

Abstract
BACKGROUND AND PURPOSE
Hyperleptinemia is commonly found in obese patients, associated with non-alcoholic steatohepatitis and hepatic fibrosis. Hepatic stellate cells (HSCs) are the most relevant effectors during hepatic fibrogenesis. We recently reported that leptin stimulated HSC activation, which was eliminated by curcumin, a phytochemical from turmeric. This study was designed to explore the underlying mechanisms, focusing on their effects on intracellular glucose in HSCs. We hypothesized that leptin stimulated HSC activation by elevating the level of intracellular glucose, which was eliminated by curcumin by inhibiting the membrane translocation of glucose transporter-4 (GLUT4) and inducing the conversion of glucose to glucose-6-phosphate (G-6-P).
EXPERIMENTAL APPROACH
Levels of intracellular glucose were measured in rat HSCs and immortalized human hepatocytes. Contents of GLUT4 in cell fractions were analysed by Western blotting analyses. Activation of signalling pathways was assessed by comparing phosphorylation levels of protein kinases.
KEY RESULTS
Leptin elevated the level of intracellular glucose in cultured HSCs, which was diminished by curcumin. Curcumin suppressed the leptin-induced membrane translocation of GLUT4 by interrupting the insulin receptor substrates/phosphatidyl inositol 3-kinase/AKT signalling pathway. Furthermore, curcumin stimulated glucokinase activity, increasing conversion of glucose to G-6-P.
CONCLUSIONS AND IMPLICATIONS
Curcumin prevented leptin from elevating levels of intracellular glucose in activated HSCs in vitro by inhibiting the membrane translocation of GLUT4 and stimulating glucose conversion, leading to the inhibition of HSC activation. Our results provide novel insights into mechanisms of curcumin in inhibiting leptin-induced HSC activation.
Keywords: hepatic fibrosis, hepatic stellate cell, leptin, non-alcoholic steatohepatitis, obesity, phytochemical
Introduction
Obesity is often accompanied by insulin resistance and hyperleptinemia, leading to the development of type 2 diabetes mellitus (T2DM) (Stefanovic et al., 2008; Yokaichiya et al., 2008). Obesity and T2DM are important risk factors for the development of non-alcoholic steatohepatitis (NASH), which is the advanced form of non-alcoholic fatty liver disease (Watanabe et al., 2008). Approximately 15–40% of NASH patients develop hepatic fibrosis (Clark, 2006). Hepatic stellate cells (HSCs) are the major cell type responsible for the over-production of extracellular matrix and for the development of hepatic fibrosis (Friedman, 2008).
The hormone leptin is mainly responsible for regulating energy homeostasis, glucose transport and a neuro-endocrine negative feedback loop (Zhang et al., 1994; Margetic et al., 2002). Binding of leptin activates its receptor and downstream signalling cascades, including the Janus kinase (JAK)/signal transducers and activators of transcription and subsequently phosphatidylinositol 3-kinase (PI3K) (Lee et al., 1996). The leptin signalling pathway cross-talks with the insulin signalling pathway mediated by activating insulin receptor substrate1/2 (IRS1/2) (Zhao et al., 2000; Benomar et al., 2006; Fruhbeck, 2006). Leptin-deficient mice fail to develop hepatic fibrosis during steatohepatitis or in response to chronic toxic liver injury (Leclercq et al., 2002), indicating the critical roles of leptin signalling pathways in mediating hepatic fibrogenesis.
The level of intracellular glucose is mainly determined by glucose supplementation, such as glucose uptake, and glucose consumption, including glucose conversion to glucose-6-phosphate (G-6-P). Glucose uptake is mainly mediated by glucose transporters (GLUTs) (Asano et al., 2004). In the liver, GLUT2 and GLUT4 are the major GLUTs responsible for glucose transport into hepatocytes (Leturque et al., 2005; Zhao and Keating, 2007). While GLUT2 transports glucose in a large range of physiological concentrations of glucose, glucose uptake by GLUT4 is extensively regulated by activated PI3K (Leturque et al., 2005; Zhao and Keating, 2007). Insulin stimulates glucose uptake into cells by activating the IRS/PI3K/AKT signalling pathway (Benomar et al., 2006) and prompting the translocation of the GLUT4 from the cytosol to the cell surface (Leney and Tavare, 2009). On the other hand, glucose consumption results in the reduction in the level of intracellular glucose. The conversion of glucose to G-6-P is mainly catalysed by glucokinase (GK), the major isoform of hexokinase in the liver (Grimsby et al., 2008). The activity of GK is reversibly inhibited by PKA and activated by AMP-activated protein kinase (AMPK) (Ekman and Nilsson, 1988; Mukhtar et al., 2008). AMPK is a well-conserved eukaryotic serine/threonine protein kinase which plays a central role in regulating cellular energy homeostasis. Leptin activates PKA in white adipocytes and in aortic endothelial cells (Yamagishi et al., 2001; Mehebik et al., 2005), and suppresses AMPK in hypothalamus (Hayes et al., 2009). High levels of glucose were found to stimulate HSC activation in vitro (Paradis et al., 2001; Sugimoto et al., 2005).
In the last two decades, advances in the understanding of genes promoting HSC activation are impressive. There are, however, few breakthroughs in therapeutic interventions for hepatic fibrogenesis. Therefore, research identifying innocuous anti-fibrogenic agents is of high priority and is urgently needed. Most evolving anti-fibrogenic therapies aim at inhibiting HSC activation. Curcumin from turmeric is one of the most extensively studied natural compounds (Joe et al., 2004; Singh and Khar, 2006) and, in the present context, has received attention as a promising dietary supplement for liver protection (O'Connell and Rushworth, 2008). We previously demonstrated that curcumin inhibited HSC activation in vitro and in vivo (Xu et al., 2003; Zheng and Chen, 2004; Fu et al., 2008). We recently reported that leptin stimulated HSC activation, which was eliminated by curcumin (Tang et al., 2009). The current study was designed to explore the underlying mechanisms, focusing on the effects of leptin and curcumin on the level of intracellular glucose in HSCs. Our results supported our initial hypothesis that leptin stimulated HSC activation by elevating the level of intracellular glucose. Curcumin eliminated the effect of leptin by inhibiting the membrane translocation of GLUT4 and inducing the conversion of glucose to G-6-P, leading to decreased intracellular glucose and inhibition of HSC activation.
Methods
HSC isolation and culture
All animal care and experimental procedures complied with the guidelines of NIH and were approved by the Institutional Animal Care and Use Committee of Saint Louis University. Animals were kept under a 12:12 h light/dark cycle and allowed free access to food and water, within the Animal Care Facilities of the Medical School, Saint Louis University. HSCs were isolated from male Sprague-Dawley, or lean wild-type Zucker (Fa/Fa), rats (200–250 g) or from male leptin receptor-deficient Zucker (fa/fa) rats (300–350 g; both from Harlan Laboratories, Inc., Indianapolis, IN, USA) by pronase–collagenase perfusion in situ prior to density gradient centrifugation, as we have described (Xu et al., 2003). Primary cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 20% of fetal bovine serum (FBS). Passaged cells were grown in DMEM with 10% of FBS. Semi-confluent HSCs with four to nine passages were used in most of experiments. Immortalized human hepatocytes (IHHs) were kindly provided by Dr Ratna Ray (Department of Pathology, Saint Louis University) (Ray et al., 2000). In some experiments, cells were cultured in serum-depleted media for 24 h before treatment, which rendered HSCs more sensitive to exogenous leptin. The cells were subsequently treated and cultured in serum-depleted media, which excluded any interference from other factors in FBS.
