

Abstract
Elongation factor (EF) Tu delivers aminoacyl-tRNAs to the actively translating bacterial ribosome in a GTP-hydrolysis-dependent process. Rapid recycling of EF-Tu, catalyzed by EF-Ts, is required for efficient protein synthesis in vivo. Here we report a combined theoretical and experimental approach aimed at identifying three-dimensional communication networks in EF-Tu. As an example, we focus on the mechanistic role of second-shell residue Asp109. We constructed full-length structural models of EF-Tu from Escherichia coli in the GDP-/GTP-bound state and performed several 10-ns-long molecular-dynamics simulations. During these simulations, the side chain of Asp109 formed a previously undetected transient hydrogen bond to His22, an invariant residue in the phosphate-binding loop (P-loop). To experimentally validate our molecular-dynamics results and further analyze the role of this hydrogen bond, we determined all rate constants for the multistep reaction between EF-Tu (wild-type and two mutants), EF-Ts, GDP, and GTP using the stopped-flow technique. This mutational analysis revealed that the side chain of Asp109 is important for acceleration of GDP, but not for GTP dissociation by EF-Ts. The possibility that the Asp109 side chain has a role in transition-state stabilization and coupling of P-loop movements with rearrangements at the base side of the nucleotide is discussed.
Introduction
During the elongation phase of translation, codon-dependent aminoacyl (aa)-tRNA delivery to the ribosome is catalyzed by elongation factor (EF) Tu in bacteria. The bound GTP is hydrolyzed upon correct codon-anticodon interaction in the ribosomal A-site. EF-Tu then undergoes a large conformational change into the inactive GDP-bound form of the factor, releasing the bound aa-tRNA, and dissociating from the ribosome. EF-Tu•GDP must be recycled from the inactive GDP-bound form to the active GTP complex before it can bind another aa-tRNA and enter the next cycle of elongation. However, the intrinsic rate of nucleotide exchange is too slow to sustain the efficient protein synthesis observed in vivo. To overcome the tight binding of the GDP-nucleotide and facilitate rapid nucleotide exchange, regeneration of EF-Tu•GTP is catalyzed by the nucleotide exchange factor EF-Ts. EF-Tu•GDP binds EF-Ts to form an unstable ternary complex that rapidly dissociates into EF-Tu•EF-Ts and GDP. After binding of GTP and the subsequent dissociation of EF-Ts from EF-Tu, EF-Tu•GTP binds another aa-tRNA and delivers it to the ribosome. All of the interactions among EF-Tu, EF-Ts, and guanine nucleotides are reversible and occur in a steady-state environment in vivo (1,2).
The crystal structure of the EF-Tu•EF-Ts complex (3) suggests that EF-Ts catalyzes nucleotide release by altering at least three of the crucial binding interactions between EF-Tu and guanine nucleotides. These crucial elements, common among GTPases, are 1), the conserved NKxD motif (residues 135–138, Escherichia coli numbering) that binds the guanine base; 2), the bound magnesium ion that coordinates the β- and γ-phosphates, stabilizing their negative charges; and 3), the P-loop, which contains the consensus element GxxxxGK(S/T) (residues 18–25) that coordinates the β-phosphate of the bound nucleotide. A previous analysis using rapid kinetic techniques revealed a 60,000-fold increase in the dissociation rate of GDP from the ternary complex EF-Tu•GDP•EF-Ts as compared to EF-Tu•GDP (2). Several aspects that contribute to the mechanism of nucleotide exchange have been studied through a combination of mutational and kinetic analyses based mainly on molecular interactions identified in the structure of the binary EF-Tu•EF-Ts complex. These studies suggest a concerted process in which the interaction between the three major nucleotide interaction sites (including the phosphate and base moieties) are altered in a well coordinated fashion involving communication between these sites to achieve efficient dissociation of GDP (1,2,4). Although this coupling is critical for efficient EF-Ts-catalyzed nucleotide exchange in EF-Tu, nothing is known about how it is achieved, given that it requires communication over >20 Å and involves amino acids separated by >100 amino acids in the primary sequence of the protein. Experimental and computational studies of many protein/protein and protein/nucleic acid complexes strongly support the view that conformational transitions in these complexes are communicated by a three-dimensional (3D) network of interresidue contacts (5–8). It is particularly difficult to identify the amino acids involved in this communication network because this is a dynamic feature involving so-called second-shell residues, which do not directly participate in interactions with the respective substrates like the nucleotide and EF-Ts in the case of EF-Tu. Nevertheless, these second-shell residues have been shown to be critical for the function and specificity of a number of enzymes due to their role in positioning catalytically active residues (10) and providing critical flexibility to secondary structure elements involved in the respective reaction (11,12). Evolutionary conservation can provide valuable information for identifying second-shell residues that participate in the 3D communications networks (13,14).
Here we report a prime example of how a combined theoretical and experimental approach can be used to successfully identify and study these 3D communication networks. Previous studies reported slow evolutionary speeds for both Asp109 and Glu152 in EF-Tu (13,14), and indicated that the latter is involved in a functionally important salt bridge with the N-terminal domain of EF-Ts, whereas the former apparently does not participate in any interactions with either EF-Tu or EF-Ts. These observations suggest an unknown functional role of the Asp side chain during the nucleotide exchange reaction that is specific for the bacterial protein. Our own molecular-dynamics (MD) simulations of the EF-Tu•GDP and EF-Tu•GTP complex revealed that a transient hydrogen (H)-bond can be formed between the aspartate residue at position 109 and a residue in the P-loop, His22 (see Fig. S1 and Fig. S2 in the Supporting Material). A subsequent detailed kinetic analysis of an Asp109-to-alanine substitution in EF-Tu revealed a functional role of this aspartate side chain specific for EF-Ts stimulated GDP but not for GTP dissociation. This is the first report, to our knowledge, of an MD-guided design of EF-Tu mutants. Our goal was to identify the functional role of second-shell residues that are members of the 3D communication network involved in the catalytic mechanism of nucleotide exchange in EF-Tu. By combining our analysis of the dynamic properties of EF-Tu in silico with a rapid kinetic analysis in vitro, we found that Asp109 plays an important role during the nucleotide exchange reaction by stabilizing the EF-Tu•GDP•EF-Ts≠ transition state and linking the interactions of EF-Ts with the base side and the phosphate side in EF-Tu. These interactions seem to be specific for the acceleration of GDP dissociation in bacteria but not in eukaryotes.
