

Abstract
BACKGROUND AND PURPOSE
Non-steroidal anti-inflammatory drugs improve inflammatory cachexia in several conditions. Thus, we have explored inhibition of cyclooxygenase-2 (COX-2) in an experimental model of rheumatoid cachexia in rabbits.
EXPERIMENTAL APPROACH
Chronic arthritis was induced in immunized rabbits by repeated intra-articular injections of ovalbumin. To increase the degree of systemic inflammation and also to induce atherosclerotic lesions, the animals were fed a hyperlipidaemic diet (2% cholesterol and 6% peanut oil) and were given an endothelial injury of the femoral artery. Rabbits were randomized to receive the COX-2 inhibitor celecoxib (10 mg·kg−1·day−1) or no treatment. After 4 weeks, sera, peripheral mononuclear cells and vessel specimens were collected.
KEY RESULTS
Inhibition of COX-2 by celecoxib modulated the systemic inflammatory response and increased total cholesterol and triglyceride levels. Celecoxib also minimized weight loss and prevented serum albumin fall. At a vascular level, celecoxib reduced COX-2 protein in the femoral arterial wall, but did not modify size or the macrophage infiltration of femoral lesions nor the percentage of rabbits with spontaneous aortic plaques.
CONCLUSIONS AND IMPLICATIONS
Our animal model induced a severe inflammatory cachexia, comparable to that of persistently active rheumatoid arthritis. The inhibition of COX-2 by celecoxib improves this state, suggesting that COX products play an important role in its development, without affecting the development or the progression of vascular lesions. Overall, these results suggest that celecoxib might be considered as a new therapeutic tool for the treatment of rheumatoid cachexia.
Keywords: rheumatoid arthritis, animal models, cachexia, PGE2 inhibition, atherosclerosis
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease, which mainly affects the synovium but also exhibits systemic manifestations, including ‘rheumatoid cachexia’ (RC) (Roubenoff et al., 1994), characterized by a lower lean mass, with or without greater fat mass (Giles et al., 2008b; Summers et al., 2008). The incidence may vary from 10% up to 2/3 of patients depending on the measure considered (Roubenoff et al., 1992; Morley et al., 2006). The consequences of this condition are physical inactivity, increasing weakness and a decreased functional status. This complication, which appears early during the course of the disease, may not be evident on clinical examination as these changes in body composition are not usually followed by modifications in body mass index (Giles et al., 2008b). The average loss of body cell mass (BCM), which comprises lean tissue mass and, to a lesser extent, visceral and immune cell mass, in these patients ranges between 13% and 15% (Roubenoff et al., 1992; 1994;). The mechanism underlying this complication in RA is not well understood, but a severe catabolic state driven by pro-inflammatory cytokines, particularly tumour necrosis factor (TNF)-α and interleukin (IL)-6, can be blamed for the loss of BCM seen in this condition (Roubenoff et al., 1994; Arshad et al., 2007). However, although BCM depletion is strongly correlated with parameters of disease activity, it is only partially restored when the disease is clinically well controlled (Walsmith et al., 2004). Together with most of these classical pro-inflammatory cytokines, there is increasing evidence about the contribution of a dys-regulated adipose tissue and its protein products (adipokines) to RC (Giles et al., 2009). Indeed, the endocrine secretory pattern of these factors by adipose tissue is markedly impaired in RA patients and might be a significant contributor to alterations of vascular function (including endothelial dysfunction), pro-thrombotic tendency and low-grade inflammation. Intriguingly, these features are also typical of visceral obesity suggesting that both extreme weight conditions (obesity or severe underweight) predispose patients to increased cardiovascular risk.
As in the general population, central obesity is a good predictor of cardiovascular risk in RA (Inaba et al., 2007). By contrast, underweight rheumatoid patients show an increase in all-cause and cardiovascular mortality, which has been partly attributed to systemic inflammation (Kremers et al., 2004; Escalante et al., 2005).
So far, a specific treatment for RC has not yet been established. Therapies focused on controlling inflammation, such as disease-modifying anti-rheumatic drugs, do not appear to reverse this condition. Furthermore, although cytokine-specific therapies such as TNF-blocking agents showed promising results in preclinical models of arthritis-induced cachexia (Granado et al., 2006), available data in humans are controversial (Marcora et al., 2006; Metsios et al., 2007). On the other hand, there are quite a few studies showing that non-steroidal anti-inflammatory drugs (NSAIDs) and particularly, cyclooxygenase (COX)-2 inhibitors, improve cachexia in patients with cancer (Lundholm et al., 2004; Mantovani et al., 2006; Lai et al., 2008; Mantovani and Madeddu, 2008). It is important to stress that these agents are widely prescribed in RA patients but data looking at their effects in RC are scarce (Arshad et al., 2007). Preclinical models of chronic arthritis in rats have been used to study different aspects of RC (Roubenoff et al., 1997; Granado et al., 2006; 2007; Martín et al., 2008) and some data point towards a beneficial effect of COX-2 inhibition in this complication (Largo et al., 2008). However, the potential implications for cardiovascular risk when vascular lesions already exist have not been simultaneously assessed.
Our group has developed an experimental model of arthritis plus atherosclerosis (Largo et al., 2008), which allows us to study the accelerated atherosclerosis and the cachexia associated with RA, in order to test the effect of pharmacological interventions for RA on both complications.
Methods
Animals
All animal care and experimental protocols for this study complied with the Spanish regulations and the Guidelines for the Care and Use of Laboratory Animals drawn up by the National Institutes of Health (USA) and were approved by the Institutional Ethics Committee of the Fundación Jiménez Díaz Hospital. Forty-four male New Zealand white rabbits were purchased when they weighed 3.0 ± 0.3 kg (Granja San Bernardo, Navarra, Spain). Rabbits were randomly assigned to three groups: healthy (n = 15), RC (RC group; n = 15) and RC rabbits treated with oral celecoxib at 10 mg·kg−1·day−1 (CXB group; n = 14). The celecoxib was in capsules (Celebrex; Pfizer) and given by oral gavage. They were fed with 85 g·day−1 of standard rabbit chow or hypercholesterolaemic diet and water ad libitum according with the experimental animal model recently published (Largo et al., 2008). A schematic representation of the experimental model of RC is shown in Figure 1.
