

Abstract
BACKGROUND AND PURPOSE
The use of opioids in treating pain is limited due to significant side effects including somnolence, constipation, analgesic tolerance, addiction and respiratory depression. Pre-clinical studies have shown that neurokinin 1 (NK1) receptor antagonists block opioid-induced antinociceptive tolerance and may inhibit opioid-induced rewarding behaviours. Here, we have characterized a bifunctional peptide with both opioid agonist and NK1 antagonist pharmacophores in a rodent model of neuropathic pain.
EXPERIMENTAL APPROACH
Rats were evaluated for behavioural responses to both tactile and thermal stimuli in either an uninjured, sham- or nerve-injured state. TY005 (Tyr-DAla-Gly-Phe-Met-Pro-Leu-Trp-O-3,5-Bn(CF3)2) was delivered spinally or systemically to assess the antinociceptive effects after acute exposure. Motor skills were evaluated using the rotarod test to determine potential sedative effects. Spinal TY005 was given chronically to sham- or nerve-injured animals to determine the development of tolerance.
KEY RESULTS
Bolus injections of TY005 produced dose-dependent antinociception in non-injured animals and alleviated nerve injury-induced thermal and tactile hypersensitivities (i.e. antihyperalgesia) more effectively than morphine. Sedative effects were not evident from the rotarod test at doses that were antihyperalgesic, nor at doses threefold higher. Repeated administration of TY005 did not lead to the development of antihyperalgesic tolerance or alter sensory thresholds.
CONCLUSIONS AND IMPLICATIONS
Collectively, the data suggest that opioid agonist/NK1 antagonist bifunctional peptides represent a promising novel approach to the management of chronic pain without the development of tolerance, reducing the need for escalation of doses and unwanted side effects associated with opiates alone.
Keywords: antinociception, neurokinin 1 receptor, neuropathic pain, opioids, substance P, tolerance
Introduction
Pain is often the most common indication of a medical problem. Neuropathic pain can result from damage to the nervous system, and common treatments include tri-cyclic antidepressants, gabapentinoids, opioids, anticonvulsants, local anaesthetics, acetaminophen, non-steroidal anti-inflammatory drugs (NSAIDs), as well as combinations of opioids and NSAIDs (Dworkin et al., 2008). While opioids are considered the gold standard of analgesic care for acute nociceptive pain, they have limited efficacy for neuropathic pain at tolerable doses (Rowbotham et al., 2003). Chronic opioid dosing can be associated with serious unwanted effects including constipation, nausea/vomiting, somnolence and respiratory depression, significantly decreasing the patients' quality of life (Reisine and Pasternak, 1996). Analgesic tolerance to opioid therapy develops in some patients with continued use, resulting in a need for dose escalation to maintain analgesic efficacy while increasing the unwanted side effects.
Pharmacological studies demonstrated the existence of opioid receptors over 25 years ago (Zaki et al., 1996). Three distinct receptors have since been characterized and are known as µ, δ and κ opioid receptors. Activation of both supraspinal and spinal µ-opioid receptors produces the analgesic effect of opioids through multiple mechanisms, including inhibiting the release of pronociceptive neurotransmitters such as substance P (SP), glutamate and calcitonin gene-related peptide (CGRP). Sustained exposure to opioids has been reported to induce neurocellular adaptations including opioid receptor internalization and long-term changes, such as receptor down-regulation (Koch and Höllt, 2008), adenylyl cyclase superactivation (Tumati et al., 2009), altered receptor trafficking and activation of descending pain facilitatory pathways from the brain stem (Vanderah et al., 2001a). These adaptations have been shown both in vitro and in vivo, and may contribute to opioid-mediated antinociceptive tolerance.
Although opioids are the most prescribed drugs to relieve pain, multiple processes can act to diminish opioid effectiveness. Firstly, a number of clinical and pre-clinical studies report that sustained opioid use can unexpectedly produce abnormally heightened pain sensations, characterized by increased sensitivity to normally non-noxious (allodynia) and noxious stimuli (hyperalgesia) (DuPen et al., 2007). Opioids can elicit heightened pain sensations in experimental models after acute (Célèrier et al., 2000) and chronic (Mao et al., 1995) administration. The mechanisms proposed to underly opioid-induced hyperalgesia (OIH) include ‘mini-withdrawals’ (Gutstein, 1996), activation of NMDA receptors (Mao et al., 1995), activation of descending pain facilitatory pathways (Vanderah et al., 2001b), altered G-protein coupling after chronic exposure (Largent-Milnes et al., 2008) and up-regulation in pronociceptive neurotransmitters (King et al., 2005). Secondly, the repeated use of opioid analgesics for chronic pain states can be rendered ineffective by the development of analgesic tolerance (Foley, 1995). Although mechanisms contributing to opioid tolerance are poorly understood, recent evidence from experimental models shows opioid analgesic tolerance may be derived from adaptations occurring at the level of the opioid receptor (Childers, 1991; Bohn et al., 2000; Wang et al., 2008) and at neural networks involved in pain transmission (Vanderah et al., 2001a). Interventions that inhibit OIH could also inhibit the behavioural development of antinociceptive tolerance (Powell et al., 2003; Vera-Portocarrero et al., 2007).
SP has been characterized as a pain-promoting neuropeptide that binds to and activates neurokinin 1 (NK1) receptors. When injected spinally in rodents, SP dose-dependently induces nocifensive behaviours such as biting, scratching and flinching (Hylden and Wilcox, 1981; Seybold et al., 1982). Increases in spinal SP levels have been shown in inflammation and after chronic opioid exposure (Malcangio et al., 2000; King et al., 2005), while redistribution of SP within the spinal dorsal horn has been reported in some models of neuropathic pain (Swamydas et al., 2004). After nerve injury, the content and release of SP are decreased in some peripheral nerve injury models, but not in others (Bisby and Keen, 1986). In models where some nerve fibres are spared, superficial dorsal horn content of SP is decreased, but dorsal root ganglion (DRG) immunoreactivity is increased (Ma and Eisenach, 2003). Swamydas et al. (2004) have shown that SP is redistributed after partial nerve transection, leading to increases in the deeper lamina of the spinal cord. Additionally, the affinity of SP for the NK1 receptor has been reported to be altered after nerve injury (Aanonsen et al., 1992). NK1 expression has been shown to be increased at protein and mRNA levels after peripheral nerve injury (Taylor and McCarson, 2004).
Sustained opioid exposure in adult rats has been shown to alter the SP–NK1 system, resulting in significant increases in SP content within the spinal cord tissue and an increase in evoked SP release in the superficial layer of the lumbar dorsal horn (Belanger et al., 2002; King et al., 2005). Sustained morphine treatment also produces an increase in NK1 immunoreactivity in lamina I–II in the spinal dorsal horn and increased internalization after mechanical pinch compared to placebo-treated rats (King et al., 2005). The evidence that sustained opioids enhance the content and evoked release of pain neurotransmitters suggests more opioid may be needed to inhibit this opioid-induced up-regulation, translating into antinociceptive tolerance. Studies from Powell et al. (2003) have shown that co-administration of morphine and a small molecule NK1 antagonist can attenuate the development of opioid antinociceptive tolerance, further indicating that the NK1 receptor and SP may contribute to the underlying mechanisms of opioid antinociceptive tolerance. Therefore, one priority would be to develop compounds that incorporate agonist activity at opioid receptors and antagonist activity at NK1 receptors. Previously, we have reported on the in vitro activity of such compounds, showing high binding affinity and strong in vitro biological activities as agonists at the opioid receptors and antagonists at NK1 receptors (Yamamoto et al., 2007). Here, we provide evidence that a single bifunctional molecule designed as an opioid agonist and NK1 antagonist dose-dependently reverses nerve injury-induced hypersensitivity and does not result in the development of antinociceptive tolerance in a rodent model of neuropathic pain.
Methods
Animals
Male, Sprague-Dawley rats (Harlan; Harlan, IN, USA) (a total of 380 rats, 175–250 g) were maintained on a 12 h light/dark cycle (lights on at 7:00h) with food and water available ad libitum. All preparations and testing were performed in accordance with the policies and recommendations of the International Association for the Study of Pain, National Institute of Health and the Institutional Animal Care and Use Committee of the University of Arizona. All terms used within the paper for receptors and related systems are in accordance with the guidelines set about by Alexander et al. (2009).
Experimental design
To evaluate the in vivo properties of the bifunctional peptide, TY005, experiments were performed using i.t. and i.v. routes of administration. Acute injection of TY005 (i.t.) was given to non-injured rats to investigate the antinociceptive properties and possible motor side effects of TY005. In separate experiments, TY005 was administered (i.t. and i.v.) to spinal nerve ligated rats to evaluate the effects of the compound on nerve injury-induced tactile allodynia and thermal hyperalgesia. Finally, the effects of multiple exposures to TY005 (i.t., twice daily) were assessed over 11 days in parallel with morphine in rats subjected to SNL and sham operations.
Surgery
Intrathecal catheter implantation
The rats were anaesthetized using ketamine/xylazine 100 mg·kg−1 i.p. (vol/vol: 80/20, Sigma-Aldrich, St Louis, MO, USA) and placed in a stereotaxic head holder. The cisterna magna was exposed, and an 8 cm catheter (PE-10, Stoelting Cat # 51151; Wood Dale, IL, USA) was implanted as described by Yaksh and Rudy (1976), terminating in the lumbar region of the spinal cord. Catheters were sutured into place and externalized at the back of the neck. Muscle and skin were closed in two layers. The animals were housed individually and allowed to recover for 5–7 days prior to baseline testing and/or spinal nerve ligation (SNL). Animals with signs of neurological deficits following surgery were not used.
