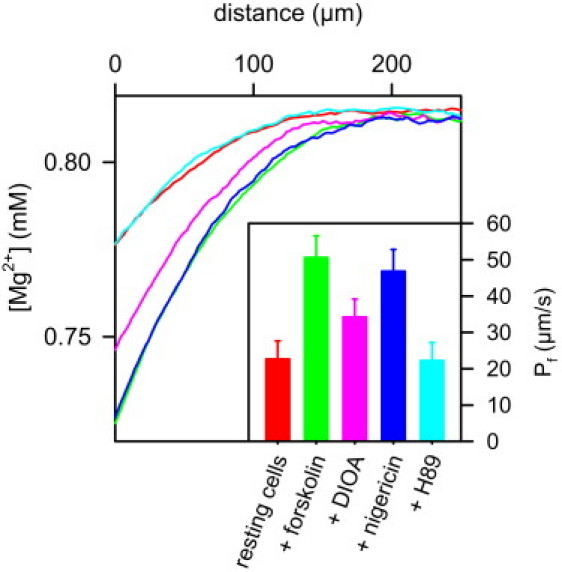
The effect of KCC1 inhibition on osmotic water flux is reversed by nigericin: As indicated by the increased Mg2+ polarization, Pf of resting cells increased upon forskolin (50 μM) stimulation. Subsequent application of 20 μM DIOA resulted in partial inhibition of water flux as indicated by the decrease in Mg2+ dilution. The inhibitory effect of DIOA was reversed by the potassium proton exchanger nigericin (30 μM). Because nigericin is too small to accommodate water molecules, this observation indicates that rescuing K+ efflux is sufficient for restoring water flux. The presence of 50 mM NH4Cl in DMEM increased intracellular and extracellular buffer capacity, thereby preventing changes of pH (7.4) which otherwise would have accompanied nigericin-mediated K+ efflux. In a final step, addition of H89 (20 μM) inhibited aquaporin-2 exocytosis. The resulting endocytic retrieval of aquaporin-2 returned Pf to its resting values (Inset). Mean values and standard deviations of Pf obtained in 10 runs of the experiment, each including subsequent additions of forskolin, DIOA, nigericin, and H89. Except 450 mM PEG600 in the basal compartment, the solutions on both sides of the epithelial monolayer were identical (5.4 mM K+ on both sides).
To test the hypothesis, we added the H+–K+ exchanger nigericin (28,29). The addition of nigericin does not change membrane potential because the flow of K+ ions along their concentration gradient generates an oppositely directed equimolar H+ flux. But it requires augmentation of extracellular and intracellular buffer capacity to lock the actual pH at the initial value. For that purpose, we added 50 mM NH4Cl (adjusted to pH 7.4) to both the basolateral and apical compartments, exploiting the fact that the basolateral and apical membranes of epithelial cells are permeable to NH3 (30). Nigericin restored the water flux as if KCC1 was still active (Fig. 5). In contrast to KCC1, nigericin is far too small to accommodate 500 water molecules. Because it does not span the membrane either, it is, thus, incapable of conducting water through the membrane. Consequently, the nigericin-mediated increase in Pf was a secondary effect mediated by K+ efflux.
If the conclusion was correct, i.e., if KCC1 inhibition resulted in an altered water flux through AQP2, water channel inhibition should restore the Pf of resting cells. If in contrast, KCC1 transports water on its own, the simultaneous removal of both water pathways by the inhibition of KCC1, and AQP2 retrieval to intracellular stores, should result in Pf values below resting ones. Consequently, we also treated the cells which were already exposed to DIOA with the phosphokinase A inhibitor H-89. H89 prevents the exocytic AQP2 insertion into the plasma membrane. Endocytic retrieval resulted in a nearly AQP2-free apical membrane (17). Pf was reduced to the resting value, providing additional evidence that KCC1 does not represent a water pathway in the resting state of IMCD cells (Fig. 5).
We repeated the experiment shown in Fig. 5 with furosemide (not shown) and obtained similar results. This observation is in line with literature data regarding the nonavailability of nonpromiscuous inhibitors. It is only in red blood cells that DIOA potently inhibits K+-CI--cotransport fluxes without side effects on NKCC (31). In epithelial cells, like in HEK293, DIOA is less specific (32). It was, thus, not surprising that the equally nonspecific furosemide mirrored the effect of DIOA on transepithelial water transport in IMCD cells. If both DIOA and furosemide act to reduce the driving force for transepithelial water movement, NKCC is unlikely to be involved. The osmotic force across the epithelium appears to be the major determinant for transcellular water flux. Inhibition of ion import by NKCC would increase the ion concentration close to the basal membrane, and hence, water flux should increase. The observed decrease is only compatible with an impairment of KCC1.