Figure 1.
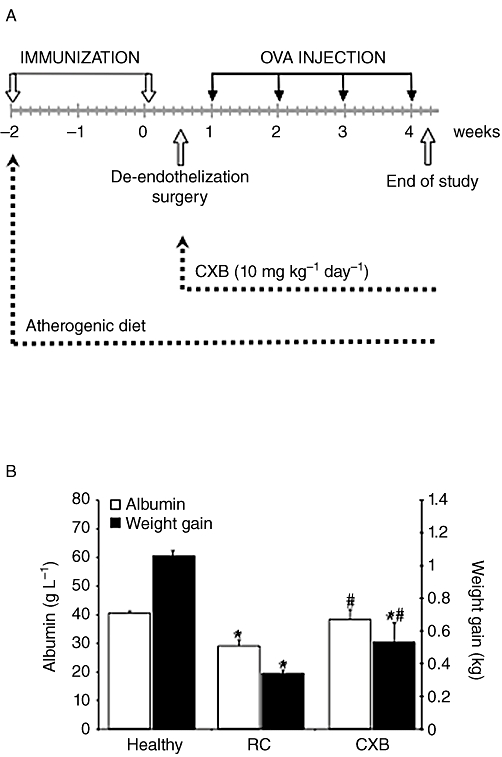
Effect of the combination of chronic antigen-induced arthritis (AIA) and atherosclerosis in parameters associated with cachexia. (A) Schematic representation of the experimental model. (B) Serum albumin levels and changes in weight-gaining healthy rabbits, rabbits with both chronic AIA and atherosclerosis, untreated (rheumatoid cachexia, RC) and treated with celecoxib (CXB). *P < 0.05 versus healthy controls; #P < 0.05 versus RC.
Body-weight measurements were recorded at baseline and at the end of the experiment, and body-weight gain was calculated. All rabbits were killed by an overdose of pentobarbital. The femoral arteries and the thoracic aortas were removed and fixed in 4% buffered paraformaldehyde, dehydrated and embedded in paraffin. Another piece of femoral artery was snap-frozen and stored at −70°C for molecular biology studies.
Biochemical measurements
At the end of the study, 10 mL blood was obtained and centrifuged to obtain serum. The total serum cholesterol, triglycerides and high-density lipoprotein (HDL) cholesterol levels were determined by enzymatic methods (Sigma-Aldrich, Inc). Serum C-reactive protein (CRP) and IL-6 levels were measured using specific commercial enzyme-linked immunosorbent assays (Alpha Diagnostic International Inc, San Antonio, TX, USA and R&D Systems Inc, Minneapolis, MN, USA respectively). Total protein levels were determined from serum total protein content by final point biuret method using DDPP Hitachi Modular automatic equipment. The sensitivity of the assay was 2 g·L−1. The intra- and inter-analysis coefficients of variation were 0.7% and 1.22% respectively. Once the total protein level was obtained, albumin was calculated as a percentage by capillary electrophoresis with Capillarys equipment (Sebia).
Isolation of peripheral blood mononuclear cells
Peripheral blood mononuclear cells (PBMC) were isolated from total blood with Lymphoprep (Hernández-Presa et al., 2002). Thereafter, cells were processed to obtain the nuclear extracts (Largo et al., 2003) or dissolved in Trizol Reagent (Roche Diagnostics) to extract total RNA from the lysates.
Electrophoretic mobility shift assay
Nuclear protein extracts pooled from mononuclear cells were prepared as described by Hernández-Presa et al. (2002), and the protein concentration in each sample was quantified by the BCA method (Thermo Scientific, Meridian Road, USA). A consensus oligonucleotide for nuclear factor-κB (NF-κB) (Promega Biotech Iberica) was end-labelled with 32P using 10 units of T4 polynucleotide kinase (Promega Biotech Iberica), and the nuclear extracts were then equilibrated for 10 min in binding buffer before adding the labelled probe (Largo et al., 2003). The specificity of the assay was tested. Samples were resolved on 4% non-denaturing acrylamide gels in Tris-borate buffer, which were exposed to X-ray film to determine the NF-κB.
RNA extraction and real-time polymerase chain reaction
Total RNA was extracted from femoral arteries or PBMC using the Trizol method (Roche Diagnostic). First strand cDNA was synthesized from 1 µg of total RNA using the High-Capacity cDNA Reverse Transcription Kit according to the manufacturer's instructions (Applied Biosystems, Stockholm, Sweden). Polymerase chain reaction primers and probes were designed by Applied Biosystems (Vidal et al., 2007) and the endogenous control of our assays was the eukaryotic 18S rRNA. Thermal cycling and florescence detection were performed on an ABI Prism 7500 Sequence Detection System with ABI Prism 7500 SDS software (Applied Biosystems, Stockholm, Sweden). Thermal cycling was carried out for 10 min at 95°C followed by 40 cycles of 15 s at 95°C and 1 min at 60°C. Gene expression values were calculated using the e−2ΔΔCt method.
Western blot analysis
Total proteins were isolated from femoral arteries with Trizol (Roche Diagnostics) and they were resolved on 10% acrylamide-SDS gels. After transfer to polyvinylidene difluoride membranes, the lysates were probed with antibodies against COX-2 (Santa Cruz Biotech) and α-tubulin (Sigma – Aldrich, Inc18). Briefly, the membranes were blocked in 5% skimmed milk in phosphate buffered saline-Tween 20 (PBS-Tween 20) for 1 h at room temperature, and incubated overnight at 4°C with the primary antibodies diluted 1/500 in PBS containing 0.3% Tween 20 and 3% bovine serum albumin. Antibody binding was detected by enhanced chemoluminiscence using peroxidise-conjugated secondary antibodies diluted 1/1000 in PBS-Tween 20, and the results were expressed in arbitrary densitometric units normalized to the α-tubulin levels.