Materials and Methods
MD simulations
The initial model for E. coli EF-Tu in the GTP-bound conformation was obtained by constructing a homology model using the Swiss-Model server (15) and the crystal structure of T. aquaticus EF-Tu•GDPNP as a template (PDB ID 1EFT). Transformation of the GDPNP molecule to GTP was done by hand. The model for E. coli EF-Tu in the GDP-bound conformation was obtained from the respective crystal structure (PDB ID 1EFC). Because the seven N-terminal residues were missing in this structure, conformations of these amino acids were assigned identically to those in the GTP-bound model. Hydrogen atoms were added to both models using psfgen in the NAMD software package (16), and histidine side chains were protonated at the ɛ-nitrogen only. Initial models were minimized in vacuum and then placed in a water box extending at least 10 Å from the protein in all directions. Water molecules present in the respective crystal structures were included in this box, and all other water molecules were added at random using the SOLVATE package in NAMD (16). Relaxation of the solvated system was achieved by minimizing the positions of water molecules, followed by protein and ligand atoms in two iterative rounds. Sodium ions were then added in random positions by the AUTOIONIZE package in VMD (17) to neutralize the total charge of the system, followed by a final all-atoms minimization. All minimizations were performed using the NAMD software package until no change in total energy was observed over 1000 steps. MD simulations were performed on solvated models with a step size of 0.5 fs using the CHARMM22 force field as implemented in the NAMD package (16) while invoking periodic boundary conditions. Minimized models were initially equilibrated at 300 K and 350 K for 150 ps at constant pressure (1 atm). Production phase simulations were performed after crossing trajectories and were carried out for 10 ns. All simulations were performed in the NAMD software package, and visualization was carried out in VMD (16,17). (NAMD and VMD were developed by the Theoretical and Computational Biophysics Group at the Beckman Institute for Advanced Science and Technology, University of Illinois at Urbana-Champaign.)
We analyzed the MD simulations using data sampled every 0.5 ps. Calculations of root mean-square deviation (RMSD) and root mean-square fluctuation (RMSF) were performed using in-house-written scripts invoked with the VMD software package (17). For the RMSD calculations, the first 10 N-terminal amino acids were excluded due to the high level of mobility exhibited throughout all simulations, consistent with the respective crystallographic studies in which the N-terminal amino acids were either not resolved or exhibited particularly large B-factors.
To assess whether H-bond formation can occur between the side chains of Asp109 and His22, we measured the distance between the two carboxylate oxygens (Oα/β) of the aspartate side chain and the proton at the Nɛ of the histidine residue over the last 5 ns of each simulation. Each oxygen atom of the carboxylate side chain on Asp109 was considered independently. Histograms were generated from the Oα/βAsp•••H-NɛHis distances measured by subsequent binning using a 0.25 Å bin size and were plotted at the bin midpoint. For both simulations, a bimodal distribution of counts was observed. To deconvolute the contribution of each state (mode), the data were fitted with the sum of two Gaussian functions (Eq. 1) using the TableCurve (Jandel Scientific, San Rafael, CA) and Prism (GraphPad Software, La Jolla, CA) software packages:
| (1) |
where A and D represent the amplitudes of the first and second Gaussian functions, respectively; B and E represent the mean O•••H distances for the two functions; and C and F represent the distribution in O•••H distance around the respective mean distance.
We determined the occupancy of each H-bonded state by integrating the Gaussian centered on 2 Å over the interval (−∞,∞) and expressing it as a percentage of the sum of both Gaussians integrated over the same interval.
Buffers and reagents
Experiments were performed in buffer A (50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 30 mM KCl, 7 mM MgCl2) at 20°C. Chemicals were purchased from Sigma or VWR. Fluorescent mant-GDP and mant-GTP were obtained from Molecular Probes (Eugene, OR).
Mutagenesis
The plasmid pEECAHis (4) containing the full-length E. coli tufA gene was obtained from M. V. Rodnina (Max Planck Institute for Biophysical Chemistry, Göttingen, Germany). The Quikchange method was then used for site-directed mutagenesis. Mutations were verified by sequencing.
Protein expression and purification, and complex formation were performed in a manner similar to that described by Gromadski et al. (2) and are described in the Materials and Methods section of the Supporting Material.
Rapid kinetic measurements
Fluorescence stopped-flow measurements were performed on a KinTek SF-2004 stopped-flow instrument (KinTek, Austin, TX). Tryptophan fluorescence was excited at 280 nm and measured after it passed a WG-305-F filter (Newport Filters, Irvine, CA). Mant-GDP/GTP fluorescence was excited indirectly using fluorescence resonance energy transfer from the single tryptophan residue proximal to the nucleotide-binding site. Mant-GDP/GTP fluorescence was measured after it passed a LG-400-F filter (Newport Filters). Experiments were carried out at 20°C in buffer A by rapidly mixing equal volumes (25 μL each) of reactants and monitoring the time courses of the fluorescence change. Fluorescence time courses were evaluated by fitting to an exponential function with the characteristic apparent time constant (kapp), the amplitude (A), and the final signal (F∞) according to the equation F = F∞ + A × exp(−kapp × t), where F is the fluorescence at time t. Calculations were performed using TableCurve (Jandel Scientific) and Prism (GraphPad Software) software. The standard deviations of the kapp-values were calculated using the same software. All elemental rate constants of nucleotide exchange except k-2 were measured directly. The value for k-2 was calculated from all other elemental rate constants based on the kinetic scheme depicted in Scheme 1. The standard deviations of the rate constants were estimated from the variation of values obtained in different experiments.