Following these considerations, blockage of IMCD K+ channels—the sole K+ export pathway alternative to KCC1—should also have an effect on water flux. The dominant channel conductance in the IMCD cells is carried by Kir2.3, i.e., it is inwardly rectified and Ba2+-sensitive (26). Due to their low abundance, K+ channel transport capacity is only ∼1/10 of that of KCC1 (27). Consequently, we expected a rather modest effect on transepithelial water flow. In line with this prediction, inhibition of K+ channels by 500 μM Ba2+ decreased Pf only by ∼10% (not shown).
The main pathway for K+ import is the Na+-K+-ATP pump. It has a K+ transport capacity close to that of KCC1 (27). The pump is exclusively located in the basolateral membrane of IMCD cells (33). Pump inhibition may lead to a decrease of salt concentration in the water layers adjacent to the basolateral membrane, because the number of sodium ions extruded exceeds the number of potassium ions imported. As a result, the apparent Pf should decrease. However, administration of the pump inhibitor ouabain had no effect on Pf. Accompanying changes in the activity of other transporters, e.g., the reduction of salt influx via NKCC, may provide an explanation. It should be mentioned that addition of oubain at concentrations >100 μM led to an increase of apparent Pf. However, it cannot be unambiguously attributed to transcellular water transport because cell morphology changed as well—probably due to a drastic decrease of IMCD cell viability. In confirmation of this hypothesis, we observed openings of tight junctions, as indicated by a decrease in the electrical resistance of the cell monolayer, which for a tight cell monolayer was equal to 4 ± 1 kΩ cm−2.
Taken altogether, the above experiments suggest that KCC1 does not provide a water pathway but acts by ion extrusion. To test whether water passively follows the extruded ions, we measured both water and potassium fluxes under the same conditions (Fig. 6). DBcAMP-treated cells were exposed to an osmotic gradient and subsequently treated with DIOA and nigericin to inhibit KCC1 and to rescue potassium efflux, respectively. DIOA decreased JW from 2.7 μmol cm−2 s−1 to 1.7 μmol cm−2 s−1. Nigericin restored JW to a level of 2.4 μmol cm−2 s−1. The potassium flux is assessed from (24)
| (13) |
Figure 6.
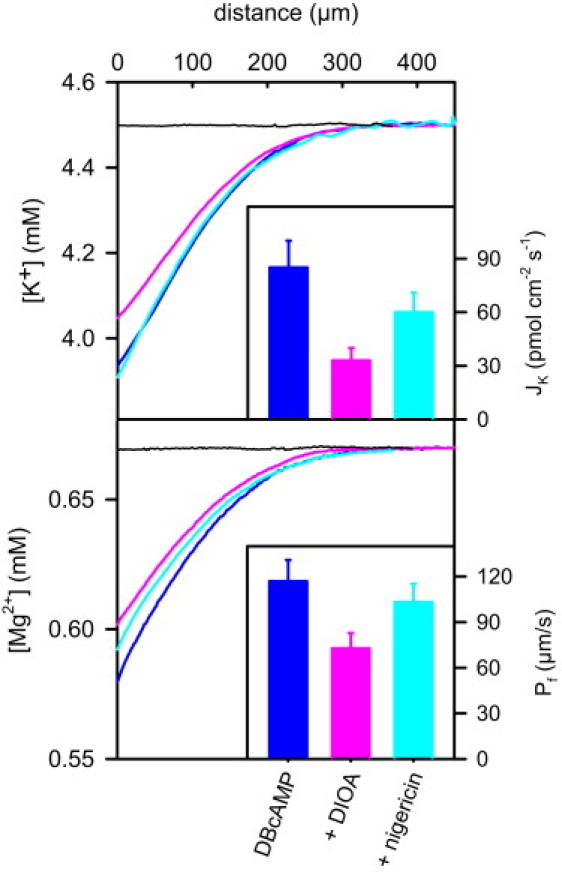
The effect of DIOA on potassium flux (Top) and water permeability (Bottom) in DBcAMP-stimulated cells. In the absence of transepithelial osmotic and K+ gradients, no water or K+ fluxes (JW and JK, respectively) were detected (black lines). JW and JK were induced by 250 mM PEG in the basal compartment. Otherwise the solutions on both sides were the same. (Top) Microelectrode measurements of K+ concentration in the immediate vicinity of the basal membrane showed a decreasing K+ ion dilution upon DIOA application. The effect was reversed by nigericin addition. Transepithelial potassium fluxes (inset) were calculated according to Eq. 6. (Bottom) As indicated by the pronounced Mg2+ polarization, the Pf of DBcAMP-stimulated cells is rather large (inset). Application of 20 μM DIOA resulted in partial inhibition of water flux from 2.6 μmol cm−2 s−1 to 1.7 μmol cm−2 s−1, as indicated by the decrease in Mg2+ dilution. Nigericin (30 μM) rescued the initial high level of water flux (2.4 μmol cm−2 s−1). DMEM contained 50 mM NH4Cl to lock intracellular pH. (Inset) Mean values and standard deviations of Pf obtained in six runs of the experiment. The ratio of DIOA-sensitive water and potassium fluxes is ∼20.000, ruling out that these fluxes are stoichiometrically or osmotically linked.