Histopathological analysis of vascular lesions
Arteries were divided into four equal fragments and embedded in a single paraffin block. These blocks were then cross-sectioned into 4 µm thick serial sections and multiple sections from each block were chosen at regular intervals and stained with haematoxylin-eosin or orcein. These sections were then analysed qualitatively to identify the zone with the most severe stenosis where morphometric and immunohistochemical studies were performed. Morphometry was performed using the Olympus semiautomatic analytic system with Micro Image software (version 1.0 for Windows). Slide photomicrographs were captured with an Olympus microscope (BH-2) connected to a video camera (Hernández-Presa et al., 2002; Vidal et al., 2007). Intima and media thickness were measured in femoral arteries and the results were expressed as intima/media thickness ratios.
Immunohistochemistry in femoral lesions
We identified macrophages in the site where the femoral lesions showed the maximal stenosis using a monoclonal anti-rabbit macrophage antibody (RAM11, Dako Corporation, Glostrup, Denmark) according to a protocol described previously (Hernández-Presa et al., 2002; Vidal et al., 2007). Tissues, previously counterstained with haematoxylin, were mounted in Pertex (Medite), and the stained area was analysed in digital photomicrographs and expressed as a percentage per square millimetre of tissue (Hernández-Presa et al., 2002; Vidal et al., 2007). The negative controls involved detection with an IgG isotype.
Data analysis
The values for lipids and the data from the morphometric analysis, immunohistochemistry, electrophoretic mobility shift assay, Western blots and real-time polymerase chain reaction are expressed as the mean ± SEM, and they were analysed using the Mann–Whitney U-test. Where multiple comparisons were performed, the Kruskal–Wallis test was used. The null hypothesis was rejected in each statistical test when the P-value was less than 0.05. All statistical analyses were performed using Windows spss version 11.0 software (SPSS, Inc, Chicago, IL, USA).
Results
Weight change and albumin
Rabbits with RC showed lower body-weight gain than healthy animals (0.34 ± 0.02 kg vs. 1.06 ± 0.03 kg, P < 0.001 in both cases). Albumin levels were lower in sera from RC rabbits compared with the healthy group (P = 0.001; Figure 1). Rabbits with RC treated with celecoxib (CXB group) showed less weight loss (P = 0.009 vs. RC) and maintained serum concentrations of albumin within the normal range (P = 0.075 vs. healthy, Figure 1).
Parameters of systemic inflammation
As described (Largo et al., 2008), the induction of atherosclerosis plus chronic arthritis leading to RC significantly increased levels of both CRP and IL-6 in serum (P < 0.001 vs. healthy animals) (Figure 2). The administration of celecoxib to rabbits with RC reduced serum IL-6 and CRP levels (P = 0.037 and P = 0.001, respectively, vs. RC) (Figure 2A).
Figure 2.
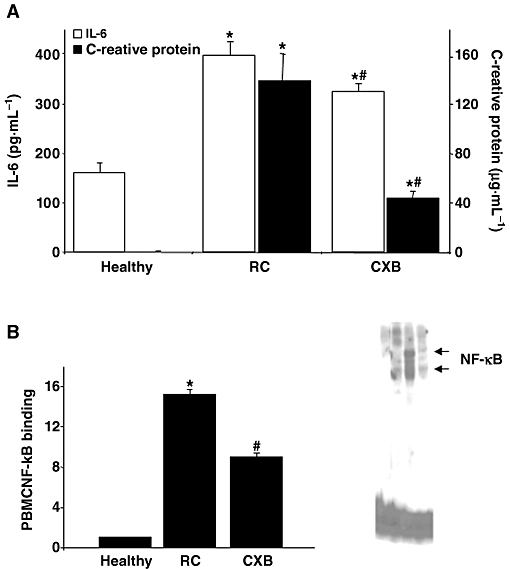
Effect of celecoxib (CXB) administration to rabbits with rheumatoid cachexia (RC) on parameters of systemic inflammation. (A) IL-6 and C-reactive protein concentrations in serum. *P < 0.01 versus healthy controls; #P < 0.05 versus non-treated (RC) rabbits. (B) Electrophoretic mobility shift assay and densitometric analysis of radio-labelled NF-κB bound to the nuclear proteins extracted from the peripheral blood mononuclear cells (PBMC). Lane 1 indicates the competitive control of the lane 3 (corresponding to a RC rabbit). *P = 0.022 versus healthy controls. #P = 0.053 versus healthy and RC.
COX-2 and CCL2 gene expression in PBMC
In previous work, we have found that PBMC from rabbits with RC show an up-regulation in the gene expression of COX-2 and the chemokine CCL2, compared with healthy animals (data not shown). Here, we looked for the gene expression of these proteins in rabbits with or without treatment, and found that celecoxib did not prevent the increase in both COX-2 (11.3 ± 3.5-fold vs. 7.8 ± 1.4-fold, P = 0.87) and CCL2 expression (2 ± 0.6-fold vs. 3.3 ± 0.6-fold, P = 0.22) on PBMC.
NF-κB activation in PBMC
NF-κB activation was determined by electrophoretic mobility shift assay in nuclear extracts from the PBMC. In these cells, there was an increase in the NF-κB activity in the RC group (P = 0.026 vs. healthy animals). The administration of celecoxib led to a reduction in NF-κB activation in PBMC in comparison with untreated animals (P = 0.053) (Figure 2B).
Lipid profile
Rabbits with RC showed lower levels of HDL and higher levels of total cholesterol and triglycerides than healthy animals. Treatment with celecoxib, increased levels of total cholesterol and triglycerides (P < 0.001 vs. RC), and decreased HDL cholesterol levels (P < 0.001, vs. RC) (Table 1).
Table 1.