Scheme 1.
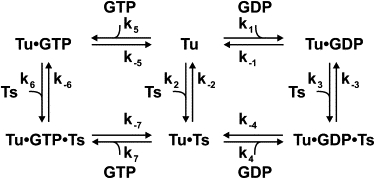
Kinetic scheme of nucleotide exchange in EF-Tu.
Calculation of k-2 for EF-Tu WT, D109A, and E152A
Using an approach similar to that described by Gromadski et al. (2), we calculated k-2-values using the law of mass action. Based on the mechanism in Scheme 1, the rate constants k-2 were calculated from rate constants of the right (GDP) branch ([k-2 = k-1k2k-3k4/(k1k3k-4)]) and left (GTP) branch ([k-2 = k-5k2k-6k7/(k5k6k-7)]), yielding two independently calculated values for k-2.
Results
Asp109 forms a transient H-bond to His22
The observation that the aspartate side chain in position 109 of EF-Tu from E. coli is conserved in >99% of the bacterial EF-Tu sequences found in the SwissProt database suggests a functional requirement for aspartate at this position. However, inspection of the available crystal structures of EF-Tu bound to the respective nucleotide (GDP and GTP) as well as to its exchange factor (EF-Ts) did not reveal any direct interactions involving the Asp109 side chain. The main-chain oxygen of Asp109 participates in a H-bond interaction with the side chain of the invariant arginine (Arg) in position 12 of EF-Ts, linking it with the equally conserved Glu152 in helix D of EF-Tu, which has been shown to be important for efficient EF-Ts-catalyzed nucleotide exchange (4,18). These observations suggest a role for the Asp109 side chain during a phase in the nucleotide exchange reaction that has not been trapped by x-ray crystallography. This most likely involves a highly dynamic transient intermediate, such as the ternary complexes EF-Tu•GDP•EF-Ts and EF-Tu•GTP•EF-Ts or their respective transition states (Scheme 1), that is not accessible by crystallographic methods. We wanted to address the functional role of Asp109 by combining MD simulations to describe the dynamic properties of the Asp side chain in silico with a subsequent analysis of the mechanistic role using rapid kinetics in vitro. To study the dynamic properties of Asp109 by MD simulations, and to circumvent effects due to sequence variations in the different organisms used to determine the respective x-ray structures, we constructed two full-length structural models of E. coli EF-Tu in complex with GTP and GDP using homology modeling, followed by hydration with explicit waters and subsequent energy minimization. Inspection of these models revealed that the side chain of Asp109 is in close proximity to His22, which would be capable of accepting an H-bond from one of the carboxylate oxygens of Asp109. However, in these energy-minimized static models the side chain of Asp109 is oriented in such a way that prevents participation in an H-bonding interaction. This is surprising because this H-bond could be established through a few minor bond rotations, consistent with an evolutionary conserved role for Asp109 as an H-bond acceptor. We wanted to know whether under more physiological conditions (which would also take into account the overall dynamic properties of the enzyme in water) this H-bond could indeed be formed in EF-Tu, which would explain the high degree of conservation of Asp109.
To address this question, we performed, based on these models, 10-ns-long MD simulations of the wild-type (WT) enzyme and an in silico mutant carrying an alanine substitution at position 109 (D109A), bound to GTP or GDP, at 300 K. The stability of EF-Tu during these simulations was judged by measuring the RMSD of the backbone atoms with respect to the initial structure throughout the whole simulation (Fig. 1 A and Fig. S3). After an initial increase in the RMSD of ∼1.5 Å, all four simulations were stable after 2 ns, yielding 8 ns of stable simulation time. When both the mutant and the WT GDP-bound complexes were compared with the respective GTP-bound complexes, the two GDP-bound complexes exhibited slightly larger fluctuations in RMSF values (Fig. 1 B), in agreement with the more open conformation of EF-Tu•GDP, which provides less intramolecular stabilization than the EF-Tu•GTP complexes (Fig. S2).
Figure 1.
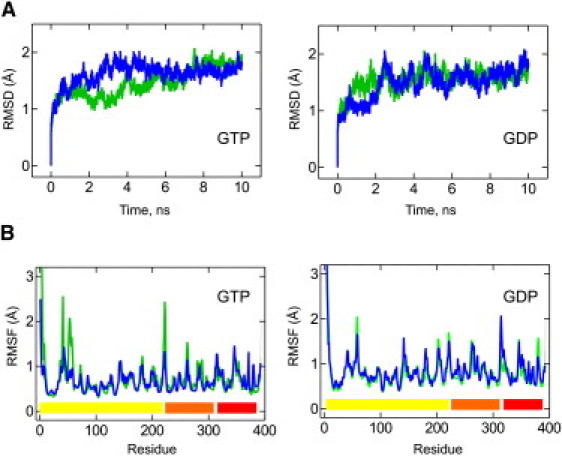
Conformational changes during MD simulation. (A) RMSDs of EF-Tu WT (green) and EF-Tu D109A (blue) from the starting conformation for the GTP and GDP conformations at 300 K. RMSDs were calculated using all backbone atoms and excluded the mobile N-terminus. (B) Cα RMSF for EF-Tu WT (green) and D109A (blue) in the GTP and GDP conformations. Colored bars at the bottom indicate the domain structure of the protein (domains 1–3 in yellow, orange, and red, respectively).
Interestingly, when we compared the RMSF of each Cα in domain 1 of EF-Tu (Fig. 1 B) between the WT and the D109A mutant simulations, we observed no significant increase in flexibility for the P-loop, including His22 in either the GTP- or GDP-bound state, indicating that the aspartate side chain is not required for its stability in these complexes. However, both complexes revealed lower RMSF values (i.e., decreased flexibility) for the majority of switch I residues (residues 44–69) in the D109A simulations, as well as consistently higher values (i.e., increased flexibility) for the N-terminal half of helix D (residues 139–144) and the preceding nucleotide specificity motif (NKxD, residues 135–138), which is slightly more pronounced in the GDP complex (Fig. 1 B).