KCC1 inhibition decreased JK from 85 pmol cm−2 s−1 to 33 pmol cm−2 s−1. Thus, the ratio ΔJW,KCC/ΔJK,KCC is equal to ∼20.000. The absolute DIOA-dependent flux values were smaller for cells deprived of DBcAMP overnight and instead stimulated by forskolin (compare Fig. 5), but the flux ratio was of the same order of magnitude (not shown). Such a large number of water molecules per cation cannot be pumped by KCC1 or explained by water passively following K+ and Cl−.
Discussion
We demonstrated that KCC1 from kidney inner medulla is unable to perform secondary active water transport despite contrasting reports about the water pumping function of KCC in the choroid plexus. Four lines of evidence have been obtained:
-
1.Contrary to what might have been expected for a continuously active water pump, no net water transport occurred through the resting cell monolayer in the absence of osmotic pressure. Dilution of Mg2+ or K+ in the vicinity of the basolateral membrane should otherwise have been observed. Although the DIOA sensitive potassium flux JK amounted to ∼0.5 nmol cm−2 s−1 (Fig. 4), it was not accompanied by water flow. This is in sharp contrast to the water flux predicted by the published 1:500 stoichiometry of K+:H2O of KCC in the choroid plexus (12). As the detection limit is ∼0.1 μmol cm−2 s−1 in this experiment, a water flux of 0.25 μmol cm−2 s−1 could hardly go unnoticed. According to
it is half as large as the remaining water flux after DIOA treatment, which led to a significant solute dilution adjacent to the cell monolayer (compare Fig. 2).(14) -
2.
The ratio of the Pf values measured before and after DIOA or furosemide addition was always equal to ∼1.5 (compare Figs. 2, 5, and 6). In contrast, for cells which differ only in the abundance of AQP2 in their apical membranes (Figs. 2 and 5), an invariant DIOA-sensitive JW was expected if K+ and H2O fluxes were stoichiometrically linked by KCC.
-
3.
The ratio of the DIOA-sensitive water and potassium fluxes is much larger than the one reported for the KCC from choroid plexus. The energy gained from dissipation of the K+ gradient cannot drive such a large number of water molecules through the membrane (see Introduction). In addition, the number of water molecules per turnover of the protein would be too large to be accommodated in any known proteinaceous cavity.
-
4.
Previously, the reduction in JW observed upon inhibition of KCC was taken as evidence for secondary active water transport (6,12,13). Our experiments show that the argumentation is misleading, because it is possible to completely restore Pf by just providing a K+ exit pathway (i.e., nigericin) without offering a water route (Figs. 5 and 6).
The effects observed upon KCC1 inhibition may also be due to water passively following K+ and Cl−. This would imply that the K+ concentration adjacent to the basal membrane is elevated above basal bulk concentration due to K+ extrusion by KCC1. In contrast, surface and bulk K+ concentrations were equal to each other in the absence of an osmotic gradient. In the presence of the gradient, we always observed K+ dilution (Fig. 6), indicating that instead of following K+, water is dragging K+. In line with this observation, the further decrease of K+ concentration expected upon KCC1 inhibition was masked by the decrease in JW which tends to elevate K+. Finally, calculation of DIOA-sensitive water and potassium fluxes (Eq. 6) revealed a ΔJW,KCC/ΔJK,KCC ratio, which is much too large to be compatible with the view that KCC1 contributes to cosm.