Lipid levels in serum from healthy, RC and CXB groups of rabbits
| (mg·L−1) | Healthy (n = 15) | RC (n = 15) | CXB (n = 14) |
|---|---|---|---|
| Total cholesterol | 500 ± 40 | 18 600 ± 680* | 20 400 ± 420*# |
| HDL | 370 ± 30 | 190 ± 20* | 80 ± 10*# |
| Triglycerides | 740 ± 100 | 900 ± 100* | 1220 ± 140*# |
Data are shown as mean ± SEM. The healthy group were untreated, the RC group had rheumatoid cachexia and the CXB group had rheumatoid cachexia and were treated with oral celecoxib (10 mg·day−1). Blood was taken at the end of the experimental period (see Figure 1A).
P < 0.05 versus healthy.
P < 0.05 versus RC.
Quantification of vascular lesions in the femoral arteries
All the animals from the groups with RC – RC and CXB groups– developed a stenotic lesion in the femoral artery characterized by a hyperplasic transformation of the intima and foam cell infiltration. The intima/media thickness ratio was quantified at the site of maximal stenosis. The administration of celecoxib did not modify this parameter in the injured femoral arteries when compared with RC animals (Figure 3). No lesions were developed in the vessels of the healthy rabbits.
Figure 3.

Quantification of vascular lesions in injured femoral arteries. Upper panel, neointimal hyperplasia. (A) Intima/media thickness ratio (IMT). *P < 0.05 versus healthy controls. Representative haematoxylin-eosin stained femoral sections of rheumatoid cachexia (RC)- and celecoxib (CXB)-treated rabbits are shown: (B) RC rabbit; (C) CXB-treated rabbit (magnification, 100×). Lower panel, macrophage detection. (D) Quantification of macrophage staining in the neointimal area. *P < 0.05 versus healthy controls. Representative samples of RAM11 immunohistochemistry in RC- and CXB-treated rabbits are shown: (E) RC rabbit; (F) CXB-treated rabbit (magnification, 100×).
Inflammatory changes in the femoral arteries
All the femoral arteries from the groups with RC (RC and CXB groups) displayed macrophage infiltration at the neointima, assessed with RAM11 staining. As inflammatory changes at the vessel wall are related to the progression of the vascular lesion, we had expected that a COX-2 inhibitor could suppress or abolish this component. However, celecoxib treatment did not decrease the macrophage density in the femoral lesion (Figure 3). We had previously found that COX-2 protein expression and CCL2 mRNA were up-regulated in femoral extracts of rabbits with RC, with regard to hyperlipidaemic rabbits (Largo et al., 2008). So we looked for the effect of celecoxib on these markers of arthritis-associated vascular damage. There was a significant fall in COX-2 protein levels in rabbits treated with celecoxib compared with RC (fivefold levels, P = 0.004), while there was no effect on the expression of CCL2 mRNA, which persisted 3.6-fold increased versus healthy specimens (Figure 4).
Figure 4.

COX-2 and CCL2 expression in the femoral artery. Top, COX-2 protein expression: (A) densitometric analysis of Western blot studies; (B) a representative Western blot of COX-2 in femoral arteries. Bottom (C and D): analysis of COX-2 (C) and CCL2 (D) mRNA expression measured by real-time polymerase chain reaction method. *P < 0.05 versus healthy controls; #P < 0.05 versus rheumatoid cachexia (RC) rabbits. CXB, celecoxib.
Atherosclerotic lesions in the aorta
We also studied segments of the thoracic aorta, as one of the additional effects of experimental arthritis upon vascular damage was an increase in the incidence of distant lesions. In this regard, aortic lesions were observed in 60% of animals from RC group and in 57% of celecoxib-treated rabbits (P > 0.05). No aortic lesions were detected in the rabbits fed with a standard diet (healthy).
The lesions in the aorta were highly variable in size and appearance, and they consisted of lipid-rich deposits adhering to the aortic wall that were infiltrated with foam cells and other mononuclear cells. The larger ones contained elastic fibres, fibroblast-like cells and collagen deposition. In some cases, fatty streaks were found inside the media layer, associated with the rupture of elastic fibres, as observed with orcein (Figure 5).
Figure 5.
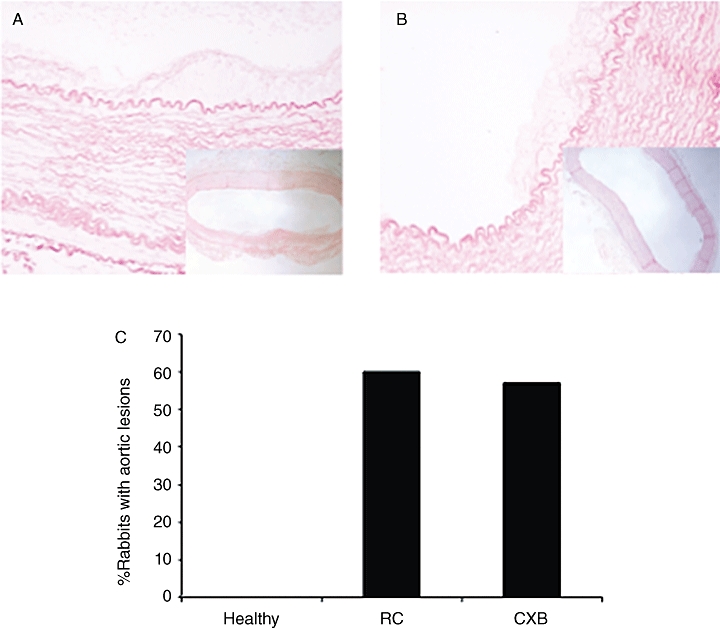
Presence of atherosclerotic plaque in the aorta of the rabbits. Low magnification photomicrographs (40×) show the distribution of plaques in representative aortas from rheumatoid cachexia (RC)- (A) and celecoxib (CXB)-treated (B) rabbits, stained with orcein. The plaques are shown in detail in high magnification (200×). (C) Graph bar. Percentage of rabbits with aortic plaque. *P < 0.05 versus healthy controls.