To investigate whether a transient H-bond can form between Asp109 and His22, we monitored the Oα/βAsp•••HHis-NɛHis distance over 5 ns of the simulation (from 5 to 10 ns). The distances between the acidic hydrogen on His22 and both carboxylate oxygens of Asp109 (Oα/βAsp) were measured in the GDP- and GTP-bound simulations and plotted as histograms (Fig. 2). The Oα/βAsp•••HHis-NɛHis distances exhibited a bimodal distribution, with the first mode centered over 2 Å and the second over 4 Å. This suggests that two major states exist for the interactions between these two side chains. Based on the frequently applied distance criterion for assigning an H-bond of <3.5 Å for the O•••H distance (19), we believe that the distribution at small Oα/βAsp•••HHis distances represents a population of conformations in which Asp109 and His22 are engaged in H-bonding with each other. Thus, the population that is observed at longer OAsp•••HHis distances and centered over 4 Å may represent non-H-bonding conformations. This is supported by the observation that the distributions representing H-bonding conformations have mean Oα/βAsp•••HHis distances ranging from 2.1 to 2.3 Å. These distances are well below the more conservative cutoff distance for H-bonding of 2.7 Å (the sum of van der Waals radii for hydrogen and oxygen). To deconvolute the partitioning between each state, and to provide a means of estimating the relative contribution of H-bonding conformations at physiological temperatures (300 K), we fit these histograms to functions with two Gaussian terms (Fig. 2, A and B). In fact, depending on the respective oxygen (α or β) and simulation (GDP- or GTP-bound state), between 81.6% and 97.5% of the H-bonding states (first mode) fall below the 2.7 Å cutoff. In comparison with the non-H-bonding state (second mode), considering both carboxylate oxygens of Asp109, WT EF-Tu exists in the H-bonding states during 19.6% (GTP) or 30.5% (GDP) of the analyzed simulation. On the basis of these MD simulations, we think it is likely that EF-Tu is indeed capable of forming an intramolecular H-bond between Asp109 and His22, and that this H-bond contributes significantly to the dynamics of the protein at 300 K. The fact that the H-bonded state is only sampled between 20% and 30% of the simulation time suggests a minor functional role in these binary complexes. Together with the evolutionary conservation of Asp109, these results suggest that the H-bonded state contributes more significantly to one of the other complexes formed during the functional cycle of EF-Tu (e.g., the EF-Ts-catalyzed nucleotide exchange (Scheme 1)). Together with the similar level of conservation of the functionally important Glu152 in helix D of EF-Tu, our findings also suggest that the H-bond between His22 and Asp109 may be important for the 3D communication network in the two ternary complexes (EF-Tu•GDP•EF-Ts and EF-Tu•GTP•EF-Ts) that are transiently formed during EF-Ts-catalyzed nucleotide exchange.
Figure 2.
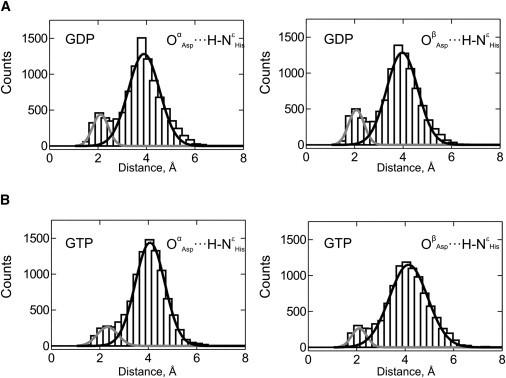
H-bonding between Asp109 and His22. Distances between the two terminal oxygen atoms (Oα and Oβ) of the Asp109 residue and the hydrogen at the Nɛ of His21 were measured during the second half of MD simulations (ns 5–10) for the EF-Tu•GDP and EF-Tu•GTP complexes and plotted as bars. Distances are shown for the Oα and Oβ oxygen atoms of the aspartate side chain in the GDP (A) and GTP (B) complexes. The superimposed Gaussian fits indicate the contribution of the H-bonded (gray) and non-bonded (black) states.
Mechanistic role of Asp109-His22 H-bonding
We then sought to determine whether a functional role for the Asp109-His22 H-bonded state could be confirmed in vitro. To that end, we constructed a mutant version of EF-Tu containing a single amino acid substitution at position 109, replacing aspartate with alanine (D109A, corresponding to our in silico mutant) to prevent the formation of the H-bond to His22. We then performed a detailed kinetic analysis of the nucleotide exchange reaction (Scheme 1) for this mutant and the WT protein, along the lines of previous studies, using rapid kinetics (2). When we compared our results with previously reported WT data (2,4,20), as well as the rate constants determined in this study, we found that replacing Asp109 with alanine did affect the rate constant for EF-Ts-stimulated GDP dissociation (k-4, a 13-fold decrease) as well as the rate constant for EF-Ts association to nucleotide-free EF-Tu (k2, a threefold decrease). Experimental results for the WT are in very good agreement with the results from previous studies (see Table 1 for a summary of the obtained rate constants, and Figs. 3 and 4 and Fig. S4 for the kinetic analysis).
Table 1.