In a search for alternative mechanisms by which KCC1 may affect transepithelial water flow, we noticed the proportionality between apparent epithelial Pf and its DIOA-mediated reduction. Effects which are proportional to Pf are commonly reminiscent of USL phenomena. Although the permeabilities of the external USLs adjacent to the apical and basal sides of the cell monolayer remained unaltered upon DIOA addition as revealed by our microelectrode based measurements, we can only estimate solute permeabilities of the USLs within the cell Pcell,s and within the lateral intercellular spaces (LIS) PLIS,s (compare Fig. 7). Because Pcell,s and PLIS,s determine how fast the dilution due to osmotic water flow of cytoplasm and the LIS is encountered by solute backdiffusion, they may affect the actual driving force for water cosm. If retention of K+ ions by KCC1 inhibition would change Pcell,s and/or PLIS,s, the accompanying change of cosm would mimic a change of Pf. To estimate the effect on Pcell,s and PLIS,s, we have to consider that cosm reflects 1), the osmotic pressure difference between the apical compartment and the cytoplasm, ΔcAP; 2), between the cytoplasm and the LIS, ΔcL; and 3), between the LIS and the basal compartment ΔcB:
| (15) |
Figure 7.
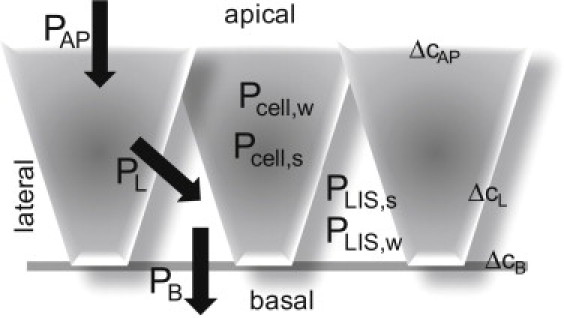
Schematic representation of the different permeabilities in the epithelial monolayer. The water permeabilities of the unstirred layers (USLs) within the cell Pcell,w and within the lateral intercellular spaces (LIS), PLIS,w, are large compared to the water permeability of the apical membrane, PAP. The same holds for the water permeabilities of the lateral and the basement membranes, PL and PB, respectively. The osmotic pressure difference between the apical compartment and the cytoplasm, ΔcAP, is much larger than the osmotic pressure differences between cytoplasm and the LIS or the LIS and the basal compartment, ΔcL and ΔcB, respectively. The intracellular and LIS solute permeabilities are indicated as Pcell,s and PLIS,s, respectively.
Due to the presence of AQP3 and AQP4 (Fig. 3), the lateral plasma membrane has a 27-fold higher water permeability PL than the apical membrane in a resting cell (34). The permeability of the basement membrane PB is at least an order-of-magnitude higher than PL (27,35). The water permeabilities of the intracellular and the LIS USLs, Pcell,w and PLIS,w, are equal to 10−5 cm2 s−1/8 μm ∼125 μm/s and 2 × 10−5 cm2 s−1/4 μm ∼500 μm/s, respectively. For the calculation of PLIS,w, we took into account that solute mobility in the LIS is very close to the mobility in the external solution (36). Because the reciprocal values of all permeabilities add to the total epithelial permeability,
| (16) |
we arrive at PAP ∼ 30 μm s−1 and PL ∼ 800 μm s−1 for a Pf of resting cells of ∼22 μm s−1. Because the water flux through all three membranes has to be the same, Eqs. 14–16 predict that ΔcAP >> ΔcL ∼ ΔcB, i.e., the osmolarity of the cytoplasm is very close to the osmolarity in the immediate vicinity of the basal compartment and ΔcAP ∼ cosm. That is, USL effects in the LIS are unlikely to be of importance.