Discussion
The excessive synthesis of pro-inflammatory cytokines, such as IL-6, TNF-α and IL-1, is thought to be the most important cause of RC (Roubenoff et al., 1992). It has been reported that high levels of these cytokines lead to an increased hepatic protein synthesis (Walsmith and Roubenoff, 2002) and to a severe protein depletion and loss of weight, attributed in part to NF-κB activation (Guttridge et al., 2000; Acharyya et al., 2007). In agreement, we found that serum albumin levels fell in parallel to the increase in systemic inflammation, and NF-κB activation in PBMC. In addition, pro-inflammatory cytokines induce the production of prostaglandin E2 and some studies also involve COX-2 activation in the induction of cachectic status by insulin-like growth factor-I axis inhibition and activation of the ubiquitin-mediated proteolytic system and muscle wasting (Ganey et al., 2001; Davis et al., 2004; Granado et al., 2007). In rabbits with cachexia, we have found COX-2 gene up-regulation at the injured tissues, that is, the synovial membrane (Largo et al., 2008) and the femoral arteries. Here, selective inhibition of COX-2 with celecoxib reduced systemic inflammation and NF-κB activation, and ameliorated the loss of weight and the reduction in circulating albumin of cachectic rabbits. As previous studies suggested, this effect could be linked to regulation of TNF-α (Konturek et al., 2006; Granado et al., 2007). Indeed, it has been reported that the inhibitory effect of NSAIDs on NF-κB signalling might be responsible for the suppression of muscle wasting induced by the activation of the ubiquitin-proteasome pathway, which has been linked to the increase in TNF-α gene expression observed in cachexia (Wyke et al., 2004).
Although increased levels of triglycerides are a common finding in most examples of cachectic status (Khovidhunkit et al., 2004), lipolysis is not increased and adipose lipogenesis is reduced in animal models of arthritis-induced cachexia (Martín et al., 2008). On the other hand, celecoxib has shown to decrease the expression of fatty acid synthase, a key enzyme in triglyceride synthesis, by down-regulating c-Jun N-terminal kinase-1 (Lu and Archer, 2007), in animal models fed a high-fat diet. Surprisingly, in our study, triglycerides were further increased in the celecoxib-treated group suggesting that this drug may reverse the effect of inflammation on lipid metabolism. In addition and, by contrast with other species, inflammation induces hypercholesterolaemia in rodents and rabbits (Cabana et al., 1983; 1996; Khovidhunkit et al., 2004). Available data about the effect of COX-2 inhibition on cholesterol metabolism are controversial. While genetic COX-2 deficiency results in hypercholesterolaemia, which is further increased on atherogenic diets (Narasimha et al., 2007), celecoxib treatment of apoE-deficient mice fed a high-fat diet did not modify plasma cholesterol levels, although a slight activation of hydroxy-methyl-glutaryl-CoA reductase, the rate-limiting enzyme in cholesterol biosynthesis, has been reported (Metzner et al., 2007). We also found a reduction in HDL levels in rabbits with RC. During inflammation, serum HDL not only decreases but can also become pro-inflammatory (Van Lenten et al., 1995). Celecoxib administration, despite decreasing pro-inflammatory cytokines, paradoxically lowered HDL and this effect was accompanied by an increase in triglycerides. Moreover, the relationship between hypertriglyceridaemia and low HDL has been previously described in acute and in non-acute phase conditions (Gotto, 1990; Cabana et al., 1996). In addition, rabbits are deficient in hepatic lipase, so HDL triglyceride hydrolysis might be further compromised (Clay et al., 1989). As we did not determine HDL composition, we cannot say if the decrease in the HDL levels was due to its enrichment in triglycerides and/or a decrease in pro-inflammatory HDL synthesis.
On the other hand, treatment with celecoxib did not worsen vascular lesions induced in rabbits with cachexia. The apparently adverse cardiovascular lipid profile developed by the celecoxib-treated group, higher triglyceride and total cholesterol levels and lower HDL cholesterol than their non-treated counterparts, did not either result in a higher incidence of atherosclerotic lesions on intact aortas. These data are consistent with the results of Metzner et al. (2007), who did not find differences in lesion sizes in apoE-/- mice on a high-fat diet when treated with coxibs while apoE-/- mice on a chow diet developed larger lesions when treated, suggesting that COX-2 inhibition may not affect the late stages of atherogenesis, once plaque generation has already been initiated. Indeed, the vascular lesions in femoral arteries of celecoxib-treated animals did show a lesser degree of inflammation as measured by COX-2 abundance. We should stress that more than lipid levels, lipoprotein content is important in the development of inflammatory vascular lesions (Van Lenten et al., 2007).
Mechanisms other than inhibition of COX-2 and prostaglandin E2 biosynthesis, such as a decrease in smooth muscle cell proliferation and neointimal hyperplasia through inhibition of protein kinase B signalling (Yang et al., 2004), have been described with celecoxib in vivo. In this context, we did not find the inhibition in CCL2 expression, which has been previously reported (Wang et al., 2005; Tegeder and Geisslinger, 2006), although there was a tendency towards a lower expression in CXB animals.
In addition, the fall in CRP levels, a parameter that has been linked to a higher cardiovascular risk, seen with celecoxib might contribute to the reduced severity of inflammatory vascular lesions. In RA, cachexia is a common aberration of body composition and it is characterized by the concurrent decrease in fat-free mass and increase in fat mass, including central obesity. This may contribute clearly to the increased morbidity as well as the mortality associated with RA. Cytokine-driven hypermetabolism and protein degradation linked to the disease causes reduction of fat-free mass, which is, at the same time, combined with a significant increase of fat mass. This status can be readily defined as ‘cachectic obesity’, a condition resembling morbid obesity and which may contribute to increased risk of cardiovascular diseases in RA patients. ‘Cachectic obesity’ could be considered a truly ‘silent killer’ of these patients (Kumar and Armstrong, 2008). In addition, the increased fat mass, which in most cases can be considered as frank obesity, is responsible of the pro-inflammatory status that perpetuates disease mechanisms associated with RA, including cardiovascular disease, immune dys-regulation and inflammatory pathways, which are, of course, capital potential targets for therapy. Interestingly, fat mass is an important contributor to CRP levels in RA (Giles et al., 2008a) and a recent study in hyperlipidaemic rats showed that celecoxib decreased intra-abdominal adipose tissue mass (Lu and Archer, 2007). Although it is tempting to do so, we cannot speculate on this as we have not performed studies of body composition.