SNL
Nerve ligation injury produces signs of neuropathic pain including tactile allodynia and thermal hypersensitivity. All nerve operations occurred 5 days after i.t. catheter implantation. The rats were anaesthetized with 2.5% isofluorane in O2 anaesthesia delivered at 2 L·min−1. The skin over the caudal lumbar region was incised and the muscles retracted. The L5 and L6 spinal nerves were exposed, carefully isolated and tightly ligated with 4-0 silk distal to the dorsal root ganglion, as we have previously reported (Vanderah et al., 2008). Control animals underwent sham surgeries in which the nerve areas were exposed, but not ligated. All animals were allowed 5–7 days to recover prior to any behavioural testing. Any animals exhibiting signs of motor deficiency or infection were killed.
Drug delivery
Intrathecal (i.t.)
Compounds were administered slowly over 30 s using a 25 µL Hamilton syringe and injector (PlasticsOne, C313I internal cannula, PlasticsOne, Roanoke, VA, USA) connected by tubing. Drugs or drug mixtures were administered in 5 µL volumes, followed by 1 µL air to follow the injection and a 9 µL saline flush. In acute experiments where one bolus injection was given, time of injection was equal to t = 0 min. Throughout the chronic i.t. experiments, the rats were given a morning injection between 8:00 h and 9:00 h, and behavioural testing performed at t =+15–20 min. An afternoon injection was given between 17:00 h and 18:00 h, but not evaluated behaviourally. This was repeated daily for 11 days. At days 12–14 after the initial exposure, all therapy was withdrawn, allowing for a ‘washout period’. Each therapy was re-introduced as a single injection at day 15 and testing performed at t =+20–30 min to more fully assess the effects of treatment.
Intravenous (i.v.)
Systemic administrations of vehicle or compounds were performed in anaesthetized rats. Solutions were prepared as an injection volume of 1 mL·kg−1. Briefly, the distal third of the tail was dipped in warm water to dilate the blood vessels. A 30.5-gauge needle was inserted into the lateral tail vein, the plunger pulled back and blood drawn to ensure needle placement. Treatment was then introduced slowly over the course of 45–60 s. A new needle/syringe combination was used for each rat, making sure no air bubbles were present prior to or during the injection. Pressure was applied to the injection site to stop any excess bleeding.
Behavioural measurements
Rotarod
Rats were trained to walk on an automated rotating rod (8 rev·min−1; Rotamex 4/8, Columbus Instrument, Columbus, OH, USA) for maximal cut-off time of 180 s (Vanderah et al., 2008). Training consisted of placement on a non-moving rod with the instrument off for 180 s, placement on the non-moving rod with the machine on for 180 s and two training sessions of 180 s each with the rod rotating. After 10 min, baseline values were recorded for each animal. If the animal reached the maximal time, a cut-off value of 180 s was assigned as the observed value. Compounds were administered as described above and assessment occurred every 15 min for the first 60 min.
SP-induced flinching, biting and scratching
Rats were implanted with an intrathecal cannula as previously stated and allowed to recover for 5–7 days. Testing was consistent with the methods by Seybold et al. (1982). SP administration (1 mM, 10 µL) was performed at t = 0 min; total doses were similar to those given by Seybold et al. (1982) at 13.47 µg per animal. Prior to testing, saline (0.9%) or naloxone (NLX) (2 mg·kg−1, s.c.) was injected at t =−15 min to block endogenous opioid and TY005 opioid activity (Sakurada et al., 1999), and TY005 (3, 10 or 30 µg in 5 µL) or 10% dimethyl sulphoxide (DMSO) (5 µL) was injected at t =−10 min. The total number of flinches, bites or scratches min-1 for the first 5 min was counted.
Tactile hypersensitivity
Rats were acclimatized in suspended, wire mesh cages for 30 min prior to baseline von Frey testing (pre- and post-nerve ligation/sham operation). The protocol for the acute exposure experiments consisted of compound or vehicle administration (t = 0), and measurement of responses to calibrated von Frey filaments (0.4–15.0 g) probed perpendicularly on the plantar surface of the left hind paw (ipsilateral to the SNL) for 7 s, at 15 min intervals for the first 60 min utilizing the up–down method used previously (Largent-Milnes et al., 2008). For the multiple exposure experiment, non-noxious tactile testing occurred 15 min after compound administration. Lifting the paw, licking the paw or vocalizing counted as positive responses to the calibrated filament. Paw withdrawal thresholds (PWTs) were calculated in g using the Dixon non-parametric test, and expressed as the mean PWT ± SEM in Prism by GraphPad. The contralateral paw was not tested, as injured animals placed more body weight on the uninjured paw when compared to the injured side.
Thermal hypersensitivity
Rats were allowed to acclimatize in Plexiglas holders for baseline testing (pre- and post-nerve ligation/exposure) for 30 min (Ugo Basile, Comerio, Italy). A mobile radiant heat source was used to direct heat to the plantar surface of the left hind paw, as described by Largent-Milnes et al. (2008). Paw withdrawal latencies (PWLs) were measured in s with an automatic shutoff of the heat source at 33 s. Antinociception experiment baselines and pre-nerve injury PWLs were established between 20 and 25 s. Post-injury baselines were obtained after the 7 day recovery period. On test days, animals were dosed, then tested using von Frey filaments every 15 min for 60 min. After multiple i.t. exposures, the rats were evaluated for responses to noxious thermal heat 20–22 min following dosing and 5–7 min after non-noxious, tactile testing. PWLs were recorded for each animal and expressed as the mean withdrawal latency ± SEM in Prism 4 by GraphPad. The contralateral paw was not tested, as injured animals placed more body weight on the uninjured paw when compared to the injured side.
Data analysis and statistical procedures
Compound activity in both non-noxious and noxious behavioural assays was calculated as follows:
Non-injury:
 |
SNL operated:
 |
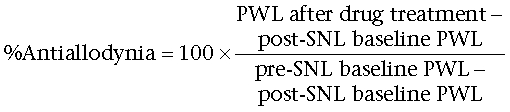 |
Sham operated:
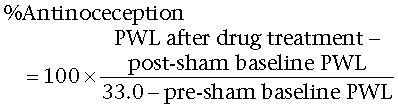 |
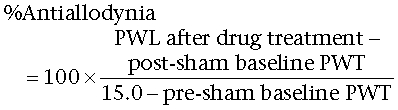 |
All data were analysed by non-parametric two-way anova (post hoc: Neuman–Kuels) in FlashCalc (Dr Michael H. Ossipov, University of Arizona, Tucson, AZ, USA). Differences were considered to be significant if P≤ 0.05. All data were plotted in GraphPad Prism 4.
Drug, chemical reagents and other materials
Compounds were dissolved in DMSO (Sigma-Aldrich, St. Louis, MO, USA, cat# D2650) and brought to volume with Millipore H2O for a final (vol/vol) of 10% DMSO (i.t.) or 20% DMSO (i.v.) unless otherwise stated. Morphine sulphate was provided by the National Institute of Drug Abuse, and dissolved in 0.9% saline. The bifunctional peptide, TY005, was synthesized in our laboratories as previously described (Yamamoto et al., 2007). SP (American Peptide, Sunnyvale, CA, USA, cat# 70-1-10) was prepared as a 1 mM solution in 0.9% saline and injected i.t. in a 10 µL volume. Beta funaltrexamine HCl (β-FNA; Tocris, Ellisville, MO, USA, cat# 0926) and naltrindole (NTI; Research Biochemicals, Inc., Natick, MA, USA, cat # N-115) were dissolved in saline and administered in a 5 µL volume i.t. NLX HCl (2 mg·kg−1, Tocris, cat #0599) was prepared in 0.9% saline (1 mL·kg−1) and injected s.c.
Results
TY005 (i.t.) produces antinociception and does not cause motor impairment
Animals were implanted with an indwelling i.t. cannula as described. After 7 days of recovery, baseline PWLs to a noxious thermal stimulus were calibrated to fall between 20.0 and 25.0 s; the mean value for all animals was found to be 21.5 ± 0.5 s (n = 48). TY005 (1, 3, 10 or 30 µg in 5 µL) or vehicle (10% DMSO in MilliPore water, 5 µL) was injected, and PWLs were recorded every 15 min for 60 min (Figure 1A). Maximal effect of TY005 occurred 15 min after the i.t. injection. The highest dose (30 µg) resulted in a maximal PWL of 32.0 ± 1.0 s. TY005 resulted in PWLs of 32.3 ± 0.7 s and 27.4 ± 1.6 s, at doses of 10 and 3 µg, respectively, at the same time-point. Each of these doses resulted in significantly higher PWLs when compared to baseline values or vehicle treatment alone (P < 0.01), but did not differ from each other. TY005 (1 µg) did not significantly alter PWL compared to baseline measurements in non-injured rats (Figure 1A).
Figure 1.