Immuno-precipitation and Western blotting analyses
Whole-cell extracts were prepared from passaged HSCs. Protein concentrations were determined by using the BCA Protein Assay Kit according to the protocol provided by the manufacturer (Pierce, Rockford, IL, USA). Immuno-precipitation was performed as we recently described (Tang et al., 2009). Electrophoresis, transblotting and immuno-detection were conducted as previously described (Xu et al., 2003). Primary antibodies against phosphoryated AMPK (p-AMPK) and total AMPK, p-PKA and total PKA were purchased from Cell Signaling Technology, Inc. (Danvers, MA, USA). Primary antibodies against p-IRS1 and total IRS1, p-IRS2 and total IRS2, p-AKT and total AKT, GLUT4, α-I(I)procollagen, α-smooth muscle actin (α-SMA), β-actin and β-tubulin and horseradish peroxidase-conjugated secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). β-Actin or β-tubulin was used in most of experiments as an internal control for equal loading. Densities of bands in Western blotting analyses were normalized with the internal invariable control. Levels of target protein bands were densitometrically determined by using Quantity One 4.4.1 (Bio-Rad, Hercules, CA, USA). Variations in the density were expressed as fold changes compared to the control in the blot.
Intracellular glucose assays
Levels of intracellular glucose were colorimetrically determined by using Glucose Assay Kit (BioVision, Mountain View, CA, USA), following the protocol provided by the manufacturer.
Measurement of intracellular G-6-P levels
The method was adapted from the colorimetric assay reported by de Koning and van Dam (1992), with minor modifications.
GK activity assays
GK activities were measured by colorimetric detection of the NADH produced by G6PDH-induced dehydrogenation of G-6-P in the presence of ATP and NAD+, as described by Perez-Echarri et al. (2009).
Preparation of cellular membrane protein extracts
The preparation of cellular membrane fraction was performed as described previously (Nishiumi and Ashida, 2007). In brief, after washed three times with PBS, HSCs were lysed with buffer A [Tris, pH 8.0, 50 mM; dithiothreitol, 0.5 mM; NP-40, 0.1% (v/v); protease inhibitors (phenylmethylsulphonyl fluoride, 1 mM; leupeptin, 5 µg·mL−1; and aprotinin, 5 µg·mL−1) and phosphatase inhibitors (NaF, 10 mM and Na3VO4, 1 mM)]. The lysates were then centrifuged at 1000× g for 10 min at 4°C. Pellets were resuspended in NP-40 free buffer A in ice for another 10 min with occasional vortex, and recentrifuged at 1000× g for 10 min at 4°C. The pellets were resuspended in buffer A and placed in ice for 1 h with occasional vortexing, and centrifuged at 16 000× g for 20 min at 4°C. The supernatant was collected as the plasma membrane fraction and stored at −80°C until use. The supernatants from the first and second spins at 1000× g were combined and centrifuged at 16 000× g for 20 min at 4°C. The resultant supernatant was collected and used as the cytosol fraction.
Statistical analyses
Results were calculated by the formula: [(Glu or G-6-P in target HSCs – Glu or G-6-P in compared HSCs)/Glu or G-6-P in compared HSCs] × 100% (n = 3). Differences between means were evaluated using an unpaired two-sided Student's t-test (P < 0.05 considered as significant). Where appropriate, comparisons of multiple treatment conditions with controls were analysed by anova with Dunnett's test for post hoc analyses.
Materials
Curcumin (purity >94%); leptin (recombinant, rat); N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinoline sulphonamide (H-89), a selective PKA inhibitor; compound C (compd C), a specific AMPK inhibitor; and 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), an AMPK activator, were purchased from Sigma (St Louis, MO, USA).
Results
Leptin caused a time- and dose-dependent increase in the level of intracellular glucose in cultured HSCs, which was attenuated by curcumin
We assumed that one of the mechanisms by which leptin stimulated HSC activation might be to elevate the level of intracellular glucose. To test the assumption, semi-confluent HSCs were serum starved for 24 h, followed by stimulation with leptin (100 ng·mL−1) for indicated times in serum-free DMEM, which contained 5.5 mM of glucose. As shown in Figure 1A, leptin caused a rapid increase in the level of intracellular glucose in a time-dependent manner, which reached a peak 30 min after the exposure to leptin. Compared with the untreated control cells, leptin significantly increased the level of intracellular glucose after 30 min treatment. This increase was gradually reduced over 24 h (Figure 1A).
Figure 1.
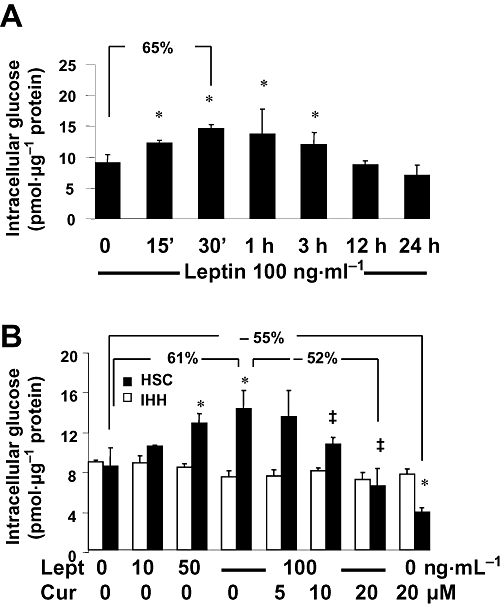
Leptin caused a time- and concentration-dependent increase in the level of intracellular glucose in cultured HSCs, which was attenuated by curcumin. Analyses of levels of intracellular glucose. Values were expressed as pmol glucose·µg−1 protein and presented as means ± SD (n = 3). *P < 0.05 versus the untreated control cells; ‡P < 0.05 versus cells treated with leptin alone at 100 ng·mL−1. Percentages were calculated by the following formula: [Glu in target HSCs – Glu in compared HSCs)/Glu in compared HSCs] × 100% (n = 3). (A) Serum-starved HSCs were treated with leptin at 100 ng·mL−1 in serum-free DMEM for indicated time-points. (B) Serum-starved HSCs were pretreated with curcumin at 0–30 µM for 1 h prior to the stimulation with or without leptin at indicated concentrations in serum-free media for additional 30 min.
To evaluate the effect of curcumin on the leptin-induced increase in the level of intracellular glucose, serum-starved HSCs or IHHs were pretreated with curcumin at indicated doses for 1 h, which blocked activating signal pathways and excluded their interference, prior to the addition of leptin at various concentrations for an additional 30 min in serum-free DMEM. Leptin caused a dose-dependent increase in the level of intracellular glucose (Figure 1B), and this leptin-induced increase was diminished by curcumin in a concentration-dependent manner (Figure 1B). For example, compared with leptin at 100 ng·mL−1 alone, curcumin at 20 µM dramatically diminished the stimulatory effect of leptin and significantly reduced the level of intracellular glucose. Interestingly, curcumin at 20 µM alone significantly reduced the level of intracellular glucose in HSCs, compared with that in cells with no treatment. However, leptin did not raise the level of cellular glucose in IHH, and curcumin had no effect on the level of cellular glucose in these hepatic cells (Figure 1B). Taken together, these results demonstrated that leptin caused a time- and dose-dependent increase in the level of intracellular glucose in cultured HSCs, which was attenuated by curcumin.
Curcumin inhibited GLUT4 membrane translocation and reduced the level of intracellular glucose in activated HSCs, by interrupting the leptin-activated IRS/PI3K/AKT signalling pathway
To elucidate the underlying mechanisms by which curcumin eliminated the effect of leptin on raising intracellular glucose, we postulated that curcumin might interrupt the leptin-activated IRS/PI3K/AKT signalling pathway in activated HSCs, leading to the inhibition of the GLUT4 membrane translocation and to the decrease of intracellular glucose. To test this postulate, serum-starved HSCs were pretreated with curcumin at indicated concentrations for 1 h prior to the stimulation with or without leptin at 100 ng·mL−1 in serum-free media for an additional 30 min. Lack of serum in the incubation excluded potential influence of factors in serum on the IRS/PI3K/AKT pathway. As shown by Western blotting analyses (Figure 2A), compared with the untreated control, leptin, as expected, significantly induced the phosphorylation of IRS1/2, PI3K and AKT, indicating activation of the IRS/PI3K/AKT signalling pathway. Curcumin prevented leptin's effects in a concentration-dependent manner.