Summarizing, the major two contributions of our results are, on one hand, the development of an experimental model, which allows us to explore further the molecular mechanisms underlying RC and to simultaneously test the effect of drugs on the cardiovascular risk associated with RA. On the other hand, our results show that the use of an inhibitor of COX-2 improves RC without worsening already established vascular lesions or facilitating new ones.
Although the published literature and available expert's guidelines do not recommend the generalized use of NSAIDs, and particularly of COX-2 inhibitors, in patients at increased cardiovascular risk, in the light of our study, this advice should not be extended to diseases with an important inflammatory component, such as active arthritis. In this context, our results are consistent with a very recent phase II non-randomized study, which confirmed that celecoxib, at a moderate dosage and for duration of less than 6 months, did not increase cardiovascular risk (Mantovani et al., 2010). Thus, the improvement of RA-associated cachexia with these drugs may decrease the related poor clinical outcomes without having a negative effect on the associated cardiovascular risk, which is a key factor of morbidity and mortality in these patients. However, we have to keep in mind that our data are limited to an experimental model. Thus, further clinical studies in this field are needed in order to consider cachexia as a new indication for NSAIDs in this group of patients.
Acknowledgments
We thank Dr Concha de la Piedra and Petra Rubio for their collaborations in biochemical and histopathological studies. This work was supported by the Spanish Ministry of Health through the Fondo de Investigación Sanitaria, Instituto de Salud Carlos III [CP03/0011, PI06/0032]; the Spanish Ministry of Science and Innovation [SAF 2006/2704]; and the Mutua Madrileña Automovilística Foundation.
Glossary
Abbreviations
- AIA
antigen-induced arthritis
- BCM
body cell mass
- BMI
body mass index
- COX-2
cyclooxygenase-2
- CRP
C-reactive protein
- EMSA
electrophoretic mobility shift assay
- HDL
high-density lipoprotein
- IL-6
interleukin-6
- mRNA
messenger ribonucleic acid
- NF-κB
nuclear factor-κB
- NSAID
non-steroidal anti-inflammatory drugs
- PBMC
peripheral blood mononuclear cells
- PBS
phosphate buffered saline
- PCR
polymerase chain reaction
- PGE2
prostaglandin E2
- RA
rheumatoid arthritis
- RC
rheumatoid cachexia
- TNF
tumour necrosis factor
Conflicts of interest
The authors have no conflicts of interest to declare.
References
- Acharyya S, Villalta SA, Bakkar N, Bupha-Intr T, Janssen PM, Carathers M, et al. Interplay of IKK/NF-kappaB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arshad A, Rashid R, Benjamin K. The effect of disease activity on fat free mass and resting energy expenditure in patients with rheumatoid arthritis versus noninflammatory arthropathies/soft tissue rheumatism. Mod Rheumatol. 2007;17:470–475. doi: 10.1007/s10165-007-0628-1. [DOI] [PubMed] [Google Scholar]
- Cabana VG, Gewurz H, Siegel JN. Inflammation-induced changes in rabbit CRP and plasma lipoproteins. J Immunol. 1983;130:1736–1742. [PubMed] [Google Scholar]
- Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J Lipid Res. 1996;37:2662–2674. [PubMed] [Google Scholar]
- Clay MA, Hopkins GJ, Ehnholm CP, Barter PJ. The rabbit as an animal model of hepatic lipase deficiency. Biochim Biophys Acta. 1989;1002:173–181. doi: 10.1016/0005-2760(89)90284-1. [DOI] [PubMed] [Google Scholar]
- Davis TW, Zweifel BS, O'Neil JM, Heuvelman DM, Abegg AL, Hendrich TO, et al. Inhibition of cyclooxigenase-2 by celecoxib reverses tumor-induced wasting. J Pharmacol Exp Ther. 2004;308:929–934. doi: 10.1124/jpet.103.063099. [DOI] [PubMed] [Google Scholar]
- Escalante A, Haas R, Del Rincón I. Paradoxical effect of body mass index on survival in rheumatoid arthritis. Arch Intern Med. 2005;165:1624–1629. doi: 10.1001/archinte.165.14.1624. [DOI] [PubMed] [Google Scholar]
- Ganey PE, Barton YW, Kinser S, Sneed RA, Barton CC, Roth RA. Involvement of cyclooxigenase-2 in the potentiation of allyl alcohol-induced liver injury by bacterial lipopolysaccharide. Toxicol Appl Pharmacol. 2001;174:113–121. doi: 10.1006/taap.2001.9183. [DOI] [PubMed] [Google Scholar]
- Giles JT, Bartlett SJ, Andersen R, Thompson R, Fontaine KR, Bathon JM. Association of body fat with C-reactive protein in rheumatoid arthritis. Arthritis Rheum. 2008a;58:2632–2641. doi: 10.1002/art.23766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles JT, Ling SM, Ferruci L, Fontaine KR, Bathon JM. Abnormal body composition phenotypes in older rheumatoid arthritis patients: association with disease characteristics and pharmacotherapies. Arthritis Rheum. 2008b;59:807–815. doi: 10.1002/art.23719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles JT, Allison M, Bingham CO, Jr 3rd, Scott WM, Jr, Bathon JM. Adiponectin is a mediator of the inverse association of adiposity with radiographic damage in rheumatoid arthritis. Arthritis Rheum. 2009;61:1248–1256. doi: 10.1002/art.24789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotto AM., Jr Interrelationship of triglycerides with lipoproteins and high density lipoproteins. Am J Cardiol. 1990;66:20A–23A. doi: 10.1016/0002-9149(90)90565-i. [DOI] [PubMed] [Google Scholar]