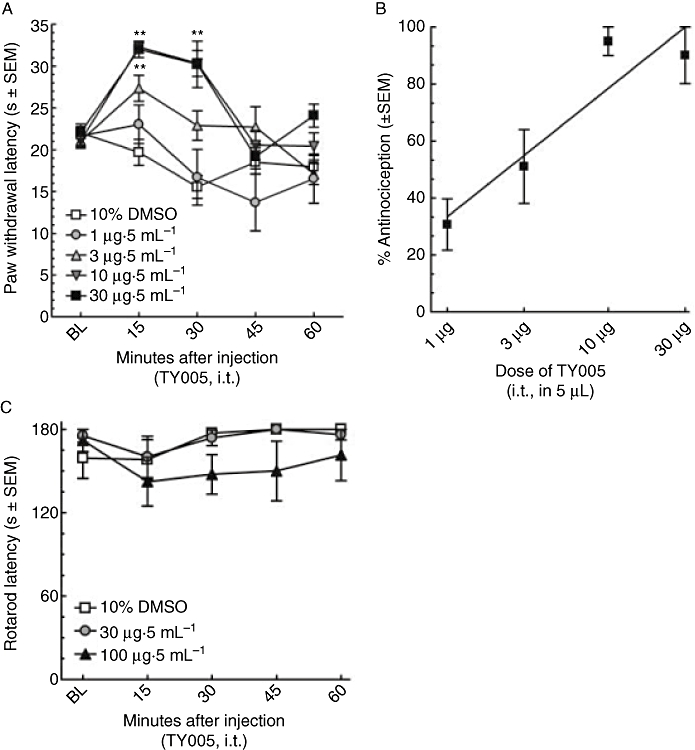
TY005 produced antinociception, but not motor impairment, in non-injured animals. (A) Intrathecal TY005 was injected at t = 0 min to non-injured animals. Mean PWL responses to noxious thermal stimulus were recorded and the time of peak effect occurred 15 min after the injection for all doses tested. Significance was observed with 10 and 30 µg when compared to vehicle and baseline latencies (*P < 0.05, anova). (B) TY005 produced antinociception in a dose-dependent manner at 15 min after i.t. injection. Data are represented as the % antinociception ± SEM. (C) Motor skills were assessed using the rotarod assay after i.t. administration of TY005. The total time spent walking on a rotating rod for a period of 180 s was recorded; data are shown as latency ± SEM. At doses threefold higher than required for maximal antinociception, there was no significant motor impairment observed (P > 0.05, anova).
% Antinociception was calculated for each dose of TY005 and a dose–response curve generated (Figure 1B). TY005 (3, 10, 30 µg) produced significant antinociception in non-injured animals compared to vehicle treatment 15 min after i.t. injection. Although the PWL after dosing with 1 µg TY005 was not statistically different from baseline or vehicle-treated values, the % activity calculated after this dose was significantly higher than the vehicle-treated group (P = 0.02) 15 min after injection (i.t.). The A50 dose to produce antinociception in response to a thermal stimulus was determined to be 2.3 µg (95% confidence interval (CI) 1.4–3.7 µg; Figure 1B).
TY005 (i.t.) was evaluated for potential motor impairment/sedation using the rotarod motor skills assay. Animals were trained to walk on a rotating rod for up to 180 s prior to compound or vehicle exposure with a mean latency of 168.7 ± 5.8 s (n = 17). After the baseline values were obtained, the animals were separated into groups of six. Vehicle-treated animals remained on the rotarod between 158.3 and 180 s throughout the time-course. The highest dose of TY005 using in the antinociception assay (30 µg) resulted in rotarod latency between 160.2 and 180 s, which was not significantly different from baseline values. A threefold higher dose (100 µg) did not result in a significant decrease in latency (142.4–180 s, P = 0.07; Figure 1C).
TY005 acts as an opioid agonist in vivo
The possibility that the antinociceptive effects of TY005 were mediated via actions at the opioid receptor was evaluated in non-injured animals. In one set of animals, the mean baseline PWL was recorded to be 22.3 ± 0.5 s (n = 20). Either saline or the non-selective opioid antagonist NLX (2 mg·kg−1, s.c.) was administered 10 min prior to i.t. TY005 (30 µg) or 10% DMSO vehicle injections (5 µL). The animals were evaluated for changes in PWL every 15 min for the first hour after the i.t. injection, corresponding to the time of peak effect of TY005 and 25 min after NLX. The PWLs of animals receiving saline–10% DMSO or NLX–10% DMSO did not differ significantly from baseline measurements (19.1 ± 2.5 s and 20.0 ± 1.20 s respectively). Animals treated with saline–TY005 (30 µg) had a significantly higher mean PWL, of 31.6 ± 1.4 s, than those treated with 10% DMSO (P = 0.004). NLX treatment 10 min before TY005 (30 µg) resulted in a PWL of 23.5 ± 3.1 s, significantly lower than that seen with TY005 treatment alone (P = 0.04). All data were converted to maximal % effect at the 15 min time-point (Figure 2A).
Figure 2.
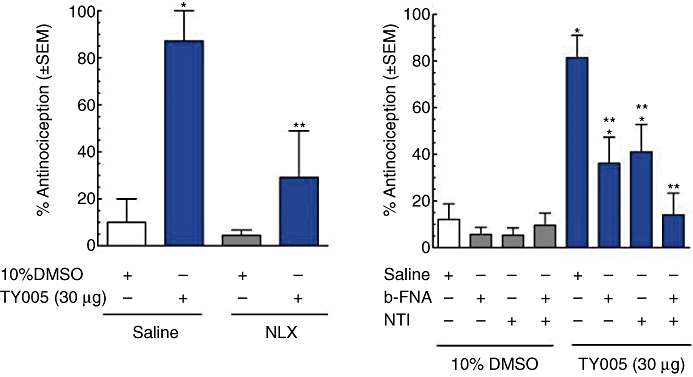
TY005 acted at opioid receptors in vivo. (A) The non-selective opioid receptor antagonist, NLX (2 mg·kg−1), given s.c. 10 min prior to an intrathecal injection of TY005, blocked the peak effect TY005-mediated antinociception (*P < 0.05). TY005 (i.t.) or vehicle (10% DMSO) was given at t = 0 min. Peak antinociceptive effect of TY005 occurred at t =+15 min, as previously shown in Figure 1. (B) Pretreatment with selective µ- and δ-opioid receptor antagonists (i.t.), β-FNA (t =−24 h; 42.5 nM, 5 µL) or NTI (t =−30 min; 66 nM, 5 µL), significantly attenuated TY005-mediated antinociception compared to saline-pretreated animals. When the two selective antagonists were combined, the TY005-mediated effect was fully blocked, similar to levels observed with the non-selective opioid antagonist NLX. *P < 0.05 compared to saline–10% DMSO-treated controls; **P < 0.01 versus saline-pretreated TY005 animals.
To further delineate the actions of TY005 at the opioid receptors, selective antagonists for the µ- and δ-opioid receptors were used. As TY005 did not have any κ-opioid receptor affinity in vitro, activity at this receptor was not evaluated. To determine the activity of TY005 at the µ-opioid receptor, a group of rats was pretreated with β-FNA (42.5 nM in 5 µL volume, i.t.) 24 h prior to TY005 (30 µg in 5 µL). Doses and times were chosen based on a previous publication by Perlikowska et al. (2009). Pretreatment with β-FNA (24 h) had no significant antinociceptive effect on vehicle-treated animals (n = 10), although the antinociceptive effect of TY005 was significantly decreased, confirming µ-opioid activity in vivo (n = 14; Figure 2B). Additionally, δ-opioid receptor activity was determined in the presence of the selective antagonist, NTI (66 nM, 5 µL i.t.; Dawson-Basoa and Gintzler, 1997) given 30 min before TY005 (30 µg in 5 µL) or 10% DMSO (5 µL). In vehicle-treated animals, NTI did not produce significant antinociception (n = 10). In the presence of NTI, the antinociception induced by i.t. TY005 was significantly lower than that induced in saline-pretreated animals (n = 14; Figure 2B). A final group of animals (n = 10) were pretreated with both β-FNA and NTI. Co-administration of the selective antagonists significantly and fully blocked TY005-mediated antinociception (P = 0.002) while not producing any significant effect in animals treated with 10% DMSO (Figure 2B).
TY005 acts as an NK1 antagonist in vivo
To assess NK1 receptor antagonism in vivo, we evaluated the effects of TY005 against SP-induced scratching and biting. Non-injured animals do not normally exhibit nocifensive behaviours in the absence of SP. However, exogenous administration of SP, i.t., produces robust flinching and biting behaviours (Hylden and Wilcox, 1981; Seybold et al., 1982). In this study, animals were pretreated at t =−15 min with either saline (1 mL·kg−1) or NLX (2 mg·kg−1, s.c.) and 10% DMSO (5 µL) or TY005 (3, 10 or 30 µg in 5 µL, i.t.; t =−10 min). All animals received 1 mM SP (10 µL, i.t.) at t = 0 min, and this induced noticeable flinching, biting and/or scratching of the hind paws. Animals (n = 27) pretreated with both saline and 10% DMSO flinched a mean total of 25.0 ± 4.2 times after SP (i.t.), but when TY005 (30 µg) was given instead of 10% DMSO, that value fell significantly (Table 1). Because TY005 contains an opioid agonist pharmacophore, NLX (2 mg·kg−1, s.c.) was given before both TY005 and SP to isolate NK1 receptor-mediated activity. NLX given to vehicle-treated animals resulted in SP inducing a number of flinches that was not significantly different from that induced in saline vehicle-treated animals (Table 1). Animals exposed to TY005 (3 µg) 10 min prior to SP, in the presence of NLX, produced significantly fewer flinches over the 5 min observation period than the SP alone and SP + NLX control groups (Table 1). Increasing the TY005 dose to 10 µg resulted in a decrease in the total number of flinches when compared to rats given only SP and TY005 (30 µg) further reduced the flinching response over a 5 min period after SP compared to vehicle (Table 1). Collectively, these data support dose-dependent antagonist activity of TY005 at the NK1 receptor in vivo.