Effect of mutations in EF-Tu on the experimentally determined rate constants of EF-Tu interaction with EF-Ts and guanine nucleotides
| Rate constant∗ | WT† | WT‡ | D109A† | E152A†/§ |
|---|---|---|---|---|
| k1 (× 106 M−1s−1) | 2.8 ± 0.1 | 2.0 ± 0.5 | 3.3 ± 0.1 | 2.0§ |
| k−1 (s−1) | 0.002 ± 0.001 | 0.002 ± 0.001 | 0.004 ± 0.001 | 0.003§ |
| k2 (× 107 M−1s−1) | 1.3 ± 0.1 | 1 ± 0.2 | 0.3 ± 0.1 | 0.08 ± 0.01† |
| k−2 (× s−1) | 0.01 | 0.03 | 0.04 | 0.01§ |
| k3 (× 107 M−1s−1) | 3 ± 1 | 6 ± 1 | 4 ± 1 | 0.46§ |
| k−3 (s−1) | 438 ± 50 | 350 ± 50 | 408 ± 64 | 170 ± 30§ |
| k4 (× 106 M−1s−1) | 7 ± 1 | 14 ± 5 | 7 ± 1 | 7.6§ |
| k−4 (s−1) | 219 ± 25 | 125 ± 25 | 16 ± 1 | 9 ± 2§ |
| k5 (× 105 M−1s−1) | 4.3 ± 0.1 | 5.0 ± 1 | 3.9 ± 0.1 | 2.5 ± 0.1† |
| k−5 (s−1) | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.05 ± 0.01 | 0.05 ± 0.01† |
| k6 (× 107 M−1s−1) | 12.4 ± 0.3 | 3 ± 0.5 | 3.3 ± 0.2 | 5.3 ± 1.6† |
| k−6 (s−1) | 60 ± 10 | 60 ± 10 | 90 ± 10 | 390 ± 90† |
| k7 (× 106 M−1s−1) | 3.9 ± 0.7 | 6 ± 1 | 1.8 ± 0.2 | 5.9 ± 2.7† |
| k−7 (s−1) | 95 ± 10 | 85 ± 10 | 50 ± 5 | 8.5 ± 1† |
Figure 3.
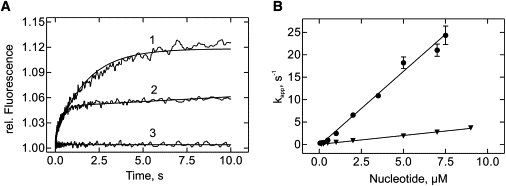
Interaction of guanine nucleotides with EF-Tu D109A in the absence of EF-Ts. (A) Time courses of mant-GTP (1 μM, line 1) or mant-GDP (1 μM, line 2) binding to nucleotide-free EF-Tu D109A (0.25 μM) measured by fluorescence resonance energy transfer between the single tryptophan in EF-Tu and the mant group (line 3 control without EF-Tu). (B) Concentration dependence of kapp. kapp-values were calculated by single-exponential fitting from time courses in panel A. Circles: mant-GDP; triangles: mant-GTP.
Figure 4.
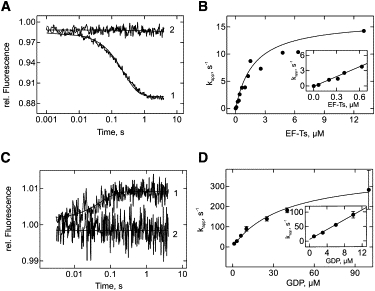
Interaction of EF-Tu D109A with EF-Ts and GDP. (A) Time courses of dissociation of EF-Tu•mant-GDP (0.15 μM) in the presence of EF-Ts (0.2 μM) and excess unlabeled GDP (25 μM, line 1), and in the absence of EF-Ts (line 2). The fluorescence of the mant group was monitored. (B) Concentration dependence of kapp on EF-Tu•mant-GDP dissociation. The kapp-values were calculated by single-exponential fitting of the time courses in A. (C) Time courses of the dissociation of EF-Tu•EF-Ts (0.5 μM) in the presence of GDP (1 μM, line 1) or absence of GDP (line 2), as monitored by the fluorescence of Trp184 in EF-Tu. (D) Concentration dependence of kapp for EF-Tu•EF-Ts dissociation in the presence of GDP. The kapp-values were calculated by single-exponential fitting of the time courses in C.
Glu152 is critical for the EF-Ts-catalyzed dissociation of both GTP and GDP
Asp109 in EF-Tu also forms an H-bond via its main-chain carbonyl to the highly conserved side chain of Arg12 in EF-Ts, which in turn participates in a salt bridge with the invariant glutamate (Glu)152 in helix D of EF-Tu. Since Asp109 and Glu152 seem to be evolutionarily linked (13,14), we wanted to know whether disruption of the salt bridge between Glu152 in EF-Tu and Arg12 in EF-Ts is also specific for the acceleration of GDP dissociation (k-4) and not GTP (k-7) dissociation from the ternary complex EF-Tu•GDP/GTP•EF-Ts. Previous studies (4) focused solely on the role of the highly conserved Glu152 on the GDP exchange mechanism, and provided no information on the respective mechanism for GTP. Values for the rate constants of the GDP branch (k1, k-1, k2, k-2, k3, k-3, k4, and k-4; summarized in Table 1) revealed effects on both the EF-Ts-stimulated GDP dissociation (k-4) and the interaction of EF-Ts with EF-Tu (k2, k-2, k3, and k-3). To assess whether these effects are also limited to the GDP-bound forms of EF-Tu, we performed a similar kinetic analysis on the previously unreported GTP branch of the nucleotide exchange mechanism (Fig. S5) using the published alanine substitution mutant of EF-Tu (4). When we compared the obtained rate constants for the GTP branch (k2, k-2, k5, k-5, k6, k-6, k7, and k-7; summarized in Table 1), we observed similar effects as for the GDP branch.
Discussion
Previous structural and biochemical studies on the mechanism of guanine nucleotide exchange in EF-Tu suggested that this process occurs through a three-part mechanism involving breakage of the Mg2+ coordination, structural changes in the P-loop (the phosphate side), and disruption of interactions with the nucleotide base (the base side). However, little is known about the timing and structural dynamics of these events. This is reflected by the somewhat surprising observation that the disruption of any of the putative key interactions in the EF-Tu•EF-Ts complex derived from the available x-ray structures of the binary complex resulted in only moderate changes in the efficiency of nucleotide exchange (1,4,18,21). EF-Ts-stimulated nucleotide exchange may therefore rely on a number of second-shell residues to induce and relay small rearrangements through a network of changes in EF-Tu that contribute synergistically to the efficient exchange of guanine nucleotides. The ability to identify these intrinsically dynamic communication networks would enhance our understanding of the design principles underlying the function and structural dynamics of biomolecular activities. However, it is inherently difficult to identify the participants of these networks and assign a specific function to them using only static structural information derived from x-ray crystallographic studies. By employing a combination of MD simulations, mutagenesis, and kinetic analysis of the nucleotide exchange mechanism using rapid kinetics, we were able to identify the second-shell residue Asp109 as just such a mechanistically important residue in vivo.