- Granado M, Priego T, Martín I, Vara E, López-Calderón A, Villanúa MA. Anti-tumor necrosis factor agent PEG-sTNFRI improves the growth hormone/insulin-like growth factor-I system in adjuvant-induced arthritic rats. Eur J Pharmacol. 2006;536:204–210. doi: 10.1016/j.ejphar.2006.02.035. [DOI] [PubMed] [Google Scholar]
- Granado M, Martín AI, Villanúa MA, López-Calderón A. Experimental arthritis inhibits the insulin-like growth factor-I axis and induces muscle wasting through cyclooxygenase-2 activation. Am J Physiol Endocrinol Metab. 2007;292:E1656–E1665. doi: 10.1152/ajpendo.00502.2006. [DOI] [PubMed] [Google Scholar]
- Guttridge DC, MayoMW MLV, Wang CY, Baldwin AS., Jr NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science. 2000;289:2363–2366. doi: 10.1126/science.289.5488.2363. [DOI] [PubMed] [Google Scholar]
- Hernández-Presa MA, Martin-Ventura JL, Ortego M, Gómez-Hernández A, Tuñón J, Hernández-Vargas P, et al. Atorvastatin reduces the expression of cyclooxygenase-2 in a rabbit model of atherosclerosis and in cultured vascular smooth muscle cells. Atherosclerosis. 2002;160:49–58. doi: 10.1016/s0021-9150(01)00547-0. [DOI] [PubMed] [Google Scholar]
- Inaba M, Tanaka K, Goto H, Usami T, Azumi K, Kubota H, et al. Independent association of increased trunk fat with increased arterial stiffening in postmenopausal patients with rheumatoid arthritis. J Rheumatol. 2007;34:290–295. [PubMed] [Google Scholar]
- Khovidhunkit W, Min-Sun K, Memon RA, Shigenaga JK, Moser AH, Feingold KR, et al. Effects of infection and inflammation on lipid and lipoprotein metabolism: mechanisms and consequences to the host. J Lipid Res. 2004;45:1169–1196. doi: 10.1194/jlr.R300019-JLR200. [DOI] [PubMed] [Google Scholar]
- Konturek PC, Rembiasz K, Burnat G, Konturek SJ, Tusinela M, Bielanski W, et al. Effects of cyclooxigenase-2 inhibition on serum and tumor gastrins and expression of apoptosis-related proteins in colorectal cancer. Dig Dis Sci. 2006;51:779–787. doi: 10.1007/s10620-006-3206-z. [DOI] [PubMed] [Google Scholar]
- Kremers HM, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Prognostic importance of low body mass index in relation to cardiovascular mortality in rheumatoid arthritis. Arthritis Rheum. 2004;50:3450–3457. doi: 10.1002/art.20612. [DOI] [PubMed] [Google Scholar]
- Kumar N, Armstrong DJ. Cardiovascular disease – the silent killer in rheumatoid arthritis. Clin Med. 2008;8:384–387. doi: 10.7861/clinmedicine.8-4-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai V, George J, Richey L, Kim HJ, Cannon T, Shores C, et al. Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck and gastrointestinal tract. Head Neck. 2008;30:67–74. doi: 10.1002/hed.20662. [DOI] [PubMed] [Google Scholar]
- Largo R, Alvarez-Soria MA, Diez-Ortego I, Calvo E, Sánchez-Pernaute O, Egido J, et al. Glucosamine inhibits IL-1β-induced NFκB activation in human osteoarthritic chondrocytes. Osteoarthritis Cartilage. 2003;11:290–298. doi: 10.1016/s1063-4584(03)00028-1. [DOI] [PubMed] [Google Scholar]
- Largo R, Sánchez-Pernaute O, Marcos ME, Moreno-Rubio J, Aparicio C, Granado R, et al. Chronic arthritis aggravates vascular lesions in rabbits with atherosclerosis: a novel model of atherosclerosis associated with chronic inflammation. Arthritis Rheum. 2008;58:2723–2734. doi: 10.1002/art.23765. [DOI] [PubMed] [Google Scholar]
- Lu S, Archer MC. Celecoxib decreases fatty acid synthase expression via down-regulation of c-Jun N-Terminal Kinase-1. Exp Biol Med. 2007;232:643–653. [PubMed] [Google Scholar]
- Lundholm K, Daneryd P, Corner U, Hyltander A, Bosaeus I. Evidence that long-term COX-treatment improves energy homeostasis and body composition in cancer patients with progressive cachexia. Int J Oncol. 2004;24:505–512. [PubMed] [Google Scholar]
- Mantovani G, Madeddu C. Cyclooxygenase-2 inhibitors and antioxidants in the treatment of cachexia. Curr Opin Support Palliat Care. 2008;2:275–281. doi: 10.1097/spc.0b013e32830f47e4. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Macciò A, Madeddu C, Gramignano G, Lusso MR, Serpe R, et al. A phase II study with antioxidants, both in the diet and supplemented, pharmaconutritional support, progestagen, and anti-cyclooxigenase-2 showing efficacy and safety in patients with cancer-related anorexia/cachexia and oxidative stress. Cancer Epidemiol Biomarkers Prev. 2006;15:1030–1034. doi: 10.1158/1055-9965.EPI-05-0538. [DOI] [PubMed] [Google Scholar]
- Mantovani G, Macció A, Madeddu C, Serpe R, Antoni G, Massa E, et al. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J Mol Med. 2010;88:85–92. doi: 10.1007/s00109-009-0547-z. [DOI] [PubMed] [Google Scholar]