Figure 2.
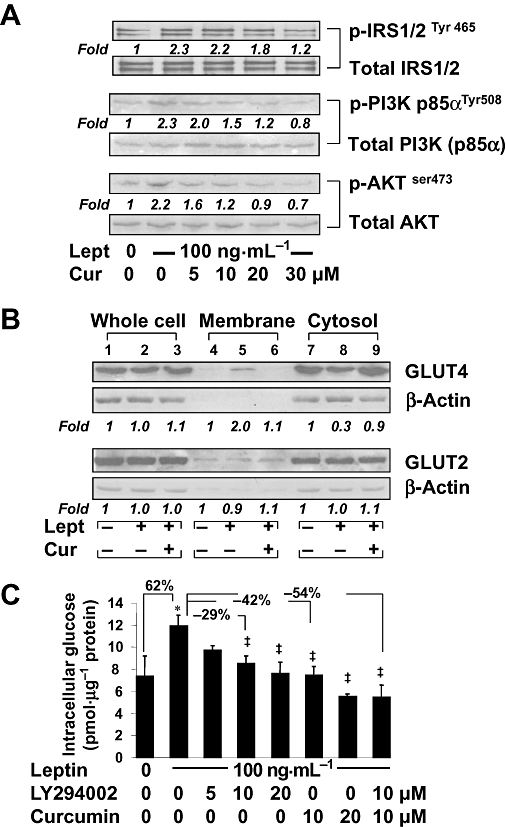
Curcumin interrupted the leptin-activated IRS/PI3K/AKT signalling pathway in cultured HSCs, leading to inhibition of the GLUT4 membrane translocation and to reduction of intracellular glucose. Serum-starved HSCs were pretreated with or without curcumin at indicated concentrations for 1 h prior to the stimulation with or without leptin (100 ng·mL−1) in serum-free media for additional 30 min. Western blotting analyses were conducted (A,B). Representative blots are shown from three independent experiments. Italic numbers beneath blots are fold changes in the densities of the bands compared to the control without treatment in the blot (n = 3), after normalization with the internal invariable control. Because of the limited space, standard deviations are not presented. (A) Detection of phosphorylated IRS1/2, PI3K and AKT. The total protein for each corresponding phospho-protein was used as an internal control for equal loading. (B) Evaluation of the abundance of GLUT2 and GLUT4, respectively, in the fractions of whole cells, membrane and cytoplasm of HSCs. (C) Serum-starved HSCs were pretreated with or without the PI3K/AKT inhibitor LY294002 at indicated concentrations, or with curcumin (20 µM), for 1 h prior to the stimulation with or without leptin (100 ng·mL−1) in serum-free media for additional 30 min. Levels of intracellular glucose were determined. Values were expressed as pmol glucose·µg−1 protein and presented as means ± SD. *P < 0.05 versus the untreated control; ‡P < 0.05 versus cells treated with leptin alone.
Further experiments (Figure 2B) revealed that these treatments had no effects on the abundance of total GLUT4 protein in whole HSC extracts. Compared with the untreated control, leptin dramatically increased the level of membrane GLUT4 and reduced the content of cytosol GLUT4, which were abolished by curcumin (20 µM). In marked contrast, leptin and/or curcumin did not affect the level of GLUT2 in any fraction of the cells (Figure 2B). These results collectively indicated that curcumin eliminated the effect of leptin on induction of GLUT4 membrane translocation in activated HSCs in vitro.
To evaluate the role of the PI3K/AKT signalling pathway on the level of intracellular glucose in HSCs, serum-starved cells were pretreated with or without LY294002, a specific PI3K/AKT inhibitor, at different concentrations as indicated, or with curcumin at 20 µM, for 1 h prior to the stimulation with or without leptin at 100 ng·mL−1 in serum-free DMEM for additional 30 min. Levels of intracellular glucose in the cells were measured. As shown in Figure 2C, compared with the untreated control, leptin significantly increased, as expected, the level of intracellular glucose in the cells. Compared with cells treated with leptin alone, inhibition of the PI3K/AKT signalling pathway by LY294002, mimicking curcumin, concentration-dependently diminished the effect of leptin and decreased the levels of intracellular glucose. These results demonstrated the critical role of the IRS/PI3K/AKT signalling pathway in controlling intracellular glucose levels. It was also of interest to observe that compared with leptin alone, LY294002 at 10 µM or curcumin at 10 µM, reduced the effect of leptin and decreased intracellular glucose. However, the combination of LY294002 (10 µM) plus curcumin (10 µM) (the last column) showed an additive effect of the two compounds in reversing the effects of leptin and reducing intracellular glucose. Taken together, these results revealed that curcumin prevented leptin from inducing GLUT4 membrane translocation and from raising intracellular glucose in activated HSCs in vitro, most likely by interrupting the leptin-activated IRS/PI3K/AKT signalling pathway.
Curcumin eliminated the inhibitory effect of leptin on the activity of GK and increased the level of intracellular G-6-P in activated HSCs in vitro
To further elucidate the underlying mechanisms by which curcumin prevented leptin from elevating intracellular glucose, we assumed that in addition to the inhibition of glucose uptake, curcumin might stimulate glucose consumption, including glucose conversion to G-6-P. It was also possible that curcumin would prevent leptin-induced inhibition of GK activity in cultured HSCs. To test these possibilities, serum-starved HSCs were pretreated with or without curcumin at indicated concentrations for 1 h prior to stimulation with leptin at different concentrations, as indicated, in serum-free DMEM for an additional 30 min. Activities of GK in the cells were measured. As shown in Figure 3A, leptin concentration-dependently reduced GK activities in the cells. The inhibitory impact of leptin was prevented by curcumin in a concentration-dependent manner. For instance, compared with leptin at 100 ng·mL−1 alone, curcumin at 20 µM reversed the effects of leptin and dramatically increased GK activity. In agreement with this observation, further experiments (Figure 3B) indicated that leptin concentration-dependently reduced intracellular G-6-P levels, which was prevented by curcumin in a concentration-dependent manner. For instance, compared with the untreated control, leptin at 100 ng·mL−1 significantly reduced the level of cellular G-6-P, and this effect of leptin was prevented by curcumin at 20 µM, leading to a marked increase in G-6-P. Note that, compared with the untreated control, curcumin at 20 µM alone significantly increased, as expected, the activity of GK and the level of intracellular G-6-P.
Figure 3.
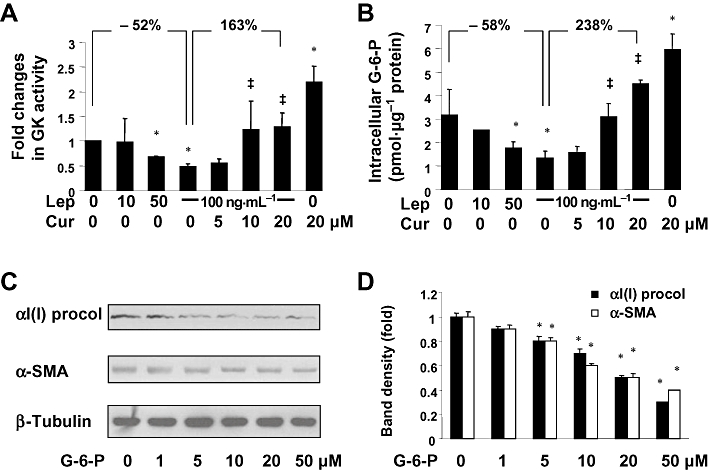
Curcumin eliminated the inhibitory effect of leptin on the activity of GK and increased intracellular G-6-P in activated HSCs in vitro. (A,B) Serum-starved HSCs were pretreated with curcumin at indicated concentrations for 1 h prior to the stimulation with or without leptin (100 ng·mL−1) in serum-free media for additional 30 min. (A) Analyses of GK activities. Values were expressed as fold changes (means ± SD), compared with the untreated control (n = 3). *P < 0.05 versus the untreated control; ‡P < 0.05 versus cells treated with leptin at 100 ng·mL−1 alone. (B) Analyses of levels of intracellular G-6-P. Values were expressed as pmol G-6-P·µg−1 protein (means ± SD) (n = 3). *P < 0.05 versus the untreated control; ‡P < 0.05 versus cells treated with leptin at 100 ng·mL−1 alone. (C) Western blotting analyses of αI(I)procollagen (procol) and α-SMA in HSCs treated with G-6-P at different concentrations as indicated in serum-depleted media for 24 h. β-Tubulin was used as an invariant control for equal loading. Representatives were presented from three independent experiments. (D) The Western blotting analyses were summarized after normalization with β-tubulin. Variations in the band density were expressed as fold changes compared to the untreated control in the blot (means ± SD., n = 3). *P < 0.05 versus the untreated control.