ΔcAP >> ΔcL ∼ ΔcB is also true for forskolin- or DBcAMP-treated cells. Our experiments show that AQP2 insertion into both the apical and basolateral membranes (Fig. 3) augments Pf only 2.2-fold or sixfold, respectively. This is also true when KCC1 is inhibited because both PAP and PL remain unaltered. That is, ΔcL cannot significantly increase even if KCC1 inhibition leads to intracellular retention of K+. If so, the increase in intracellular osmolyte concentration should be compensated by cell swelling. In turn, intracellular solute mobility increases, and with it, Pcell,s as well (37). The thereby-facilitated back-diffusion decreases the osmotic effect of all flow-induced concentration changes; i.e., the water flux induced enhancement of intracellular solute concentration adjacent to the basolateral membrane is less pronounced and less solute is swept away from the apical membrane. Consequently, KCC1 inhibition may only increase transcellular water flux because the actual osmotic driving force ΔcAP increases with Pcell,s. Because this is the opposite of what we observed experimentally, we can rule out intracellular USL as a possible source of the effects observed upon KCC1 inhibition.
By exclusion of all other possibilities, we arrive at the conclusion that DIOA— or furosemide-mediated KCC1 inhibition— decreases PAP. Because AQP2 is not known to be a target of DIOA, and because exclusive application of DIOA to the apical compartment (result not shown) did not alter Pf, the drug should exert an indirect effect on exocytic AQP2 apical membrane insertion or its endocytic retrieval. The most plausible mechanism is provided by the above-discussed swelling which occurs in response to K+ retention. Cell swelling is known to increase vesicle recycling (38). A 100-fold increase in the rate of endocytosis, as observed in intestine 407 cells, could easily account for the decreased abundance of AQP2 in the apical membrane of IMCD cells and the decrease in PAP (and, thus in Pf). An increased rate of exocytosis does not confound the analysis because vesicles not bearing AQP2 may be involved. This mechanism would also explain why nigericin restores Pf to 100%. It restores cell volume by providing an exit route for K+. In turn, the rates of exocytosis and endocytosis return to normal as does, consequently, the AQP2 concentration in the apical membrane.
While the effect of DIOA on the AQP2 membrane abundance awaits experimental verification, we have unambiguously shown that the renal KCC1 does not perform secondary active water transport. Doubts concerning the existence of molecular water pumps have previously been raised. In addition to insufficient copy and turnover numbers (3), methodological flaws arising from giant unstirred layers inside the most commonly used Xenopus oocytes were put forward (9). Because any inward-directed solute flux leads to solute accumulation close to the inner oocyte membrane and because increased viscosity of the cytoplasm and lack of convection hamper solute advection, water was envisioned to flow passively along the osmotic gradient instead of being cotransported by the sodium glucose cotransporter SGLT1 (10).
In our model of epithelial cell monolayers on porous support, intracellular USLs play a minor role. Nonetheless, we came to the same conclusion that transport of water is not a general property of cotransporters. However, based on renal KCC1 alone, it is impossible to exclude that other cotransporters may still perform secondary active water transport. Therefore, we currently perform steady-state investigations of water flow through the sodium sugar cotransporter, SGLT1. Together with this study, these experiments will provide more general evidence as to whether molecular machines are able to pump water against the osmotic gradient.
Acknowledgments
We thank Andrea Geelhaar and Jenny Eichhorst (both Forschungsinstitut für Molekulare Pharmakologie, Berlin) for excellent technical assistance. We also thank Quentina Beatty (Johannes Kepler Universität, Linz) for editorial help.
The project was supported by the Deutsche Forschungsgemeinschaft (grant Nos. Po 533/8 and Kl 1415/3-2).
Footnotes
Rustam Mollajew's present address is Zentrum für Physiologie und Pathophysiologie, Abteilung Neuro- und Sinnesphysiologie, Universität Göttingen, Humboldtallee 23, D-37073 Göttingen, Germany.