Table 1.
TY005 acts as an NK1 antagonist in vivo: effects on SP-induced flinching
| Treatment | Mean no. of flinches in 5 min (±SEM) | N | P value to saline–DMSO | P value to NLX–DMSO |
|---|---|---|---|---|
| Sa–10% DMSO–SP | 25.0 (±4.3) | 27 | – | – |
| Sa–TY00530–SP | 11.2 (±3.6) | 18 | 0.02* | – |
| NLXb–10% DMSO–SP | 20.1 (±2.2) | 30 | 0.30 | – |
| NLXb–TY0053–SP | 5.8 (±1.8) | 12 | 0.006** | 0.001 |
| NLXb–TY00510–SP | 3.0 (±1.0) | 12 | 0.0002** | 0.001 |
| NLXb–TY00530–SP | 2.1 (±0.9) | 18 | 0.00005** | 0.0001 |
S: 0.9% saline (1 mL·kg−1, s.c.)
NLX: (2 mg·kg−1, s.c.) given 15–17 min before SP. DMSO (10%) or TY005 (dose indicated by subscripts; µg in 5 µL; i.t.) was administered 10 min prior to SP (10 µL of 1 mM; i.t.). P values were determined using one-way anova (FlashCalc) with Newman–Keuls post hoc analysis, and significance was assumed if
P ≤ 0.05
P < 0.01.
Intrathecal TY005 reverses SNL-induced hypersensitivities
TY005 (1–30 µg in 5 µL) was given as an acute bolus i.t. to rats that had undergone L5/L6 SNL 7 days earlier. Following administration, behavioural measurements of tactile and thermal hypersensitivities were obtained every 15 min for the first hour. Responses were compared to pre-injury and post-injury PWTs, and those of vehicle-treated and i.t. morphine-treated animals. Morphine was used for comparison with the peptide as it is used clinically for the treatment of neuropathic pain.
Prior to and after injury, all animals were evaluated for mechanical response to non-noxious probing of the left hind paw with calibrated von Frey filaments. The mean PWT before SNL was 15.0 ± 0.0 g. Seven days after nerve ligation, the mean withdrawal threshold was 3.0 ± 0.3 g, indicating the development of tactile allodynia (n = 58). The animals were separated at random into i.t. treatment groups: 10% DMSO vehicle, morphine (10 µg) and TY005 (1, 3, 10 and 30 µg). TY005 (30 µg) significantly attenuated SNL-induced tactile allodynia 15 and 30 min after a single i.t. injection, with the mean PWTs being increased to 14.7 ± 0.3 g and 13.4 ± 0.8 g, respectively (n = 6, Figure 3A). The 10 µg dose of TY005 resulted in a similar reversal of allodynia with mean PWTs of 10.1 ± 1.5 g and 6.6 ± 1.5 g, both 15 and 30 min following administration (n = 9). TY005 (3 µg) had a significant effect on SNL-induced allodynia only 15 min after peptide administration (n = 6); similarly, the lowest experimental dose of the bifunctional peptide evaluated (1 µg, n = 6) resulted in significant anti-allodynia (Figure 3A). Vehicle treatment did not result in a statistically significant reversal of SNL-induced allodynia at any time-point after injection (n = 26, P = 0.33; Figure 3A). Similarly morphine administration (10 µg, i.t.) did not significantly attenuate SNL-induced allodynia at any of the time-points tested (n = 6; P = 0.83; Figure 3A). From the PWTs obtained 15 min after TY005 (i.t.), a dose–response curve was generated (Figure 3C). The A50 value was calculated to be 6.1 µg with a 95% CI of 4.2–9.0 µg (r2= 0.99).
Figure 3.
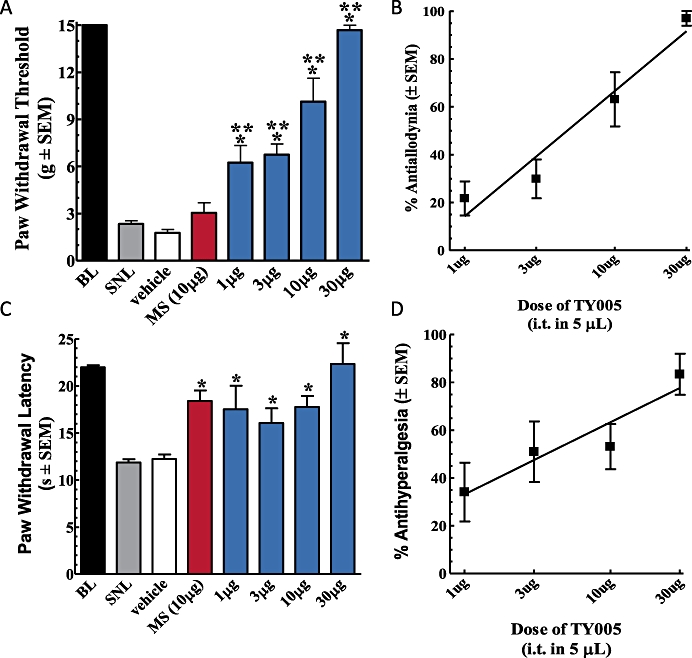
Intrathecal TY005 attenuated nerve injury-induced tactile and thermal hypersensitivities in a dose-dependent manner. Vehicle, TY005 (1, 3, 10, 30 µg), or morphine (10 µg) was injected spinally in SNL animals in a 5 µL volume. Non-noxious probing of the hind paw with von Frey filaments or noxious heat stimulation was performed at 15 min time-points. The peak effects of TY005 and morphine on the responses to (A) non-noxious and (B) noxious stimulation were observed 15 min after i.t. injection. The % activity of TY005 was calculated for each dose and behavioural test, and a graded dose–response curve generated for (C) anti-allodynic and (D) antihyperalgesic effects. Acute i.t. administration of TY005 significantly attenuated SNL-induced hypersensitivities dose-dependently when compared to vehicle-treated (*P < 0.05) and morphine-treated animals (**P < 0.05).
In non-injured rats, noxious infrared stimulation results in a mean PWL of 21.6 ± 0.3 s. Seven days after peripheral nerve ligation, the same stimulus produced a mean PWL of 12.0 ± 0.3 s, indicating the development of thermal hyperalgesia (n = 71). The animals were separated into treatment groups at random. TY005 (30 µg) significantly attenuated the enhanced sensitivity to a thermal stimulus compared to post-injury, baseline values at both 15 min (22.3 ± 2.2 s) and 30 min (19.8 ± 3.6 s) after i.t. administration (n = 6; Figure 3B). The 10 µg dose of TY005 resulted in a significantly higher PWL only 15 min after the bolus injection (n = 11), and the same was observed for TY005 (3 and 1 µg) (n = 10; Figure 3B). Morphine-treated animals withdrew the hind paw in response to the thermal stimulus with a mean latency similar to that of the TY005-treated animals15 min post-i.t. injection (n = 10), whereas vehicle treatment did not result in a significant change in PWL when compared to post-injury baseline values (Figure 3B).
A dose–response curve was generated from data collected 15 min after the i.t. injection of TY005. The % antihyperalgesic activity for each dose was calculated and presented in Figure 3D. The A50 value was determined to be 4.0 µg in 5 µL (95% CI 1.6–9.7 µg; r2= 0.88). The dose of morphine used resulted in 69.9 ± 10.5% antihyperalgesic activity (data not shown).
Systemic TY005 acutely reverses nerve injury-induced hypersensitivities
Seven days after nerve injury, TY005 was given i.v. into the tail vein as an acute bolus and behavioural measurements of tactile and thermal hypersensitivities recorded. For both tactile and thermal measurements, three doses were evaluated (3, 10 and 30 mg·kg−1) and compared to baseline values and vehicle-treated control animals.
Mean PWTs in response to non-noxious tactile probing with von Frey filaments before and after nerve ligation for all animals were 14.6 ± 0.4 g and 2.5 ± 0.2 g, respectively (n = 23). Nerve-injured animals given TY005 (30 mg·kg−1) withdrew the hind paw at the maximal threshold of 15.0 ± 0.0 g 15 min following the tail vein injection, and 8.6 ± 1.6 g at the 30 min time-point (n = 6, P < 0.0001; Figure 4A). PWTs had returned to post-injury values 45 min after administration of TY005. Similarly, treatment with 10 mg·kg−1 TY005 resulted in PWTs significantly higher than post-injury values 15 and 30 min after injection; 11.2 ± 2.2 g and 7.7 ± 2.2 g, respectively (n = 5, P < 0.001). The lowest dose of TY005 evaluated, 3 mg·kg−1, significantly increased the PWT of injured animals only at 15 min after i.v. administration (n = 6; Figure 4A). SNL-induced tactile allodynia was not altered in animals treated with vehicle (20% DMSO, 1 mL·kg−1) throughout the duration of the experiment (n = 6, P > 0.05).
Figure 4.