To evaluate the effect of G-6-P on HSC activation, serum-starved HSCs were treated with exogenous G-6-P at different concentrations, as indicated, for 24 h. As shown by Western blotting analyses (Figure 3C,D), G-6-P concentration-dependently reduced the abundance of αI(I) procollagen and α-SMA, the two unique markers for activated HSCs in the cells, indicating the inhibition by G-6-P of HSC activation. Taken together, these results suggested that curcumin prevented the inhibitory effect of leptin on GK activity and increased the level of intracellular G-6-P in activated HSCs in vitro, leading to an overall reduction in the level of intracellular glucose.
Curcumin inhibited PKA activity, which facilitated the increase in the activity of GK and the reduction in the level of intracellular glucose in cultured HSCs
To explore the mechanisms by which curcumin eliminated the inhibitory effect of leptin on the activity of GK, we assumed that curcumin increased GK activity by inhibiting PKA, resulting in the reduction in the level of intracellular glucose. To test this assumption, serum-starved HSCs were pretreated with curcumin at indicated concentrations for 1 h prior to the stimulation with or without leptin at 100 ng·mL−1 in serum-free DMEM for additional 30 min. As shown by Western blotting analyses (Figure 4A), compared with the untreated control, leptin increased the level of phosphorylated PKA (p-PKA) in cultured HSCs. Curcumin dose-dependently diminished the effect of leptin and reduced the level of p-PKA.
Figure 4.
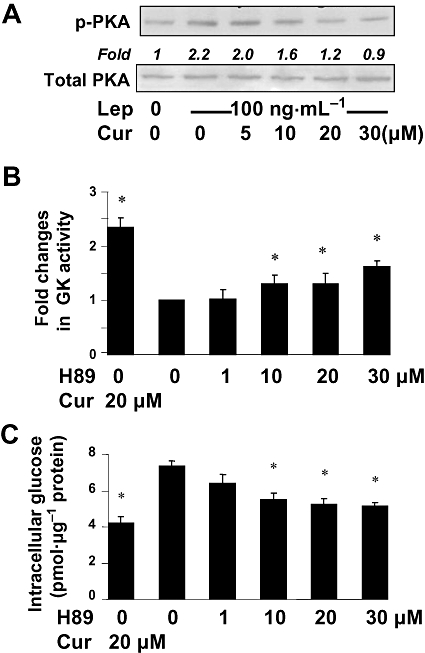
Curcumin inhibited PKA activity, which facilitated the increase in the activity of GK and the reduction in intracellular glucose in cultured HSCs. (A) Serum-starved HSCs were pretreated with curcumin at indicated concentrations for 1 h prior to the stimulation with or without leptin (100 ng·mL−1) in serum-free media for additional 30 min. Levels of p-PKA were evaluated by Western blotting analyses. Total PKA was used as an invariant control for equal loading. Representative blots are presented from three independent experiments. Italic numbers beneath blots were fold changes in the densities of the bands compared to the control without treatment in the blot (n = 3), after normalization with the internal invariable control. (B,C) Serum-starved HSCs were pretreated with the PKA inhibitor H89 at indicated concentrations, or with curcumin (20 µM), in serum-free media for 1 h prior to the addition of leptin (100 ng·mL−1) for additional 30 min. *P < 0.05 versus the untreated control (the second column). (B) Analyses of GK activities. Values were expressed as fold changes compared with the untreated control (means ± SD) (n = 3). (C) Determination of levels of intracellular glucose. Values were expressed as pmol glucose·µg−1 protein (means ± SD) (n = 3).
H89, a specific inhibitor of PKA (Chijiwa et al., 1990), was used to confirm the role of PKA in the elevation of GK activity and in the reduction of the level of intracellular glucose. Serum-starved HSCs were pretreated with H89 at indicated concentrations, or with curcumin at 20 µM, in serum-free DMEM for 1 h prior to the stimulation with leptin (100 ng·mL−1) for additional 30 min. GK activity (Figure 4B) and intracellular glucose levels (Figure 4C) were measured. Compared with the untreated control, the PKA inhibitor H89, like curcumin, concentration-dependently increased GK activities and reduced levels of intracellular glucose. Taken together, our results indicated that curcumin inhibited PKA activity, which facilitated the increase in the activity of GK and the reduction of intracellular glucose in cultured HSCs.
Curcumin activated AMPK activity, which resulted in the increase in the activity of GK and the reduction in the level of intracellular glucose in cultured HSCs
Further experiments were conducted to explore additional mechanisms by which curcumin increased the activity of GK and reduced the level of intracellular glucose in HSCs. We presumed that leptin inhibited AMPK activity in HSCs, which was prevented by curcumin, and that the curcumin effect facilitated the increase in the activity of GK and the conversion of glucose to G-6-P, leading to the reduction in the level of intracellular glucose. To test the presumptions, serum-starved HSCs were treated with leptin at different concentrations, or pretreated with curcumin at 0–30 µM for 1 h prior to the stimulation with leptin (100 ng·mL−1), in serum-free media for additional 30 min. As shown by Western blotting analyses (Figure 5), leptin concentration-dependently reduced the levels of phosphorylated AMPK (p-AMPK) (Figure 5A), while curcumin blocked the inhibitory effect of leptin (100 ng·mL−1) and elevated p-AMPK in a concentration-dependent manner (Figure 5B).
Figure 5.