Supporting Material
References
- 1.Spring K.R. Routes and mechanism of fluid transport by epithelia. Annu. Rev. Physiol. 1998;60:105–119. doi: 10.1146/annurev.physiol.60.1.105. [DOI] [PubMed] [Google Scholar]
- 2.King L.S., Kozono D., Agre P. From structure to disease: the evolving tale of aquaporin biology. Nat. Rev. Mol. Cell Biol. 2004;5:687–698. doi: 10.1038/nrm1469. [DOI] [PubMed] [Google Scholar]
- 3.Spring K.R. Epithelial fluid transport—a century of investigation. News Physiol. Sci. 1999;14:92–98. doi: 10.1152/physiologyonline.1999.14.3.92. [DOI] [PubMed] [Google Scholar]
- 4.Zeuthen T., MacAulay N. Cotransporters as molecular water pumps. Int. Rev. Cytol. 2002;215:259–284. doi: 10.1016/s0074-7696(02)15012-1. [DOI] [PubMed] [Google Scholar]
- 5.Loo D.D., Wright E.M., Zeuthen T. Water pumps. J. Physiol. 2002;542:53–60. doi: 10.1113/jphysiol.2002.018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeuthen T. Secondary active transport of water across ventricular cell membrane of choroid plexus epithelium of Necturus maculosus. J. Physiol. 1991;444:153–173. doi: 10.1113/jphysiol.1991.sp018871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zeuthen T., Hamann S., la Cour M. Cotransport of H+, lactate and H2O by membrane proteins in retinal pigment epithelium of bullfrog. J. Physiol. 1996;497:3–17. doi: 10.1113/jphysiol.1996.sp021745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loo D.D., Zeuthen T., Wright E.M. Cotransport of water by the Na+/glucose cotransporter. Proc. Natl. Acad. Sci. USA. 1996;93:13367–13370. doi: 10.1073/pnas.93.23.13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duquette P.P., Bissonnette P., Lapointe J.Y. Local osmotic gradients drive the water flux associated with Na+/glucose cotransport. Proc. Natl. Acad. Sci. USA. 2001;98:3796–3801. doi: 10.1073/pnas.071245198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gagnon M.P., Bissonnette P., Lapointe J.Y. Glucose accumulation can account for the initial water flux triggered by Na+/glucose cotransport. Biophys. J. 2004;86:125–133. doi: 10.1016/S0006-3495(04)74090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charron F.M., Blanchard M.G., Lapointe J.Y. Intracellular hypertonicity is responsible for water flux associated with Na+/glucose cotransport. Biophys. J. 2006;90:3546–3554. doi: 10.1529/biophysj.105.076745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zeuthen T. Cotransport of K+, Cl− and H2O by membrane proteins from choroid plexus epithelium of Necturus maculosus. J. Physiol. 1994;478:203–219. doi: 10.1113/jphysiol.1994.sp020243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zeuthen T. Water-transporting proteins. J. Membr. Biol. 2010;234:57–73. doi: 10.1007/s00232-009-9216-y. [DOI] [PubMed] [Google Scholar]
- 14.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol. Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 15.Liapis H., Nag M., Kaji D.M. K-Cl cotransporter expression in the human kidney. Am. J. Physiol. 1998;275:C1432–C1437. doi: 10.1152/ajpcell.1998.275.6.C1432. [DOI] [PubMed] [Google Scholar]
- 16.Nielsen S., Frøkiaer J., Knepper M.A. Aquaporins in the kidney: from molecules to medicine. Physiol. Rev. 2002;82:205–244. doi: 10.1152/physrev.00024.2001. [DOI] [PubMed] [Google Scholar]
- 17.Lorenz D., Krylov A., Maric K. Cyclic AMP is sufficient for triggering the exocytic recruitment of aquaporin-2 in renal epithelial cells. EMBO Rep. 2003;4:88–93. doi: 10.1038/sj.embor.embor711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stefan E., Wiesner B., Klussmann E. Compartmentalization of cAMP-dependent signaling by phosphodiesterase-4D is involved in the regulation of vasopressin-mediated water reabsorption in renal principal cells. J. Am. Soc. Nephrol. 2007;18:199–212. doi: 10.1681/ASN.2006020132. [DOI] [PubMed] [Google Scholar]
- 19.Maric K., Oksche A., Rosenthal W. Aquaporin-2 expression in primary cultured rat inner medullary collecting duct cells. Am. J. Physiol. 1998;275:F796–F801. doi: 10.1152/ajprenal.1998.275.5.F796. [DOI] [PubMed] [Google Scholar]
- 20.Maric K., Wiesner B., Rosenthal W. Cell volume kinetics of adherent epithelial cells measured by laser scanning reflection microscopy: determination of water permeability changes of renal principal cells. Biophys. J. 2001;80:1783–1790. doi: 10.1016/S0006-3495(01)76148-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klussmann E., Maric K., Rosenthal W. Protein kinase A anchoring proteins are required for vasopressin-mediated translocation of aquaporin-2 into cell membranes of renal principal cells. J. Biol. Chem. 1999;274:4934–4938. doi: 10.1074/jbc.274.8.4934. [DOI] [PubMed] [Google Scholar]