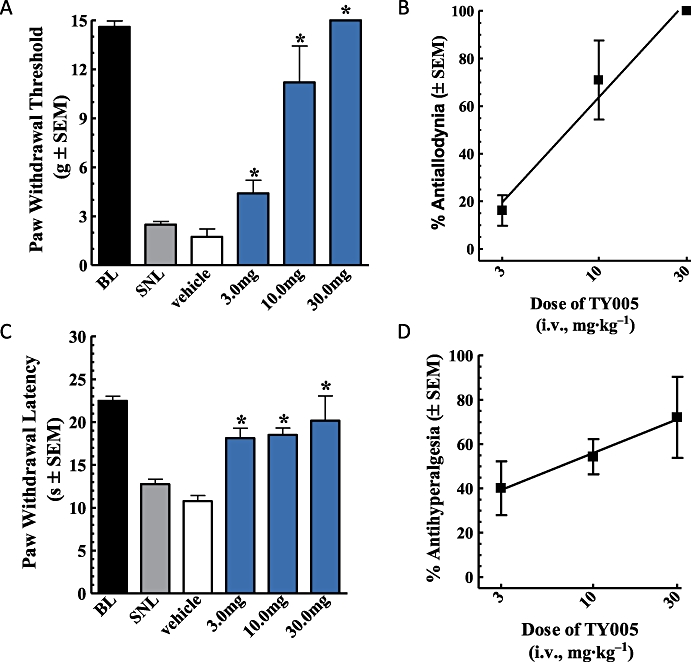
Intravenous TY005 attenuated nerve injury-induced tactile and thermal hypersensitivities in a dose-dependent manner. Vehicle or TY005 (3, 10 or 30 mg·kg−1) was injected systemically in SNL animals. Non-noxious probing of the hind paw with von Frey filaments or noxious heat stimulation was performed every 15 min. TY005 attenuated pain-like responses 15 min after i.v. injection, (A) tactile and (C) thermal. The % activity of TY005 was calculated for each dose, and a graded dose–response curve generated for both (B) anti-allodynic and (D) antihyperalgesic effects. Acute systemic administration of TY005 significantly attenuated SNL-induced hypersensitivities dose-dependently when compared to vehicle treated (*P < 0.05).
A dose–response curve for i.v. TY005 was generated using data from the 15 min time-point. The % anti-allodynic activity of each dose was calculated and the results presented in Figure 4B. The A50 value was determined to be 7.0 mg·kg−1 (95% CI 5.1–9.5 mg·kg−1; r2= 0.98).
In a separate group of animals, the acute effect of TY005 (i.v.) on thermal hyperalgesia was evaluated. As in previous experiments, baseline PWLs were recorded prior to and after peripheral nerve ligation. The mean pre-injury baseline was 22.5 ± 0.5 s; the post injury mean withdrawal latency was 12.8 ± 0.6 s, indicating the development of hypersensitivity to a noxious thermal stimulus (n = 23, Figure 4C). After i.v. administration of TY005 (30 mg·kg−1), PWLs at the 15 min (20.2 ± 2.9 s) and 30 min (17.0 ± 2.0 s) time-points were significantly higher than the post-injury baseline (n = 5, P = 0.004; Figure 4C). Treatment of SNL rats with 10 mg·kg−1 TY005 resulted in PWLs of 18.5 ± 0.8 s and 14.7 ± 1.0 s 15 and 30 min after the i.v. injection, respectively; only the 15 min measurement was significantly different from post-injury baseline values (n = 6, P = 0.003; Figure 4C). The peak time of effect of TY005 (3 mg·kg−1) was observed 15 min post-administration, with a mean PWL significantly higher than that of the untreated or vehicle-treated injury (n = 6; Figure 4C). In vehicle control animals, PWLs were not different from post-injury baseline values over the course of the experiment (n = 6, Figure 4C).
The ability of TY005 to attenuate SNL-induced thermal hyperalgesia, 15 min after its i.v. administration, was calculated as % activity compared to vehicle-treated controls; a dose–response curve was constructed and is shown as Figure 4D. The A50 valued was calculated to be 6.5 mg·kg−1 (95% CI 2.2–19.7 mg·kg−1; r2= 0.99).
TY005 (i.t.) retains antihypersensitivity effects after multiple exposures
In both sham-operated and SNL animals, TY005 was compared to vehicle-treated controls and morphine-treated rats for effects after multiple exposures. Prior to surgery and 7 days after sham or nerve ligation, baseline tactile and thermal PWTs and latencies were recorded. TY005, morphine or vehicle was administered twice daily for 11 days afterwards, and behavioural measurements were obtained 15 min after the morning injection. It is important to note that at days 12–14 and 16–18, no drug was given. At day 15, each treatment was given as a bolus to animals previously exposed to multiple intrathecal injections (Figure 5A).
Figure 5.
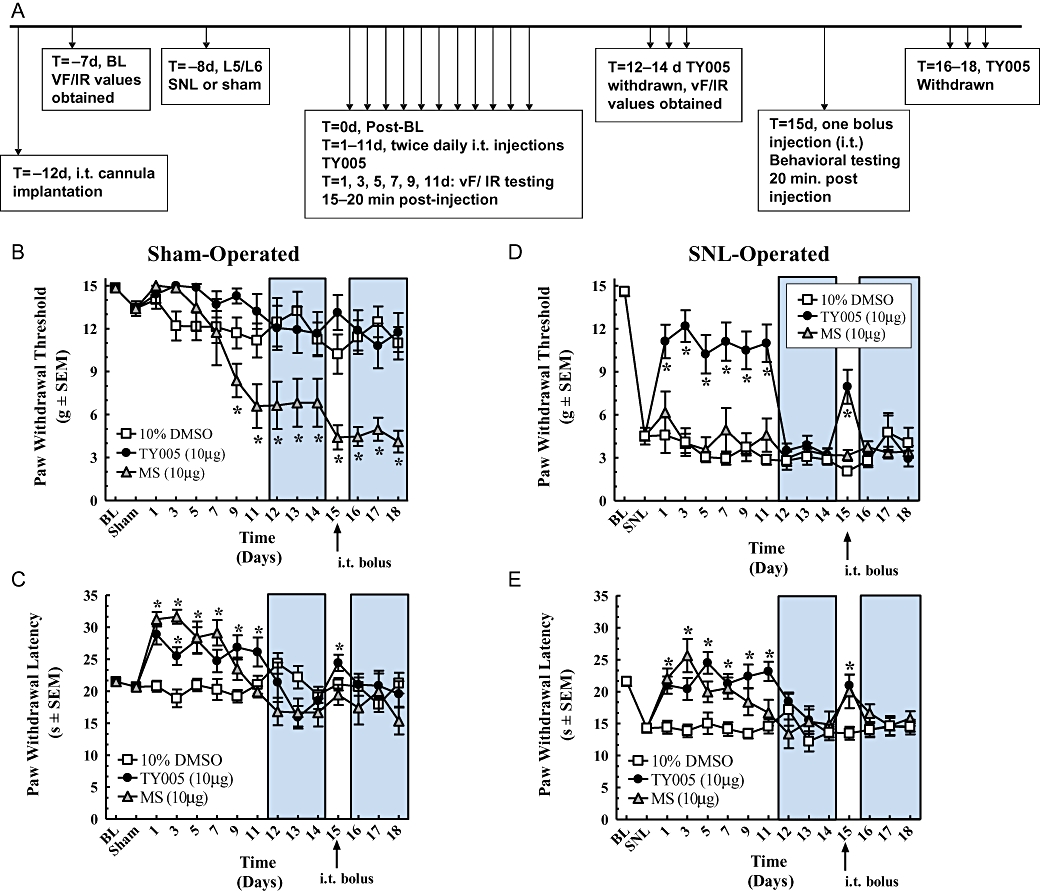
Chronic intrathecal TY005 did not produce antinociceptive tolerance in nerve-injured nor paradoxical hypersensitivity in sham-operated animals. (A) Time line of experiment. All animals were treated with 5 µL of either vehicle (10% DMSO), TY005 or morphine (MS) twice daily for 11 days, given 3 days washout (shaded areas on graph), and challenged again at day 15. Behavioural measurements were obtained at 15 min post-injection (tactile; B and D) or at 20 min post-injection (thermal; C and E). (B) PWTs of sham-operated animals; (C) PWLs of sham-operated animals; (D) PWTs of SNL-operated animals; (E) PWLs of SNL-operated animals. Significance was set at *P < 0.05 compared to baseline values.
In sham-operated animals, the mean baseline PWT before surgery was 14.9 ± 0.2 g. The mean post-sham PWT value was significantly lower than the pre-sham value at 13.4 ± 0.5 g (n = 41, P = 0.007). Animals given twice daily i.t. injections of 10% DMSO (5 µL) did not withdraw the hind paw at thresholds significantly different from the post-sham threshold across the 18 day testing period (n = 20, P = 0.18; Figure 5B). In rats treated with i.t. morphine (10 µg in 5 µL), PWTs are shown in Figure 5B. Significantly lower PWTs were achieved by the ninth day of twice daily morphine (i.t.), and these thresholds ranged between 8.4 ± 1.2 g and 4.1 ± 0.8 g for the remainder of the experiment (n = 7, P < 0.001; Figure 5B), indicating the development of allodynia to morphine alone. TY005 (10 µg in 5 µL) did not significantly decrease PWT at days 1–11, nor at day 15 after the i.t. injection (n = 14, P = 0.10; Figure 5B). At days 12–14 and 16–18, when no TY005 was administered, the animals withdrew the paw at thresholds between 12.1 ± 1.6 g and 10.8 ± 1.6 g, which was significantly lower than pre-sham and post-sham thresholds only at day 18 (P = 0.0004). Collectively, these data show twice daily injections of morphine (10 µg in 5 µL) leads to the development of tactile allodynia in sham-operated animals, while animals receiving TY005 did not develop tactile allodynia.