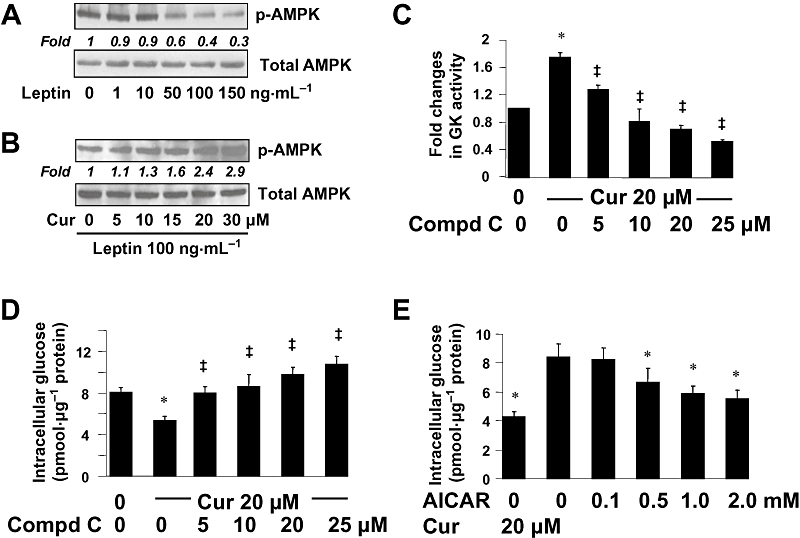
Curcumin activated AMPK activity, which resulted in the increase in the activity of GK and in the reduction in the level of intracellular glucose in cultured HSCs. (A,B) Serum-starved HSCs were pretreated with or without curcumin (0–30 µM) for 1 h prior to the stimulation with leptin at indicated concentrations in serum-free media for additional 30 min. Levels of p-AMPK were evaluated by Western blotting analyses. Total AMPK was used as an invariant control for equal loading. Representative blots are presented from three independent experiments. Italic numbers beneath blots were fold changes in the densities of the bands compared to the control without treatment in the blot (n = 3), after normalization with total AMPK. (A) Cells were stimulated with leptin only. (B) Cells were pretreated with curcumin prior to the stimulation with leptin (100 ng·mL−1). (C,D) Serum-starved HSCs were pretreated with or without curcumin at 20 µM for 1 h prior to the exposure to the AMPK inhibitor compd C at various concentrations in serum-depleted media for additional 30 min. (C) Analyses of GK activities. Values were expressed as fold changes compared with the untreated control (means ± SD) (n = 3). *P < 0.05 versus the untreated control; ‡P < 0.05 versus cells treated with curcumin alone. (D) Analyses of levels of intracellular glucose. Values were expressed as pmol glucose·µg−1 protein (means ± SD) (n = 3). *P < 0.05 versus the untreated control; ‡P < 0.05 versus cells treated with curcumin alone. (E) HSCs were treated with the AMPK activator AICAR at various concentrations, or with curcumin at 20 µM, for 1.5 h. Levels of intracellular glucose were determined. Values were expressed as pmol glucose·µg−1 protein (means ± SD) (n = 3). *P < 0.05 versus the untreated control (the second column).
To determine the impact of AMPK on regulating the level of intracellular glucose, serum-starved HSCs were pretreated with or without curcumin at 20 µM for 1 h prior to the exposure to the specific AMPK inhibitor compd C at various concentrations in serum-depleted media for additional 30 min. GK activity assays (Figure 5C) demonstrated that, compared with the untreated control, curcumin significantly increased, as expected, the activity of GK. The AMPK inhibitor compd C concentration-dependently blocked the stimulatory role of curcumin and reduced the activity of GK, confirming that AMPK positively regulated GK activity in HSCs. Further analyses of intracellular glucose in HSCs after this treatment (Figure 5D) revealed that compared with the untreated control, curcumin reduced, as expected, the level of intracellular glucose. Inhibition of AMPK activity by compd C concentration-dependently diminished the effects of curcumin and, consequently, increased intracellular glucose (Figure 5D).
To confirm the role of AMPK in the reduction of the level of intracellular glucose, HSCs were treated with the selective AMPK activator AICAR at various concentrations, or with curcumin at 20 µM, in serum-depleted media for 1.5 h. As illustrated in Figure 5E, activation of AMPK by AICAR, mimicking the effects of curcumin, concentration-dependently reduced the level of intracellular glucose in cultured HSCs. Taken together, these results indicated that in addition to the inhibition of PKA, curcumin also activated AMPK activity, both of which facilitated the increase of GK activity and the reduction in the level of intracellular glucose in cultured HSCs.
The roles of curcumin in altering the levels of intracellular glucose and G-6-P in HSCs in vitro were dependent on leptin receptors
To clarify whether the roles of curcumin in altering the levels of intracellular glucose and G-6-P were dependent on leptin receptors, additional experiments were conducted using HSCs from natural leptin receptor-deficient Zucker (fa/fa) rats and HSCs from lean wild-type Zucker (Fa/Fa) rats. Serum-starved HSCs were pretreated with or without curcumin (20 µM) for 1 h prior to the addition of leptin (100 ng·mL−1) in serum-free DMEM for additional 30 min. Levels of intracellular glucose and G-6-P were analysed. As shown in Figure 6, compared with the untreated control, leptin significantly increased the level of glucose and reduced the content of G-6-P in lean Zucker HSCs (Fa/Fa). On the other hand, compared with the untreated control, curcumin alone significantly lowered intracellular glucose (Figure 6A) and increased intracellular G-6-P (Figure 6B) in the same cells. Further, curcumin blocked the effects of leptin in reducing intracellular glucose (Figure 6A) and increasing the content of intracellular G-6-P (Figure 6B) in Fa/Fa HSCs. However, in leptin receptor-deficient HSCs from fa/fa rats, curcumin was no longer able to affect intracellular glucose or G-6-P, irrespective of leptin treatment. These results indicated that the ability of curcumin to alter intracellular glucose and G-6-P in HSCs in vitro was dependent on the presence of leptin receptors.
Figure 6.
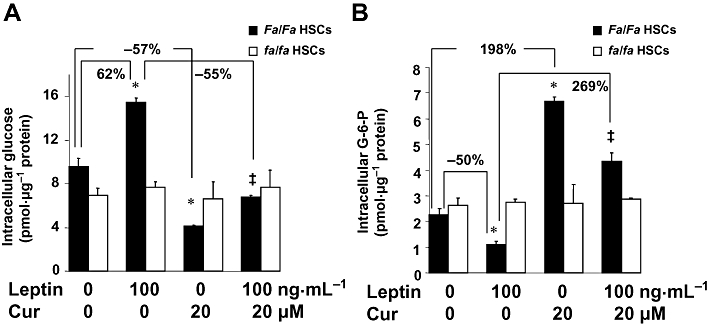
The roles of curcumin in altering the levels of intracellular glucose and G-6-P in HSCs in vitro were dependent on leptin receptor. Serum-starved HSCs from natural leptin receptor-deficient Zucker (fa/fa) rats and HSCs from lean wild-type Zucker (Fa/Fa) rats were pretreated with or without curcumin (20 µM) for 1 h prior to the addition of leptin (100 ng·mL−1) for additional 30 min in serum-free DMEM. Levels of intracellular glucose and G-6-P were analysed. Values were expressed as pmol glucose or G-6-P·µg−1 protein, and presented as means ± SD (n = 3). *P < 0.05 versus the untreated control cells; ‡P < 0.05 versus cells treated with leptin alone. (A) Analyses of levels of intracellular glucose. (B) Analyses of levels of intracellular G-6-P.
Discussion
Elevated levels of plasma leptin, or hyperleptinemia, correlate with hyperphagia, insulin resistance and other symptoms of the metabolic syndrome, which are often associated with NASH and hepatic fibrogenesis (Friedman, 2004; Yokaichiya et al., 2008). Earlier studies have shown that abnormal levels of leptin stimulate HSC activation (Saxena et al., 2002; Aleffi et al., 2005; Choudhury et al., 2006). We recently showed that curcumin blocked the effect of leptin on the activation of HSCs by interrupting leptin signalling (Tang et al., 2009). However, the underlying mechanisms remain largely undefined. In the current report, we demonstrated that curcumin prevented leptin from raising intracellular glucose in activated HSCs in vitro, leading to inhibition of HSC activation. Curcumin interrupted the leptin-activated IRS/PI3K/AKT signalling pathway, leading to inhibition of the membrane translocation of GLUT4. In addition, curcumin stimulated GK activity by inhibiting PKA activity and activating AMPK activity, resulting in increased conversion of glucose to G-6-P.