- Marcora SM, Chester KR, Mittal G, Lemmey AB, Maddison PJ. Randomized phase 2 trial of anti-tumor necrosis factor therapy for cachexia in patients with early rheumatoid arthritis. Am J Clin Nutr. 2006;84:1463–1472. doi: 10.1093/ajcn/84.6.1463. [DOI] [PubMed] [Google Scholar]
- Martín AI, Castillero E, Granado M, López-Menduiña M, Villanúa MA, López-Calderón A. Adipose tissue loss in adjuvant arthritis is associated with a decrease in lipogenesis, but not with an increase in lipolysis. J Endocrinol. 2008;197:111–119. doi: 10.1677/JOE-07-0491. [DOI] [PubMed] [Google Scholar]
- Metsios GS, Stavropoulos-Kalinoglou A, Douglas KMJ, Koutedakis Y, Nevill AM, Panoulas VF, et al. Blockade of tumour necrosis factor-α in rheumatoid arthritis: effects on components of rheumatoid cachexia. Rheumatology. 2007;46:1824–1827. doi: 10.1093/rheumatology/kem291. [DOI] [PubMed] [Google Scholar]
- Metzner J, Popp L, Marian C, Schmidt R, Manderscheid C, Renne C, et al. The effects of COX-2 selective and non-selective NSAIDs on the iniciation and progression of athrosclerosis in ApoE-/- mice. J Mol Med. 2007;85:623–633. doi: 10.1007/s00109-007-0162-9. [DOI] [PubMed] [Google Scholar]
- Morley JE, Thomas DR, Wilson MMG. Cachexia: Pathophysiology and clinical relevance. Am J Clin Nutr. 2006;83:735–743. doi: 10.1093/ajcn/83.4.735. [DOI] [PubMed] [Google Scholar]
- Narasimha A, Watanabe J, Lin JA, Hama S, Langenbach R, Navab M, et al. A novel anti-atherogenic role for COX-2: potencial mechanism for the cardiovascular side effects of COX-2 inhibitors. Prostaglandins Other Lipid Mediat. 2007;84:24–33. doi: 10.1016/j.prostaglandins.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubenoff R, Roubenoff RA, Ward LM, Holland SM, Hellmann DB. Rheumatoid cachexia: depletion of lean body mass in rheumatoid artritis. Possible association with tumor necrosis factor. J Rheumatol. 1992;19:1505–1510. [PubMed] [Google Scholar]
- Roubenoff R, Roubenoff RA, Cannon JG, Kehayias JJ, Zhuang H, Dawson-Hughes B, et al. Rheumatoid cachexia: cytokine-driven hypermetabolism accompanying reduced body cell mass in chronic inflammation. J Clin Invest. 1994;93:2379–2386. doi: 10.1172/JCI117244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roubenoff R, Freeman LM, Smith DE, Abad LW, Dinarello CA, Kehayias JJ. Adjuvant arthritis as a model of inflammatory cachexia. Arthritis Rheum. 1997;40:534–539. doi: 10.1002/art.1780400320. [DOI] [PubMed] [Google Scholar]
- Summers GD, Deighton CM, Rennie MJ, Booth AH. Rheumatoid cachexia: a clinical perspective. Rheumatology (Oxford) 2008;47:1124–1131. doi: 10.1093/rheumatology/ken146. [DOI] [PubMed] [Google Scholar]
- Tegeder I, Geisslinger G. Cardiovascular risk with cyclooxygenase inhibitors: general problem with substance specific differences. Naunyn Schmiedebergs Arch Pharmacol. 2006;373:1–17. doi: 10.1007/s00210-006-0044-7. [DOI] [PubMed] [Google Scholar]
- Van Lenten BJ, Hama SY, de Beer FC, Stafforini DM, McIntyre TM, Prescott SM, et al. Anti-inflammatory HDL becomes pro-inflammatory during the acute phase response. J Clin Invest. 1995;96:2758–2767. doi: 10.1172/JCI118345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lenten BJ, Wagner AC, Navab M, Anantharamaiah GM, Hama S, Reddy ST, et al. Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol-fed rabbits. J Lipid Res. 2007;48:2344–2353. doi: 10.1194/jlr.M700138-JLR200. [DOI] [PubMed] [Google Scholar]
- Vidal C, Gómez-Hernández A, Sánchez-Galán E, González A, Ortega L, Gómez-Gerique JA, et al. Licofenole, a balanced inhibitor of cyclooxigenase and lypoxygenase reduces inflammation in a rabbit model of atherosclerosis. J Pharmacol Exp Ther. 2007;320:108–116. doi: 10.1124/jpet.106.110361. [DOI] [PubMed] [Google Scholar]
- Walsmith J, Roubenoff R. Cachexia in rheumatoid arthritis. Int J Cardiol. 2002;85:89–99. doi: 10.1016/s0167-5273(02)00237-1. [DOI] [PubMed] [Google Scholar]
- Walsmith J, Abad L, Kehayias J, Roubenoff R. Tumor necrosis factor-α production is associated with less body cell mass in women with rheumatoid arthritis. J Rheumatol. 2004;31:23–29. [PubMed] [Google Scholar]
- Wang K, Tarakji K, Zhou Z, Zhang M, Forudi F, Zhou X, et al. Celecoxib, a selective cyclooxygenase-2 inhibitor, decreases monocyte chemoattractant protein-1 expression and neointimal hyperplasia in the rabbit atherosclerotic balloon injury model. J Cardiovasc Pharmacol. 2005;45:61–67. doi: 10.1097/00005344-200501000-00011. [DOI] [PubMed] [Google Scholar]
- Wyke SM, Russell ST, Tisdale MJ. Induction of proteasome expression in skeletal muscle is attenuated by inhibitors of NF-kappa B activation. Br J Cancer. 2004;91:1742–1750. doi: 10.1038/sj.bjc.6602165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW, et al. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signalling. Circulation. 2004;110:301–308. doi: 10.1161/01.CIR.0000135467.43430.16. [DOI] [PubMed] [Google Scholar]