Role of Asp109 during EF-Ts-catalyzed nucleotide exchange
The highly conserved Asp109 interacts with Arg12 of EF-Ts through the main-chain carbonyl group. The strict conservation as an aspartate residue in bacterial and mitochondrial EF-Tu sequences indicates that the side chain plays a functional role during EF-Ts-mediated nucleotide exchange in bacteria but not in eukaryotes. Although no direct interaction of the side chain with other residues has been reported (3,22–24), mutation of Asp109 has a significant effect on the EF-Ts-mediated GDP (but not GTP) dissociation. In contrast, disruption of the salt bridge between Glu152 and Arg12 affects the rate of nucleotide dissociation for GDP (4) and GTP (this study), and significantly alters the association rates of EF-Ts to the respective binary complexes and nucleotide-free EF-Tu. Arg12 also interacts via an H-bond with the main-chain carbonyl oxygen of Asp109, thereby connecting Glu152 and Asp109 in the EF-Tu•EF-Ts complex (Fig. 5). The use of MD simulations in this study allowed us for the first time, to our knowledge, to identify and assign a role for the highly conserved side-chain Asp109. As part of the dynamic properties of EF-Tu, Asp109 is able to participate in a previously unidentified H-bond with His22 located in the P-loop, thus connecting a secondary shell residue (Asp109) with residues at the phosphate side in the nucleotide-binding pocket (His22). This effectively establishes a communication path between helix D and the P-loop. However, this H-bond is only observed for a fraction of the simulation time (20–30%) in either the EF-Tu•GDP or EF-Tu•GTP complex, suggesting sampling of a functional interaction between the two residues, which becomes important upon formation of the transient ternary complex (EF-Tu•nucleotide•EF-Ts) or an intermediate on the reaction path. This is supported by the close approach of the two side chains (3.09 Å) in the EF-Tu•EF-Ts complex (PDB-ID 1EFU) and the reduced affinity of EF-Ts (k2) for the nucleotide-free EF-Tu (D109A), as well as the increased flexibility of the N-terminal half of helix D and the NKxD specificity motif in the D109A simulations.
Figure 5.
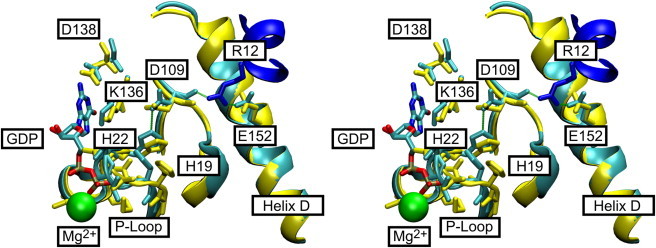
Structure of the nucleotide-binding pocket (stereo view). (A) H-bonding interactions between Asp109 and His22: detailed view of the nucleotide interactions in the EF-Tu•GDP (yellow) and EF-Tu•EF-Ts (cyan for EF-Tu and blue for EF-Ts) complexes. Residues involved in nucleotide binding are highlighted. Arg12 of EF-Ts is depicted in dark blue, and Mg2+ is represented by a green sphere. H-bonds of the proposed H-bond relay are indicated in green. The figure was prepared with VMD (17) using coordinate sets 1EFC and 1EFU in the PDB (3,24).
Our detailed kinetic analysis of the nucleotide exchange mechanism (Scheme 1) in the absence (D109A) and presence (WT) of the predicted H-bond (Table 1) revealed that only the rate constants for GDP (and not GTP) dissociation from the EF-Tu•GDP•EF-Ts complex (k-4, lowered 13-fold) and EF-Tu/EF-Ts association (k2, lowered threefold) were affected. Based on these rate constants and the respective equilibrium dissociation constants (KD), we can calculate the change in Gibbs free energy (ΔG) of step 2 (interaction of EF-Tu and EF-Ts) and step 4 (interaction of GDP with the EF-Tu•EF-Ts complex) for both the WT and the D109A mutant of EF-Tu as follows: ΔG2,WT = −51.1 kJ/mol and ΔG2,D109A = −44.2 kJ/mol; ΔG4,WT = −25.3 kJ/mol and ΔG4,D109A = −31.6 kJ/mol. The fact that only k-4 and k2 differ (decreased) between the D109A mutant and the WT enzyme can be explained by a destabilization of the transition states for the two reactions resulting in higher transition state energies upon removal of the H-bond between Asp109 and His22 (depicted by the length of the arrows of the respective rate constants in Fig. 6). On the other hand, the rate constants for the reverse reactions (k4 and k-2) are not changed, suggesting that the difference in the Gibbs free energies between the destabilized transition states and the mutant EF-Tu•EF-Ts complex is not affected. The latter can be explained by a destabilization of the EF-Tu•EF-Ts complex to a similar extent as the destabilization of the respective transition states (Fig. 6). Consequently, the Gibbs free energy ΔG4,D109A should be larger and ΔG2,D109A should be smaller than in the WT case. This is consistent with the calculated values for these ΔGs (vide supra). It is furthermore reflected by the respective ΔΔGs (ΔΔG4 = 6.4 kJ/mol and ΔΔG2 = −7.0 kJ/mol) between the WT and mutant, which correspond well with the energy contribution of a single H-bond being removed. The fact that the observed changes in ΔΔGs indeed reflect the removal of a single H-bond demonstrates the power of this combined MD and experimental approach. It also indicates the importance of this H-bond for the mechanism and the molecular dynamics of the protein (vide infra). Furthermore, if the H-bond was part of a degenerate communication network within the protein, other members of the network would compensate for its loss, and it would have been difficult to measure the difference on a specific rate constant, further supporting the significance of this finding.