After the tactile PWTs had been recorded, the animals were given 5–7 min to acclimatize for thermal testing. Before the sham nerve ligation, the mean thermal PWL was 21.5 ± 0.4 s, after the sham operation, the mean PWL was 20.7 ± 0.6 s, which was not significantly different (n = 41, P = 0.26; Figure 5C). Vehicle (10% DMSO) treatment did not produce a significant increase or decrease in PWL over the course of the experiment (n = 20, P = 0.29). Rats receiving morphine had mean PWLs between 31.3 ± 1.1 s and 23.5 ± 1.8 s on days 1–9, demonstrating antinociception (n = 7, P = 0.001). After 11 days of treatment with morphine, the PWL was 19.2 ± 1.0 s, a value not significantly different from post-sham values (P > 0.05), indicating loss of antinociceptive efficacy, but not the development of hyperalgesia in this paradigm. At day 15, a bolus injection of morphine (10 µg in 5 µL, i.t.) did not result in a significantly different PWL compared to the post-sham baseline, indicating antinociceptive tolerance (19.4 ± 1.6 s, P = 0.45; Figure 5C). In the group of animals receiving TY005, antinociception was evident throughout the 11 day treatment time-course with PWLs ranging from 28.9 ± 1.6 s to 24.7 ± 1.8 s (n = 14, P < 0.001; Figure 5C). The rats were challenged with an i.t. bolus of TY005 at day 15, and the resulting mean PWL of 24.4 ± 1.3 s recorded further indicating the development of antinociception after multiple exposures (P = 0.006, Figure 5C). These data demonstrate that the in vivo antinociceptive activity of TY005 after multiple exposure is maintained in sham-operated rats and when challenged after 3 treatment-free days, while the antinociceptive actions of morphine are not.
SNL animals were prepared and exposed to the same treatment paradigm as sham-operated animals to evaluate potential tolerance to the antihyperalgesic and anti-allodynic efficacy of TY005. Tactile PWTs before and after nerve ligation were 14.6 ± 0.3 g and 4.5 ± 0.6 g, respectively (n = 42, P < 0.001). Repeated vehicle treatment did not result in a significant increase or decrease in PWTs throughout the duration of the experiment (n = 14, P = 0.38; Figure 5D). Nerve-injured rats given repeated injections of morphine (10 µg in 5 µL) did not have PWTs significantly different from the mean post-SNL threshold or those of vehicle-treated animals across the full time-course of the experiment (n = 12, P = 0.17; Figure 5D). TY005 (10 µg in 5 µL), administered twice daily for 11 days, produced an increase in PWT between 11.1 ± 1.2 g and 10.2 ± 1.4 g at days 1–11 with a peak effect occurring at day 3 (12.2 ± 1.1 g). When values were compared to post-SNL thresholds and vehicle-treated animals, the effects of TY005 were significantly higher (n = 16, P < 0.001; Figure 5D). When challenged at day 15, the animals receiving TY005 withdrew the hind paw at a mean threshold of 8.0 ± 1.2 g, which was significantly higher than post-SNL and vehicle treatment alone (P = 0.001), but lower than the pre-injury baseline (P = 1.3 × 10−5). Compared to morphine, TY005, retained significant anti-allodynic activity after multiple exposures and with a single bolus challenge after a 3 day washout period in SNL animals.
As in the sham experiment, SNL animals were allowed to acclimatize for 5–7 min between tactile and thermal testing. Prior to injury, the rats removed the hind paw from the thermal stimuli at a mean latency of 21.6 ± 0.4 s. After the SNL, the mean PWL was 14.3 ± 0.6 s, indicating the development of thermal hyperalgesia (n = 39, P < 0.001). The PWL of rats treated with vehicle did not differ significantly from the post-SNL value (n = 14, P = 0.38). Twice daily, i.t. morphine (10 µg in 5 µL) treatment resulted in a significant increase in PWL compared to post-injury latencies at days 1 through 7 with values ranging from 20.0 ± 1.6 s to 25.6 ± 2.6 s (n = 9, P = 0.001; Figure 5E). The peak effect of morphine was observed at day 3 of treatment (25.6 ± 2.6 s), which was not only significantly higher than post-injury values but also pre-injury PWLs (P = 0.004), although not as pronounced as in non-injured rats. At day 9, morphine did not produce a significant increase in PWL compared to vehicle (P = 0.06), and this was further observed at day 11 when the mean PWL in the morphine group was 16.7 ± 2.0 s (P = 0.14), demonstrating a decrease in antihyperalgesic efficacy. The morphine challenge at day 15, after 3 treatment-free days, resulted in a PWL of 20.0 ± 2.6 s (P = 0.002). Animals treated with TY005 withdrew the hind paw at significantly longer latencies compared to the post-injury value and vehicle-treated rats on all days of treatment (n = 16, Figure 5E); the peak antihyperalgesic effect of TY005 was observed at day 5 of treatment and corresponded to the PWL of 24.5 ± 1.7 s (Figure 5E). After 11 days, TY005 treatment did not result in the development of antinociceptive tolerance in SNL-operated rats. When treatment was discontinued for 3 days, PWLs returned to post-injury levels (days 12–14). TY005 (10 µg in 5 µL, i.t.) was administered as a challenge at day 15, and the resulting mean PWL was 21.0 ± 1.7 s; this was significantly higher than vehicle-treated animals, but not when compared to morphine treatment (P = 0.004 and P = 0.75, respectively).
Discussion
The present study demonstrates the in vivo activity of a rationally designed peptide that takes advantage of two distinct mechanisms in order to attenuate acute and chronic pain. We have shown in non-injured rats that targeting the µ- and δ-opioid receptors while blocking the NK1 receptor results in antinociception, as well as antihyperalgesia in a rodent model of neuropathic pain. More importantly, unlike morphine, spinal TY005 fully attenuated nerve injury-induced tactile allodynia. Finally, we demonstrated that multiple i.t. administrations of our multifunctional peptide did not result in the development of antinociceptive tolerance nor sedation.
Acute nociceptive pain arising from tissue injury or noxious input is reasonably well controlled with opioids. Neuroplastic adaptations within pain pathways, from injury or chronic opioid exposure, can lead to an alteration of transmission and ultimately a change in processing of the pain signal (Vanderah, 2007; Seybold, 2009). Neuropathic pain arising from injuries to peripheral nerves is often intractable clinically, and much effort has been devoted to understanding the underlying mechanisms for this pain state. Treatments for neuropathic pain have been outlined and include tri-cyclic antidepressants, selective 5-hydroxytryptamine (serotonin) re-uptake inhibitors, α2δ1 subunit calcium channel blockers, local anaesthetics and opioids (Dworkin et al., 2008). Despite the advances that have been made in understanding the mechanisms underlying neuropathic pain and the therapeutic agents available, opioids remain one of few options for many patients. However, the presence of adverse side effects limits the effectiveness of opioids at tolerable doses. In some patients receiving long-term opioids, antinociceptive tolerance or opioid-induced hyperesthesias may develop, thus requiring higher doses to maintain efficacy (DuPen et al., 2007). To address impeded pain relief and side effects occurring with extended exposure to currently available therapies, we have characterized the effects of a single peptide comprised of two pharmacophores, an opioid agonist and an NK1 antagonist, in vitro (Yamamoto et al., 2007), and here, in rat models of acute and neuropathic pain.
The ability of acute opioid therapy to achieve high levels of pain relief is well documented. Opioid agonists (e.g. morphine) have the ability to modulate release of all pain neurotransmitters by indirectly blocking calcium channels and indirectly opening pre- and postsynaptic potassium channels (for review see Vanderah, 2007). There have been reports, however, that short-term application of opioid agonists, such as remifentanil, can lead to the development of hyperalgesia after acute withdrawal (Angst et al., 2003), and may induce long-term potentiation of c-fibres (Drdla et al., 2009). In the latter study, the duration of action, peak effect and potency obtained were within the range of those observed after i.t. administration of remifentanil (<10 min, 0.1–10 µg in 10 µL) and alfentanil (10–30 min, 10–100 µg in 10 µL) (Buerkle and Yaksh, 1996). Although these phenomena were not observed in the present study, they may be of interest in future investigations with i.t. TY005, or its derivatives, given its similarities to the short-acting opioids. Furthermore, it is important to determine the stability of this peptide in the CNS as its metabolites may also be active at the individual receptor subtypes.
Previous studies using NK1 receptor antagonists alone for the treatment of pain have produced mixed results. Depending on the stimulus and the intensity thereof, both antinociception with very little effect on acute nociception have been reported with NK1 antagonists (Garces et al., 1993; Rupniak et al., 1993; Hill, 2000). The lack of specificity of the effects of these NK1 antagonists in these models suggests that other pain neurotransmitters, including glutamate and CGRP, are able to induce nociception (Hill, 2000; Woodcock et al., 2007).