Higher or abnormal levels of intracellular glucose could be a deleterious factor and impair cellular functions (Jellinger, 2007), including stimulating expression of connective tissue growth factor, one of the downstream effectors of TGF-β1, in HSCs (Paradis et al., 2001), and inducing oxidative stress (Sugimoto et al., 2005). The level of intracellular glucose in cells is mainly determined by the balance between glucose supplementation, such as glucose uptake, and glucose consumption, including glucose conversion to G-6-P. In the present report, we showed that curcumin inhibited leptin-induced translocation of GLUT4 from the cytosol to the cell surface through interrupting the IRS1&2/PI3K/AKT signalling pathway, most likely leading to a reduction in glucose uptake and, hence, in the level of intracellular glucose in activated HSCs in vitro. Our results were consistent with other studies. Thus, activation of the JAK2/IRS/PI3K/AKT signalling pathway was necessary for leptin and insulin to stimulate GLUT4 membrane translocation and glucose uptake in cells (Berti et al., 1997; Niswender et al., 2001; Duan et al., 2004). We further demonstrated in this report that curcumin blocked the effects of leptin on inhibiting GK activity by reducing PKA activity and activating AMPK, leading to increased conversion of glucose to G-6-P and to a reduction in intracellular glucose. Our results showed the opposite effects of PKA and AMPK on the activity of GK and on the level of intracellular glucose in activated HSCs in vitro. Exogenous G-6-P inhibited the production of α-SMA and αI(I)procollagen in HSCs. Extracellular G-6-P might be transported into cells, as has been shown earlier (Kristiansen et al., 1994). The roles of curcumin were mimicked and confirmed by the inhibition of PKA by its inhibitor, H89, and by the activation of AMPK by its activator, AICAR.
Other studies have indicated that H89 at 10 µM could significantly inhibit PKA activity, while it has no effect on PKC activity (Wieprecht et al., 1994). However, it was noticed that H89 at concentrations up to 30 µM was not as effective as curcumin at 20 µM in increasing the activity of GK and in reducing the level of intracellular glucose (Figure 4B,C). This observation suggested that besides the inhibition of PKA, there might be additional mechanisms by which curcumin increased the activity of GK and consequently decreased intracellular glucose in HSCs. Curcumin might possess broader spectrum of capacities on altering activities of protein kinases. Curcumin was shown not only to inhibit PKA, but also to activate AMPK in HSCs. Our results are consistent with earlier observations. Curcumin inhibited PKA activity (Hasmeda and Polya, 1996) and activated AMPK activity in 3T3-L1 adipocytes (Lee et al., 2009). The phosphorylation of GK by PKA resulted in a reduction in the activity of GK (Ekman and Nilsson, 1988). The activity of GK was also regulated by AMPK by phosphorylation of glucokinase regulatory protein, which resulted in the dissociation and translocation of GK from the nucleus into the cytoplasm, leading to the activation of GK (Mukhtar et al., 2008).
Results in this report indicated that the roles of curcumin in altering the levels of intracellular glucose and G-6-P in HSCs in vitro were dependent on leptin receptor. The result was supported by our finding that curcumin rapidly reduced the level of phosphorylated Ob-R induced by leptin in HSCs in vitro (Tang et al., 2009). In addition, curcumin showed an acute action in HSCs and rapidly reduced the phosphorylation levels of IRS1/2, PI3K and AKT, leading to reduction in GLUT4 membrane translocation and altered phosphorylation levels of PKA and AMPK. The mechanisms underlying this aspect of curcumin action remain largely undefined. Accumulating evidence has indicated the role of calcium in regulating activities of protein tyrosine kinases and phosphatases (Nilsson et al., 2002; Tani and Matsumoto, 2004; Gerthoffer, 2005), but the effect of curcumin on the level of Ca2+ is still controversial. Curcumin induced a marked depletion of Ca2+ in Caki cells (Bae et al., 2003). We recently observed that curcumin dramatically and instantly reduced the level of cellular Ca2+ in cultured HSCs (unpublished observations). In contrast, other reports showed that curcumin elevated the level of Ca2+ (Su et al., 2006), and inhibited Ca2+ mobilization (Wang et al., 2006) in colon cancer cells. Curcumin inhibited overall ATPase activity and Ca2+ transport (Sumbilla et al., 2002). It is possible that curcumin could show differing effects on the level of Ca2+, depending on cell types. Further experiments are necessary to elucidate the mechanisms by which curcumin acts as a protein tyrosine kinase inhibitor and/or a phosphatase activator in regulating the phosphorylation levels of the IRS/PI3K/AKT pathway, PKA and AMPK in HSCs.
A critical question was raised during our initial experiments as to whether curcumin would disturb glucose uptake in hepatocytes and/or muscle cells, which would exacerbate hyperglycemia and diabetes. To answer this question, we evaluated the impact of curcumin on intracellular glucose in IHHs. Results in Figure 1 clearly indicated that curcumin had no effects on the level of intracellular glucose in these hepatocytes, and suggested that curcumin, at these concentrations, would not exacerbate hyperglycemia and diabetes. Our results were in agreement with reports from other groups that curcumin had anti-hyperglycemic and anti-diabetic effects in T2DM rat models (Pari and Murugan, 2005; Seo et al., 2008), and in diabetic human subjects (Srinivasan, 1972). In the present context, it should be noted that curcumin exerted different effects on regulating expression of genes depending on cell types (Syng-Ai et al., 2004). Curcumin induced gene expression of LDL receptors (LDLRs) in hepatoma cell line HepG2 (Peschel et al., 2007). On the other hand, curcumin suppressed LDLR gene expression in cultured HSCs, which attenuated the stimulatory impact of LDL on the activation of HSCs (Kang and Chen, 2009). These observations collectively suggested that curcumin could exert a range of effects on regulating gene expression, on controlling the level of intracellular Ca2+ and intracellular glucose, all dependent on the cell types involved.
Based on our observations, a simplified model is proposed to explain the inhibitory effects of curcumin on leptin-induced HSC activation (Figure 7). An increase in the level of leptin stimulates the phosphorylation of leptin receptor and activates the downstream IRS/PI3K/AKT signalling pathway in HSCs, which induces the GLUT4 translocation from the cytosol to the plasma membrane, leading to increased glucose uptake. On the other hand, leptin inhibits AMPK activity and induces the activation of PKA, which suppresses GK activity, leading to the prevention of the conversion from intracellular glucose to G-6-P. These effects collectively increase the level of intracellular glucose and stimulate HSC activation. Curcumin prevents leptin from raising intracellular glucose by reducing phosphorylation of leptin receptors, interrupting the IRS/PI3K/AKT signalling pathway, inhibiting GLUT4 membrane translocation and stimulating glucose conversion to G-6-P by activating GK, all effects resulting in the inhibition of leptin-induced HSC activation. We would point out that this model does not exclude any other mechanisms by which curcumin might inhibit leptin-induced HSC activation. Our results in this report shed novel insights into mechanisms of curcumin in inhibiting leptin-induced HSC activation, and provide a therapeutic candidate for the treatment of obesity- and NASH-associated hepatic fibrogenesis.
Figure 7.
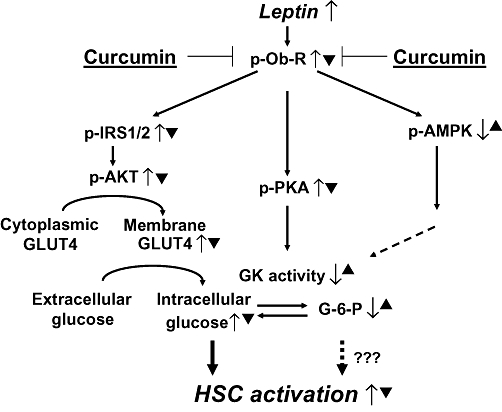
A simplified model for explaining the roles of curcumin in eliminating the stimulatory effects of leptin on the activation of HSCs, focusing on the level of intracellular glucose. ‘↑’ indicates the actions of leptin, while ‘▴’ represents the effects of curcumin.
Acknowledgments
The work was supported by grant RO1 DK 047995 from NIH/NIDDK to A.C. We are grateful to Harlan Laboratories, Inc. for generously providing Zucker rats.