Figure 6.

Gibbs free-energy diagram depicting the transition-state stabilizing effect of the H-bond between Asp109 and His22. Gibbs free-energy diagram for the two reactions described by the equilibrium constants K2 and K4 (Scheme 1) of the WT (black) and D109A mutant (gray) of EF-Tu, respectively. Removal of the H-bond between His22 and Asp109 results (as calculated from the experimentally determined rate constants summarized in Table 1) in destabilization of the Tu•Ts complex and the two transition states (Tu•Ts≠ and Tu•GDP•Ts≠) to a similar extent, as reflected by the comparable ΔΔGs for both reactions (ΔΔG4 = 6.4 kJ/mol and ΔΔG2 = 7.0 kJ/mol) consistent with the energy for a single H-bond. This increase in energy of the transition states and the Tu•Ts complex results in decreased rate constants for k-4 and k2, and no changes for the rate constants of k4 and k-2, as indicated by the length of the respective arrows (gray versus black).
To understand the role of this H-bond for the nucleotide exchange reaction, it is important to view the observed destabilization of the EF-Tu D109A complexes as a stabilization of the respective complexes with the WT protein. A recent kinetic analysis of the nucleotide exchange reaction in yeast supported a different mechanism of nucleotide exchange and, moreover, different structural requirements for the nucleotide-binding properties of the eukaryotic factor (eEF1A) compared with bacterial EF-Tu (25). Dissociation of the bound nucleotide is up to 100 times faster for eEF1A than for EF-Tu (0.13 s−1 vs. 0.002 s−1 for GDP, and 0.1 s−1 vs. 0.02 s−1 for GTP). These differences may also be reflected by the different mode of interaction between eEF1A and its nucleotide exchange factor, eEF1B. Asp109 may therefore be important for overcoming the additional stabilizations of GDP-binding observed in the prokaryotic EF-Tu•GDP complex (as indicated by the lower dissociation constant) compared to their eukaryotic counterparts eEF1A•GDP. This in turn suggests that prokaryotes require this H-bond (and therefore have maintained it during evolution) to overcome the additional stabilization of GDP binding by stabilizing the EF-Tu•EF-Ts complex and in particular the two transition states.
The results we obtained by combining MD simulations with a detailed rapid kinetics analysis provide the first step toward unraveling the 3D communication network that facilitates 60,000-fold and 3,000-fold accelerations of the nucleotide dissociation rate in the transient ternary complexes EF-Tu•GDP•EF-Ts and EF-Tu•GTP•EF-Ts, respectively. This is particularly important because although the eukaryotic ternary complexes have been successfully crystallized (26), no structural information is available for the bacterial factors, due to the low stability of these ternary complexes.
Role of His22 for efficient nucleotide release
His22 is located two amino acid residues downstream of the crucial amino acids Val20 and Asp21, which are involved in flipping of the main-chain carbonyl (between Val20 and Asp21; Fig. S6) in EF-Tu upon EF-Ts interaction (3). This main-chain carbonyl is positioned in the EF-Tu•EF-Ts complex such that its partial negative charge as well as the resulting steric clash will disfavor the binding of the β-phosphate of GDP (Fig. S6). Hence, formation of the H-bond between Asp109 and His22 implies a function in constraining the flexibility of the peptide backbone upstream of His22, effectively forming the pivot point that is required for flipping the peptide backbone upon intrusion of the conserved Phe81 of EF-Ts and the subsequent displacement of helices B and C in EF-Tu. The N-terminal end of helix C contains the strictly conserved Met112, a residue that is in close proximity to the conserved His19 and His22 and is moved by several Ångströms, providing additional flexibility for the P-loop between His19 and His 22. Movement of these two helices results in widening of the crevice in which Ile17 is buried. Ile17 is the first amino acid upstream of the P-loop sequence and which shows a different orientation in the EF-Tu•GDP and the EF-Tu•EF-Ts complex. Relieving the spatial constraints on the N-terminal side of the P-loop, in particular at Ile17, allows the P-loop to be more flexible and explore a larger conformational space. Along with stabilizing the C-terminal side of the P-loop through additional H-bonding interactions (His22-Asp109), this will ultimately lead to the flip of the peptide bond between Val20 and Asp21, and subsequently promote dissociation of the bound nucleotide. This is consistent with the observation that removal of this H-bond destabilizes the transition state of step 4 of the nucleotide exchange mechanism (EF-Tu•EF-Ts•GDP≠), but not the ternary complex Tu•GDP•EF-Ts itself (vide supra).
Conclusions
By using evolutionary analysis in combination with MD simulations, we were able to identify Asp109 as a second-shell residue that serves as a relay point (the Glu152-Arg12-Asp109-His22 relay) in the 3D communication network underlying the protein dynamics that are responsible for facilitating efficient nucleotide exchange in bacterial EF-Tus. By combining these computational approaches with the determination of the rate constants governing this interaction using rapid kinetic techniques, we were able to further define the role of Asp109 as a requirement for efficient GDP exchange in EF-Tu by contributing to the stabilization of the respective transition state in prokaryotes. We believe that Asp109 is important for linking structural rearrangements at the base side of the binding pocket with structural rearrangements at the phosphate side of the binding pocket. In this study we show that very short MD simulations (which should be carefully interpreted with respect to energetics, due to limitations in sampling and possible inaccuracies in the force field), confirmed by experimental data, can be used to develop likely enzyme mechanisms. Our approach clearly demonstrates the power of combining in silico and in vitro studies to identify the dynamic 3D communication pathways underlying the efficient function of proteins.
Acknowledgments
We thank C. Knudsen (Åarhus, Denmark) for providing the plasmid coding for the EF-Ts-intein construct, R. Quaghebeur for expert technical assistance, and U. Kothe for a critical reading of the manuscript and valuable suggestions.