Studies on the effects of peripheral nerve injury have reported both decreases and increases in SP immunoreactivity in the dorsal root ganglion and ipsilateral dorsal horn, while inflammation leads to an increased SP concentration within the dorsal horn of the spinal cord (review by Seybold, 2009). Likewise, both decreases and increases in SP release have been observed after nerve injury (Malcangio et al., 2000; Hughes et al., 2007). Despite these differing results and the seeming lack of effect at producing acute antinociception, spinal NK1 antagonists have been reported to alleviate both injury-induced thermal and tactile hypersensitivities (Cumberbatch et al., 1998; Cahill and Coderre, 2002).
Given the conflicting literature and our own results showing increases in both SP content and release, as well as NK1 internalization after chronic opioid therapy (King et al., 2005), in the present study, we evaluated the efficacy of the spinal and systemic administration of TY005, a bifunctional peptide, in a model of peripheral neuropathic pain. SNL rapidly induces both thermal hyperalgesia and tactile allodynia; TY005 was found to completely attenuate the tactile allodynia in this model, whereas morphine alone (i.t.) had less than 20% anti-allodynic effect, as previously reported (Lee et al., 1995); although other groups have demonstrated greater attenuation of nerve injury-induced allodynia after morphine (i.t.) (Zhang et al., 2005). Systemic administration of morphine has been shown to attenuate L5/L6 tactile allodynia, yet spinal administration of morphine did not have a significant effect in this model (Bian et al., 1995). We found that TY005 attenuated SNL-induced hypersensitivities to the same levels after both systemic and intrathecal administration. These results suggest that TY005 may cross the blood brain barrier, although plasma half-life and metabolic stability still need to be determined. Given the linked pharmacophore design of TY005 (Yamamoto et al., 2007), it is possible that the resulting metabolites may have effects that outlast the parent peptide, which may be responsible for the in vivo activity of TY005 after systemic administration. Future studies are ongoing to investigate further TY derivatives and their CNS penetration.
The present data support the notion that incorporation of an opioid agonist and NK1 antagonist into one compound will provide a useful therapy for neuropathic pain states. It could be a useful as an alternative to morphine, because spinal TY005 appears to be more effective at attenuating mechanical allodynia than morphine while retatining antinociceptive efficacy. Recent findings in rodent models have suggested that SP and NK1 receptor activation have a role in temporomandibular joint pain and inflammation (Takeda et al., 2005), and this has been supported by further studies where similar mechanisms have been proposed (Torsney and MacDermott 2006; Zhang et al., 2008).
Sustained administration of opioids can lead to the development of antinociceptive tolerance (Ossipov et al., 2005), so higher doses need to be administered to achieve adequate pain relief. Repeated exposure of opioids can also produce OIH both pre-clinically and clinically (Vanderah et al., 2001b; Dworkin et al., 2008), which may be the behavioural representation of antinociceptive tolerance as a result of increases in excitatory neurotransmitters (Gu et al., 2005; Vanderah, 2007). The results presented here suggest that an opioid agonist in combination with an NK1 antagonist does not result in OIH. These accord with previous findings, which showed that morphine-induced antinociceptive tolerance and hyperalgesia could be attenuated or reversed by co-treatment with an NK1 antagonist or prevented by deleting cells expressing the NK1 receptor in the spinal dorsal horn using a conjugated neurotoxin (Powell et al., 2003; Vera-Portocarrero et al., 2007).
The use of a single compound, rationally designed to target multiple systems, such as TY005, introduces an alternative approach to the co-administration of individual agents alone as in the study by Powell et al. (2003). The standard approach in the management of clinical pain looks to adjunctive therapies of available agents to optimize pain relief (Dworkin et al., 2008). However, the agents available must be balanced for each patient, with regard to efficacy and safety within the current treatment schedule. The use of designed, multivalent, chimeric molecules, like TY005, has advantages over a cocktail of individual drugs for ease of administration, a simple ADME property, no drug–drug interaction and the potential to achieve a higher local concentration. Also, because the expressions of the NK1 and opioid receptors show a significant degree of anatomical overlap in the central nervous system, effects on both receptors could lead to synergies in potency and efficacy.
Conclusions
In this study, we have provided the initial evidence that a peptidomimetic compound introduces an alternative to the enhanced synergy of combined pain therapeutics. By designing compounds that target multiple mechanisms of action for both antinociceptive efficacy, as well as side effects, we have produced a novel compound that addresses some aspects of the nerve injury state and sustained opioid pathophysiology. This is the first report demonstrating in vivo behavioural studies with a compound designed to have agonist and antagonist activity at three distinctly different receptors (µ-, δ-opioid agonist and NK1 antagonist) involved in neuropathic pain. Our data validate the concept of targeting multiple mechanisms within the pain pathways, and show that a multifunctional peptide design is efficacious in rodent models of acute and neuropathic pain. They also indicate that a multimodal approach to pain therapy may decrease or attenuate the unwanted side effects and antinociceptive tolerance associated with chronic opioid exposure.
Acknowledgments
The work was supported by grants from the USDHS, National Institute on Drug Abuse, DA13449 and DA06284. This paper is dedicated to our late colleague, Regents' Professor Henry I. Yamamura.
Glossary
Abbreviations
- β-FNA
beta-funaltrexamine
- CGRP
calcitonin gene-related peptide
- CI
confidence interval
- DMSO
dimethyl sulphoxide
- DRG
dorsal root ganglion
- NK1
neurokinin 1 receptor
- NSAIDs
non-steroidal anti-inflammatory drugs
- NTI
naltrindole
- OIH
opioid-induced hyperalgesia
- PWL
paw withdrawal latency
- PWT
paw withdrawal threshold
- SNL
spinal nerve ligation
- SP
substance P
Conflict of interest
None.
References
- Aanonsen LM, Kajander KC, Bennett GJ, Seybold VS. Autoradiographic analysis of 125I-substance P binding in rat spinal cord following chronic constriction injury of the sciatic nerve. Brain Res. 1992;596:259–268. doi: 10.1016/0006-8993(92)91556-t. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angst MS, Koppert W, Pahl I, Clark DJ, Schmelz M. Short-term infusion of the mu-opioid agonist remifentanil in humans causes hyperalgesia during withdrawal. Pain. 2003;106:49–57. doi: 10.1016/s0304-3959(03)00276-8. [DOI] [PubMed] [Google Scholar]
- Belanger S, Ma W, Chabot JG, Quirion R. Expression of calcitonin gene-related peptide, substance P and protein kinase C in cultured dorsal root ganglion neurons following chronic exposure to mu, delta and kappa opiates. Neuroscience. 2002;115:441–453. doi: 10.1016/s0306-4522(02)00452-9. [DOI] [PubMed] [Google Scholar]
- Bian D, Nichols ML, Ossipov MH, Lai J, Porreca F. Characterization of the antiallodynic efficacy of morphine in a model of neuropathic pain in rats. Neuroreport. 1995;6:1981–1984. doi: 10.1097/00001756-199510010-00007. [DOI] [PubMed] [Google Scholar]
- Bisby MA, Keen P. Regeneration of primary afferent neurons containing substance P-like immunoreactivity. Brain Res. 1986;365:85–95. doi: 10.1016/0006-8993(86)90725-0. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Buerkle H, Yaksh TL. Comparison of the spinal actions of the mu-opioid remifentanil with alfentanil and morphine in the rat. Anesthesiol. 1996;84:94–102. doi: 10.1097/00000542-199601000-00012. [DOI] [PubMed] [Google Scholar]
- Cahill CM, Coderre TL. Attenuation of hyperalgesia in a rat model of neuropathic pain after intrathecal pre- or post-treatment with a neurokinin-1 antagonist. Pain. 2002;95:277–285. doi: 10.1016/S0304-3959(01)00410-9. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Rivat C, Jun Y, Laulin JP, Larcher A, Reynier P, et al. Long-lasting hyperalgesia induced by fentanyl in rats: preventive effect of ketamine. Anesthesiol. 2000;92:465–472. doi: 10.1097/00000542-200002000-00029. [DOI] [PubMed] [Google Scholar]
- Childers SR. Opioid receptor-coupled second messenger systems. Life Sci. 1991;48:1991–2003. doi: 10.1016/0024-3205(91)90154-4. [DOI] [PubMed] [Google Scholar]
- Cumberbatch MJ, Carlson E, Wyatt A, Boyce S, Hill RG, Rupniak NM. Reversal of behavioural and electrophysiological correlates of experimental peripheral neuropathy by the NK1 receptor antagonist GR205171 in rats. Neuropharmacol. 1998;37:1535–1543. doi: 10.1016/s0028-3908(98)00125-7. [DOI] [PubMed] [Google Scholar]