Glossary
Abbreviations
- AICAR
5-aminoimidazole-4-carboxamide 1-β-d-ribofuranoside
- AMPK
AMP-activated protein kinase
- DAPI
4′-6-diamidino-2-phenylindole
- DMEM
Dulbecco's modified Eagle's medium
- DMSO
dimethyl sulphoxide
- FBS
fetal bovine serum
- G-6-P
glucose-6-phosphate
- GK
glucokinase
- GLUT4
glucose transporter-4
- HSCs
hepatic stellate cells
- IRS
insulin receptor substrate
- JAK
Janus kinase
- MAPK
mitogen-activated protein kinase
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- Ob-R
leptin receptor
- PI3K
phosphatidylinositol 3-kinase
- α-SMA
α-smooth muscle actin
Conflict of interest
None to declare.
References
- Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- Asano T, Ogihara T, Katagiri H, Sakoda H, Ono H, Fujishiro M, et al. Glucose transporter and Na+/glucose cotransporter as molecular targets of anti-diabetic drugs. Curr Med Chem. 2004;11:2717–2724. doi: 10.2174/0929867043364360. [DOI] [PubMed] [Google Scholar]
- Bae JH, Park JW, Kwon TK. Ruthenium red, inhibitor of mitochondrial Ca2+ uniporter, inhibits curcumin-induced apoptosis via the prevention of intracellular Ca2+ depletion and cytochrome c release. Biochem Biophys Res Commun. 2003;303:1073–1079. doi: 10.1016/s0006-291x(03)00479-0. [DOI] [PubMed] [Google Scholar]
- Benomar Y, Naour N, Aubourg A, Bailleux V, Gertler A, Djiane J, et al. Insulin and leptin induce Glut4 plasma membrane translocation and glucose uptake in a human neuronal cell line by a phosphatidylinositol 3-kinase-dependent mechanism. Endocrinology. 2006;147:2550–2556. doi: 10.1210/en.2005-1464. [DOI] [PubMed] [Google Scholar]
- Berti L, Kellerer M, Capp E, Haring HU. Leptin stimulates glucose transport and glycogen synthesis in C2C12 myotubes: evidence for a P13-kinase mediated effect. Diabetologia. 1997;40:606–609. doi: 10.1007/s001250050722. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, et al. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- Choudhury J, Mirshahi F, Murthy KS, Yager DR, Sanyal AJ. Physiologic concentrations of leptin increase collagen production by non-immortalized human hepatic stellate cells. Metabolism. 2006;55:1317–1322. doi: 10.1016/j.metabol.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40(Suppl 1):S5–S10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- Duan C, Li M, Rui L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem. 2004;279:43684–43691. doi: 10.1074/jbc.M408495200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekman P, Nilsson E. Phosphorylation of glucokinase from rat liver in vitro by protein kinase A with a concomitant decrease of its activity. Arch Biochem Biophys. 1988;261:275–282. doi: 10.1016/0003-9861(88)90342-6. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Modern science versus the stigma of obesity. Nat Med. 2004;10:563–569. doi: 10.1038/nm0604-563. [DOI] [PubMed] [Google Scholar]
- Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruhbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Zheng S, Lin J, Ryerse J, Chen A. Curcumin protects the rat liver from CCl4-caused injury and fibrogenesis by attenuating oxidative stress and suppressing inflammation. Mol Pharmacol. 2008;73:399–409. doi: 10.1124/mol.107.039818. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT. Signal-transduction pathways that regulate visceral smooth muscle function. III. Coupling of muscarinic receptors to signaling kinases and effector proteins in gastrointestinal smooth muscles. Am J Physiol Gastrointest Liver Physiol. 2005;288:G849–G853. doi: 10.1152/ajpgi.00530.2004. [DOI] [PubMed] [Google Scholar]
- Grimsby J, Berthel SJ, Sarabu R. Glucokinase activators for the potential treatment of type 2 diabetes. Curr Top Med Chem. 2008;8:1524–1532. doi: 10.2174/156802608786413483. [DOI] [PubMed] [Google Scholar]
- Hasmeda M, Polya GM. Inhibition of cyclic AMP-dependent protein kinase by curcumin. Phytochemistry. 1996;42:599–605. doi: 10.1016/0031-9422(96)00091-x. [DOI] [PubMed] [Google Scholar]
- Hayes MR, Skibicka KP, Bence KK, Grill HJ. Dorsal hindbrain 5′-adenosine monophosphate-activated protein kinase as an intracellular mediator of energy balance. Endocrinology. 2009;150:2175–2182. doi: 10.1210/en.2008-1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger PS. Metabolic consequences of hyperglycemia and insulin resistance. Clin Cornerstone. 2007;8(Suppl 7):S30–S42. doi: 10.1016/s1098-3597(07)80019-6. [DOI] [PubMed] [Google Scholar]
- Joe B, Vijaykumar M, Lokesh BR. Biological properties of curcumin – cellular and molecular mechanisms of action. Crit Rev Food Sci Nutr. 2004;44:97–111. doi: 10.1080/10408690490424702. [DOI] [PubMed] [Google Scholar]
- Kang Q, Chen A. Curcumin suppresses expression of low-density lipoprotein (LDL) receptor, leading to the inhibition of LDL-induced activation of hepatic stellate cells. Br J Pharmacol. 2009;157:1354–1367. doi: 10.1111/j.1476-5381.2009.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Koning W, van Dam K. A method for the determination of changes of glycolytic metabolites in yeast on a subsecond time scale using extraction at neutral pH. Anal Biochem. 1992;204:118–123. doi: 10.1016/0003-2697(92)90149-2. [DOI] [PubMed] [Google Scholar]
- Kristiansen S, Wojtaszewski JF, Juel C, Richter EA. Effect of glucose-6-phosphate and pH on glucose transport in skeletal muscle plasma membrane giant vesicles. Acta Physiol Scand. 1994;150:227–233. doi: 10.1111/j.1748-1716.1994.tb09680.x. [DOI] [PubMed] [Google Scholar]
- Leclercq IA, Farrell GC, Schriemer R, Robertson GR. Leptin is essential for the hepatic fibrogenic response to chronic liver injury. J Hepatol. 2002;37:206–213. doi: 10.1016/s0168-8278(02)00102-2. [DOI] [PubMed] [Google Scholar]
- Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, et al. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996;379:632–635. doi: 10.1038/379632a0. [DOI] [PubMed] [Google Scholar]
- Lee YK, Lee WS, Hwang JT, Kwon DY, Surh YJ, Park OJ. Curcumin exerts antidifferentiation effect through AMPKalpha-PPAR-gamma in 3T3-L1 adipocytes and antiproliferatory effect through AMPKalpha–COX-2 in cancer cells. J Agric Food Chem. 2009;57:305–310. doi: 10.1021/jf802737z. [DOI] [PubMed] [Google Scholar]
- Leney SE, Tavare JM. The molecular basis of insulin-stimulated glucose uptake: signalling, trafficking and potential drug targets. J Endocrinol. 2009;203:1–18. doi: 10.1677/JOE-09-0037. [DOI] [PubMed] [Google Scholar]
- Leturque A, Brot-Laroche E, Le Gall M, Stolarczyk E, Tobin V. The role of GLUT2 in dietary sugar handling. J Physiol Biochem. 2005;61:529–537. doi: 10.1007/BF03168378. [DOI] [PubMed] [Google Scholar]
- Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407–1433. doi: 10.1038/sj.ijo.0802142. [DOI] [PubMed] [Google Scholar]
- Mehebik N, Jaubert AM, Sabourault D, Giudicelli Y, Ribiere C. Leptin-induced nitric oxide production in white adipocytes is mediated through PKA and MAP kinase activation. Am J Physiol Cell Physiol. 2005;289:C379–C387. doi: 10.1152/ajpcell.00320.2004. [DOI] [PubMed] [Google Scholar]