- 22.Klussmann E., Lorenz D., Rosenthal W. Cyclic AMP-mediated aquaporin-2 translocation: identification of protein kinase A anchoring proteins and the role of the small GTPases of the Rho family. In: Hohmann S., Nielsen S., editors. Molecular Biology and Physiology of Water and Solute Transport. Kluwer Academic/Plenum Publishers; London: 2000. pp. 145–149. [Google Scholar]
- 23.Pohl P., Saparov S.M., Antonenko Y.N. The effect of a transmembrane osmotic flux on the ion concentration distribution in the immediate membrane vicinity measured by microelectrodes. Biophys. J. 1997;72:1711–1718. doi: 10.1016/S0006-3495(97)78817-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pohl P., Saparov S.M. Solvent drag across gramicidin channels demonstrated by microelectrodes. Biophys. J. 2000;78:2426–2434. doi: 10.1016/S0006-3495(00)76786-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Krylov A.V., Pohl P., Hill W.G. Water permeability of asymmetric planar lipid bilayers: leaflets of different composition offer independent and additive resistances to permeation. J. Gen. Physiol. 2001;118:333–340. doi: 10.1085/jgp.118.4.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Millar I.D., Taylor H.C., Robson L. A Kir2.3-like K+ conductance in mouse cortical collecting duct principal cells. J. Membr. Biol. 2006;211:173–184. doi: 10.1007/s00232-006-0036-z. [DOI] [PubMed] [Google Scholar]
- 27.Weinstein A.M. A mathematical model of the inner medullary collecting duct of the rat: pathways for Na and K transport. Am. J. Physiol. 1998;274:F841–F855. doi: 10.1152/ajprenal.1998.274.5.F841. [DOI] [PubMed] [Google Scholar]
- 28.Pressman B.C. Properties of ionophores with broad range cation selectivity. Fed. Proc. 1973;32:1698–1703. [PubMed] [Google Scholar]
- 29.Pohl P., Saparov S.M., Antonenko Y.N. The size of the unstirred layer as a function of the solute diffusion coefficient. Biophys. J. 1998;75:1403–1409. doi: 10.1016/S0006-3495(98)74058-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saparov S.M., Liu K., Pohl P. Fast and selective ammonia transport by aquaporin-8. J. Biol. Chem. 2007;282:5296–5301. doi: 10.1074/jbc.M609343200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garay R.P., Nazaret C., Cragoe E.J., Jr. Demonstration of a [K+,Cl−]-cotransport system in human red cells by its sensitivity to [(dihydroindenyl)oxy]alkanoic acids: regulation of cell swelling and distinction from the bumetanide-sensitive [Na+,K+,Cl−]-cotransport system. Mol. Pharmacol. 1988;33:696–701. [PubMed] [Google Scholar]
- 32.Gillen C.M., Forbush B., 3rd Functional interaction of the K-Cl cotransporter (KCC1) with the Na-K-Cl cotransporter in HEK-293 cells. Am. J. Physiol. 1999;276:C328–C336. doi: 10.1152/ajpcell.1999.276.2.C328. [DOI] [PubMed] [Google Scholar]
- 33.Barlet-Bas C., Arystarkhova E., Doucet A. Are there several isoforms of Na,K-ATPase α subunit in the rabbit kidney? J. Biol. Chem. 1993;268:11512–11515. [PubMed] [Google Scholar]
- 34.Strange K., Spring K.R. Cell membrane water permeability of rabbit cortical collecting duct. J. Membr. Biol. 1987;96:27–43. doi: 10.1007/BF01869332. [DOI] [PubMed] [Google Scholar]
- 35.Welling L.W., Grantham J.J. Physical properties of isolated perfused renal tubules and tubular basement membranes. J. Clin. Invest. 1972;51:1063–1075. doi: 10.1172/JCI106898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xia P., Bungay P.M., Spring K.R. Diffusion coefficients in the lateral intercellular spaces of Madin-Darby canine kidney cell epithelium determined with caged compounds. Biophys. J. 1998;74:3302–3312. doi: 10.1016/S0006-3495(98)78037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jacobson K., Wojcieszyn J. The translational mobility of substances within the cytoplasmic matrix. Proc. Natl. Acad. Sci. USA. 1984;81:6747–6751. doi: 10.1073/pnas.81.21.6747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Wijk T., Tomassen S.F.B., Tilly B.C. Increased vesicle recycling in response to osmotic cell swelling. Cause and consequence of hypotonicity-provoked ATP release. J. Biol. Chem. 2003;278:40020–40025. doi: 10.1074/jbc.M307603200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.