- Dawson-Basoa M, Gintzler AR. Involvement of spinal cord delta opiate receptors in the antinociception of gestation and its hormonal simulation. Brain Res. 1997;757:37–42. doi: 10.1016/s0006-8993(97)00092-9. [DOI] [PubMed] [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkühler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- DuPen A, Shen D, Ersek M. Mechanisms of opioid-induced tolerance and hyperalgesia. Pain Manag Nurs. 2007;8:113–121. doi: 10.1016/j.pmn.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. J Pain. 2008;9:105–121. doi: 10.1016/j.jpain.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Foley KM. Misconceptions and controversies regarding the use of opioids in cancer pain. Anticancer Drugs. 1995;3:4–13. doi: 10.1097/00001813-199504003-00002. [DOI] [PubMed] [Google Scholar]
- Garces YI, Rabito SF, Minshall RD, Sagen J. Lack of potent antinociceptive activity by substance P antagonist CP-96,345 in the rat spinal cord. Life Sci. 1993;52:353–360. doi: 10.1016/0024-3205(93)90148-v. [DOI] [PubMed] [Google Scholar]
- Gu G, Kondo I, Hua XY, Yaksh TL. Resting and evoked spinal substance P release during chronic intrathecal morphine infusion: parallels with tolerance and dependence. J Pharmacol Exp Ther. 2005;314:1362–1369. doi: 10.1124/jpet.105.087718. [DOI] [PubMed] [Google Scholar]
- Gutstein HB. The effects of pain on opioid tolerance: how do we resolve the controversy? Pharmacol Rev. 1996;48:403–407. [PubMed] [Google Scholar]
- Hill R. NK1 (substance P) receptor antagonists – why are they not analgesic in humans? Trends Pharmacol Sci. 2000;21:244–246. doi: 10.1016/s0165-6147(00)01502-9. [DOI] [PubMed] [Google Scholar]
- Hughes DI, Scott DT, Riddell JS, Todd AJ. Upregulation of substance P in low-threshold myelinated afferents is not required for tactile allodynia in the chronic constriction injury and spinal nerve ligation models. J Neurosci. 2007;27:2035–2044. doi: 10.1523/JNEUROSCI.5401-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 1981;217:212–215. doi: 10.1016/0006-8993(81)90203-1. [DOI] [PubMed] [Google Scholar]
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, et al. Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain. 2005;116:276–288. doi: 10.1016/j.pain.2005.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Höllt V. Role of receptor internalization in opioid tolerance and dependence. Pharmacol Ther. 2008;117:199–206. doi: 10.1016/j.pharmthera.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Largent-Milnes TM, Guo W, Wang HY, Burns LH, Vanderah TW. Oxycodone plus ultra-low-dose naltrexone attenuates neuropathic pain and associated mu-opioid receptor-Gs coupling. J Pain. 2008;9:700–713. doi: 10.1016/j.jpain.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YW, Chaplan SR, Yaksh TL. Systemic and supraspinal, but not spinal, opiates suppress allodynia in a rat neuropathic pain model. Neurosci Lett. 1995;199:111–114. doi: 10.1016/0304-3940(95)12034-2. [DOI] [PubMed] [Google Scholar]
- Ma W, Eisenach JC. Intraplantar injection of a cyclooxygenase inhibitor ketorlac reduced immunoreactivities of substance P, calcitonin gene-related peptide, and dynorphin in the dorsal horn of rats with nerve injury or inflammation. Neuroscience. 2003;121:681–690. doi: 10.1016/s0306-4522(03)00497-4. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Ramer MS, Boucher TJ, McMahon SB. Intrathecally injected neurotrophins and the release of substance P from the rat isolated spinal cord. Eur J Neurosci. 2000;12:139–144. doi: 10.1046/j.1460-9568.2000.00890.x. [DOI] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Experimental mononeuropathy reduces the antinociceptive effects of morphine: implications for common intracellular mechanisms involved in morphine tolerance and neuropathic pain. Pain. 1995;61:353–364. doi: 10.1016/0304-3959(95)00022-K. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Porreca F. Underlying mechanisms of pronociceptive consequences of prolonged morphine exposure. Biopolymers. 2005;80:319–324. doi: 10.1002/bip.20254. [DOI] [PubMed] [Google Scholar]
- Perlikowska R, do-Rego JC, Cravezic A, Fichna J, Wyrebska A, Toth G, et al. Synthesis and biological evaluation of cyclic endomorphin-2 analogs. Peptides. 2009 doi: 10.1016/j.peptides.2009.12.002. doi: 10.1016/j.peptides.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Powell KJ, Quirion R, Jhamandas K. Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci. 2003;18:1572–1583. doi: 10.1046/j.1460-9568.2003.02887.x. [DOI] [PubMed] [Google Scholar]
- Reisine T, Pasternak G. Opioid analgesics and antagonists. In: Hardman JG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. Goodman and Gilman's the Pharmacological Basis of Therapeutics. 9th edn. New York: McGraw-Hill; 1996. pp. 521–577. [Google Scholar]
- Rowbotham MC, Twilling L, Davies PS, Reisner L, Taylor K, Mohr D. Oral opioid therapy for chronic peripheral and central neuropathic pain. N Engl J Med. 2003;348:1223–1232. doi: 10.1056/NEJMoa021420. [DOI] [PubMed] [Google Scholar]
- Rupniak NM, Boyce S, Williams AR, Cook G, Longmore J, Seabrook GR, et al. Antinociceptive activity of NK1 receptor antagonists: non-specific effects of racemic RP67580. Br J Pharmacol. 1993;110:1607–1613. doi: 10.1111/j.1476-5381.1993.tb14008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurada T, Yuhki M, Inoue M, Sakurada C, Tan-No K, Ohba M, et al. Opioid activity of sendide, a tachykinin NK receptor antagonist. Eur J Pharmacol. 1999;369:261–266. doi: 10.1016/s0014-2999(99)00078-3. [DOI] [PubMed] [Google Scholar]
- Seybold VS. The role of peptides in central sensitization. Handb Exp Pharmacol. 2009;194:451–491. doi: 10.1007/978-3-540-79090-7_13. [DOI] [PubMed] [Google Scholar]
- Seybold VS, Hylden JLK, Wilcox GL. Intrathecal substance P and somatostatin in rats: behaviors indicative of sensation. Peptides. 1982;3:49–54. doi: 10.1016/0196-9781(82)90141-3. [DOI] [PubMed] [Google Scholar]
- Swamydas M, Skoff AM, Adler JE. Partial sciatic nerve transection causes redistribution of pain-related peptides and lowers withdrawal threshold. Exp Neurol. 2004;188:444–451. doi: 10.1016/j.expneurol.2004.04.018. [DOI] [PubMed] [Google Scholar]
- Takeda M, Tanimoto T, Nasu M, Ikeda M, Kadoi J, Matsumoto S. Activation of NK1 receptor of trigeminal root ganglion via substance P paracrine mechanism contributes to the mechanical allodynia in the temporomandibular joint inflammation in rats. Pain. 2005;116:375–385. doi: 10.1016/j.pain.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Taylor BK, McCarson KE. Neurokinin-1 receptor gene expression in the mouse dorsal horn increases with neuropathic pain. J Pain. 2004;5:71–76. doi: 10.1016/j.jpain.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Torsney C, MacDermott AB. Disinhibition opens the gate to pathological pain signaling in superficial neurokinin 1 receptor-expressing neurons in rat spinal cord. J Neurosci. 2006;26:1833–1843. doi: 10.1523/JNEUROSCI.4584-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumati S, Yamamura HI, Vanderah TW, Roeske WR, Varga EV. Sustained morphine treatment augments capsaicin-evoked calcitonin gene-related peptide release from primary sensory neurons in a protein kinase A- and Raf-1-dependent manner. J Pharmacol Exp Ther. 2009;330:810–817. doi: 10.1124/jpet.109.151704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW. Pathophysiology of pain. Med Clin North Am. 2007;91:1–12. doi: 10.1016/j.mcna.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan TP, Jr, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain. 2001a;92:5–9. doi: 10.1016/s0304-3959(01)00311-6. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001b;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW, Largent-Milnes T, Lai J, Porreca F, Houghten RA, Menzaghi F, et al. Novel d-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral kappa-opioid receptors. Eur J Pharmacol. 2008;583:62–72. doi: 10.1016/j.ejphar.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, et al. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45. doi: 10.1016/j.pain.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HY, Frankfurt M, Burns LH. High-affinity naloxone binding to filamin a prevents mu opioid receptor-Gs coupling underlying opioid tolerance and dependence. PLoS One. 2008;3:e1554. doi: 10.1371/journal.pone.0001554. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Woodcock J, Witter J, Dionne RA. Stimulating the development of mechanism-based, individualized pain therapies. Nat Rev Drug Discov. 2007;6:703–710. doi: 10.1038/nrd2335. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Chronic catheterization of the spinal subarachnoid space. Physiol Behav. 1976;17:1031–1036. doi: 10.1016/0031-9384(76)90029-9. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Davis P, Ma SW, Navratilova E, Moye S, et al. Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for delta/mu opioid receptor agonists and neurokinin-1 receptor antagonists. J Med Chem. 2007;50:2779–2786. doi: 10.1021/jm061369n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Opioid receptor types and subtypes: the delta receptor as a model. Annu Rev Pharmacol Toxicol. 1996;36:379–401. doi: 10.1146/annurev.pa.36.040196.002115. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Conklin DR, Li X, Eisenach JC. Intrathecal morphine reduces allodynia after peripheral nerve injury in rats via activation of a spinal A1 adenosine receptor. Anesthesiology. 2005;102:416–420. doi: 10.1097/00000542-200502000-00027. [DOI] [PubMed] [Google Scholar]
- Zhang SH, Sun QX, Seltzer Z, Cao DY, Wang HS, Chen Z, et al. Paracrine-like excitation of low-threshold mechanoceptive C-fibers innervating rat hairyskin is mediated by substance P via NK-1 receptors. Brain Res Bull. 2008;75:138–145. doi: 10.1016/j.brainresbull.2007.08.003. [DOI] [PubMed] [Google Scholar]