This work was supported by grants from the National Science and Engineering Research Council of Canada, the Alberta Ingenuity Fund, and the Canadian Foundation for Innovation. Supercomputer time was provided by the WestGrid/Compute Canada Resource Allocation Committee.
Supporting Material
References
- 1.Dahl L.D., Wieden H.J., Knudsen C.R. The importance of P-loop and domain movements in EF-Tu for guanine nucleotide exchange. J. Biol. Chem. 2006;281:21139–21146. doi: 10.1074/jbc.M602068200. [DOI] [PubMed] [Google Scholar]
- 2.Gromadski K.B., Wieden H.J., Rodnina M.V. Kinetic mechanism of elongation factor Ts-catalyzed nucleotide exchange in elongation factor Tu. Biochemistry. 2002;41:162–169. doi: 10.1021/bi015712w. [DOI] [PubMed] [Google Scholar]
- 3.Kawashima T., Berthet-Colominas C., Leberman R. The structure of the Escherichia coli EF-Tu.EF-Ts complex at 2.5 A resolution. Nature. 1996;379:511–518. doi: 10.1038/379511a0. [DOI] [PubMed] [Google Scholar]
- 4.Wieden H.J., Gromadski K., Rodnina M.V. Mechanism of elongation factor (EF)-Ts-catalyzed nucleotide exchange in EF-Tu. Contribution of contacts at the guanine base. J. Biol. Chem. 2002;277:6032–6036. doi: 10.1074/jbc.M110888200. [DOI] [PubMed] [Google Scholar]
- 5.Boehr D.D. During transitions proteins make fleeting bonds. Cell. 2009;139:1049–1051. doi: 10.1016/j.cell.2009.11.031. [DOI] [PubMed] [Google Scholar]
- 6.Boehr D.D., Nussinov R., Wright P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eisenmesser E.Z., Millet O., Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 8.Sethi A., Eargle J., Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reference deleted in proof.
- 10.Röthlisberger D., Khersonsky O., Baker D. Kemp elimination catalysts by computational enzyme design. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 11.Drawz S.M., Bethel C.R., Bonomo R.A. The role of a second-shell residue in modifying substrate and inhibitor interactions in the SHV β-lactamase: a study of ambler position Asn276. Biochemistry. 2009;48:4557–4566. doi: 10.1021/bi9003292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tomatis P.E., Rasia R.M., Vila A.J. Mimicking natural evolution in metallo-β-lactamases through second-shell ligand mutations. Proc. Natl. Acad. Sci. USA. 2005;102:13761–13766. doi: 10.1073/pnas.0503495102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaucher E.A., Das U.K., Benner S.A. The crystal structure of eEF1A refines the functional predictions of an evolutionary analysis of rate changes among elongation factors. Mol. Biol. Evol. 2002;19:569–573. doi: 10.1093/oxfordjournals.molbev.a004113. [DOI] [PubMed] [Google Scholar]
- 14.Gaucher E.A., Miyamoto M.M., Benner S.A. Function-structure analysis of proteins using covarion-based evolutionary approaches: elongation factors. Proc. Natl. Acad. Sci. USA. 2001;98:548–552. doi: 10.1073/pnas.98.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwede T., Kopp J., Peitsch M.C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y., Yu N.J., Spremulli L.L. Mutational analysis of the roles of residues in Escherichia coli elongation factor Ts in the interaction with elongation factor Tu. J. Biol. Chem. 1998;273:4556–4562. doi: 10.1074/jbc.273.8.4556. [DOI] [PubMed] [Google Scholar]
- 19.Andersen C.A., Rost B. Secondary structure assignment. In: Gu J., Bourne P.E., editors. Structural Bioinformatics. John Wiley & Sons; Hoboken, NJ: 2009. pp. 419–484. [Google Scholar]
- 20.Schümmer T., Gromadski K.B., Rodnina M.V. Mechanism of EF-Ts-catalyzed guanine nucleotide exchange in EF-Tu: contribution of interactions mediated by helix B of EF-Tu. Biochemistry. 2007;46:4977–4984. doi: 10.1021/bi602486c. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y., Li X., Spremulli L.L. Role of the conserved aspartate and phenylalanine residues in prokaryotic and mitochondrial elongation factor Ts in guanine nucleotide exchange. FEBS Lett. 1996;391:330–332. doi: 10.1016/0014-5793(96)00789-2. [DOI] [PubMed] [Google Scholar]
- 22.Jeppesen M.G., Navratil T., Nyborg J. Crystal structure of the bovine mitochondrial elongation factor Tu.Ts complex. J. Biol. Chem. 2005;280:5071–5081. doi: 10.1074/jbc.M411782200. [DOI] [PubMed] [Google Scholar]
- 23.Kjeldgaard M., Nissen P., Nyborg J. The crystal structure of elongation factor EF-Tu from Thermus aquaticus in the GTP conformation. Structure. 1993;1:35–50. doi: 10.1016/0969-2126(93)90007-4. [DOI] [PubMed] [Google Scholar]
- 24.Song H., Parsons M.R., Phillips S.E. Crystal structure of intact elongation factor EF-Tu from Escherichia coli in GDP conformation at 2.05 A resolution. J. Mol. Biol. 1999;285:1245–1256. doi: 10.1006/jmbi.1998.2387. [DOI] [PubMed] [Google Scholar]
- 25.Gromadski K.B., Schümmer T., Rodnina M.V. Kinetics of the interactions between yeast elongation factors 1A and 1Bα, guanine nucleotides, and aminoacyl-tRNA. J. Biol. Chem. 2007;282:35629–35637. doi: 10.1074/jbc.M707245200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen G.R., Valente L., Nyborg J. Crystal structures of nucleotide exchange intermediates in the eEF1A-eEF1Bα complex. Nat. Struct. Biol. 2001;8:531–534. doi: 10.1038/88598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.