- Mukhtar MH, Payne VA, Arden C, Harbottle A, Khan S, Lange AJ, et al. Inhibition of glucokinase translocation by AMP-activated protein kinase is associated with phosphorylation of both GKRP and 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Am J Physiol Regul Integr Comp Physiol. 2008;294:R766–R774. doi: 10.1152/ajpregu.00593.2007. [DOI] [PubMed] [Google Scholar]
- Nilsson BO, Gomez MF, Sward K, Hellstrand P. Regulation of Ca2+ channel and phosphatase activities by polyamines in intestinal and vascular smooth muscle – implications for cellular growth and contractility. Acta Physiol Scand. 2002;176:33–41. doi: 10.1046/j.1365-201X.2002.01013.x. [DOI] [PubMed] [Google Scholar]
- Nishiumi S, Ashida H. Rapid preparation of a plasma membrane fraction from adipocytes and muscle cells: application to detection of translocated glucose transporter 4 on the plasma membrane. Biosci Biotechnol Biochem. 2007;71:2343–2346. doi: 10.1271/bbb.70342. [DOI] [PubMed] [Google Scholar]
- Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. doi: 10.1038/35101657. [DOI] [PubMed] [Google Scholar]
- O'Connell MA, Rushworth SA. Curcumin: potential for hepatic fibrosis therapy? Br J Pharmacol. 2008;153:403–405. doi: 10.1038/sj.bjp.0707580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis V, Perlemuter G, Bonvoust F, Dargere D, Parfait B, Vidaud M, et al. High glucose and hyperinsulinemia stimulate connective tissue growth factor expression: a potential mechanism involved in progression to fibrosis in nonalcoholic steatohepatitis. Hepatology. 2001;34:738–744. doi: 10.1053/jhep.2001.28055. [DOI] [PubMed] [Google Scholar]
- Pari L, Murugan P. Effect of tetrahydrocurcumin on blood glucose, plasma insulin and hepatic key enzymes in streptozotocin induced diabetic rats. J Basic Clin Physiol Pharmacol. 2005;16:257–274. doi: 10.1515/jbcpp.2005.16.4.257. [DOI] [PubMed] [Google Scholar]
- Perez-Echarri N, Perez-Matute P, Marcos-Gomez B, Martinez JA, Moreno-Aliaga MJ. Effects of eicosapentaenoic acid ethyl ester on visfatin and apelin in lean and overweight (cafeteria diet-fed) rats. Br J Nutr. 2009;101:1059–1067. doi: 10.1017/S0007114508048307. [DOI] [PubMed] [Google Scholar]
- Peschel D, Koerting R, Nass N. Curcumin induces changes in expression of genes involved in cholesterol homeostasis. J Nutr Biochem. 2007;18:113–119. doi: 10.1016/j.jnutbio.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Ray RB, Meyer K, Ray R. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology. 2000;271:197–204. doi: 10.1006/viro.2000.0295. [DOI] [PubMed] [Google Scholar]
- Saxena NK, Ikeda K, Rockey DC, Friedman SL, Anania FA. Leptin in hepatic fibrosis: evidence for increased collagen production in stellate cells and lean littermates of ob/ob mice. Hepatology. 2002;35:762–771. doi: 10.1053/jhep.2002.32029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo KI, Choi MS, Jung UJ, Kim HJ, Yeo J, Jeon SM, et al. Effect of curcumin supplementation on blood glucose, plasma insulin, and glucose homeostasis related enzyme activities in diabetic db/db mice. Mol Nutr Food Res. 2008;52:995–1004. doi: 10.1002/mnfr.200700184. [DOI] [PubMed] [Google Scholar]
- Singh S, Khar A. Biological effects of curcumin and its role in cancer chemoprevention and therapy. Anticancer Agents Med Chem. 2006;6:259–270. doi: 10.2174/187152006776930918. [DOI] [PubMed] [Google Scholar]
- Srinivasan M. Effect of curcumin on blood sugar as seen in a diabetic subject. Indian J Med Sci. 1972;26:269–270. [PubMed] [Google Scholar]
- Stefanovic A, Kotur-Stevuljevic J, Spasic S, Bogavac-Stanojevic N, Bujisic N. The influence of obesity on the oxidative stress status and the concentration of leptin in type 2 diabetes mellitus patients. Diabetes Res Clin Pract. 2008;79:156–163. doi: 10.1016/j.diabres.2007.07.019. [DOI] [PubMed] [Google Scholar]
- Su CC, Lin JG, Li TM, Chung JG, Yang JS, Ip SW, et al. Curcumin-induced apoptosis of human colon cancer colo 205 cells through the production of ROS, Ca2+ and the activation of caspase-3. Anticancer Res. 2006;26:4379–4389. [PubMed] [Google Scholar]
- Sugimoto R, Enjoji M, Kohjima M, Tsuruta S, Fukushima M, Iwao M, et al. High glucose stimulates hepatic stellate cells to proliferate and to produce collagen through free radical production and activation of mitogen-activated protein kinase. Liver Int. 2005;25:1018–1026. doi: 10.1111/j.1478-3231.2005.01130.x. [DOI] [PubMed] [Google Scholar]
- Sumbilla C, Lewis D, Hammerschmidt T, Inesi G. The slippage of the Ca2+ pump and its control by anions and curcumin in skeletal and cardiac sarcoplasmic reticulum. J Biol Chem. 2002;277:13900–13906. doi: 10.1074/jbc.M111155200. [DOI] [PubMed] [Google Scholar]
- Syng-Ai C, Kumari AL, Khar A. Effect of curcumin on normal and tumor cells: role of glutathione and bcl-2. Mol Cancer Ther. 2004;3:1101–1108. [PubMed] [Google Scholar]
- Tang Y, Zheng S, Chen A. Curcumin eliminates leptin's effects on hepatic stellate cell activation via interrupting leptin signaling. Endocrinology. 2009;150:3011–3020. doi: 10.1210/en.2008-1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tani E, Matsumoto T. Continuous elevation of intracellular Ca2+ is essential for the development of cerebral vasospasm. Curr Vasc Pharmacol. 2004;2:13–21. doi: 10.2174/1570161043476492. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Q, Ives KL, Evers BM. Curcumin inhibits neurotensin-mediated interleukin-8 production and migration of HCT116 human colon cancer cells. Clin Cancer Res. 2006;12:5346–5355. doi: 10.1158/1078-0432.CCR-06-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Yaginuma R, Ikejima K, Miyazaki A. Liver diseases and metabolic syndrome. J Gastroenterol. 2008;43:509–518. doi: 10.1007/s00535-008-2193-6. [DOI] [PubMed] [Google Scholar]
- Wieprecht M, Wieder T, Geilen CC. N-[2-bromocinnamyl(amino)ethyl]-5-isoquinolinesulphonamide (H-89) inhibits incorporation of choline into phosphatidylcholine via inhibition of choline kinase and has no effect on the phosphorylation of CTP : phosphocholine cytidylyltransferase. Biochem J. 1994;297(Pt 1):241–247. doi: 10.1042/bj2970241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Fu Y, Chen A. Activation of peroxisome proliferator-activated receptor-gamma contributes to the inhibitory effects of curcumin on rat hepatic stellate cell growth. Am J Physiol Gastrointest Liver Physiol. 2003;285:G20–G30. doi: 10.1152/ajpgi.00474.2002. [DOI] [PubMed] [Google Scholar]
- Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;276:25096–25100. doi: 10.1074/jbc.M007383200. [DOI] [PubMed] [Google Scholar]
- Yokaichiya DK, Galembeck E, Torres BB, Da Silva JA, de Araujo DR. Insulin and leptin relations in obesity: a multimedia approach. Adv Physiol Educ. 2008;32:231–236. doi: 10.1152/advan.00014.2007. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007;8:113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao AZ, Shinohara MM, Huang D, Shimizu M, Eldar-Finkelman H, Krebs EG, et al. Leptin induces insulin-like signaling that antagonizes cAMP elevation by glucagon in hepatocytes. J Biol Chem. 2000;275:11348–11354. doi: 10.1074/jbc.275.15.11348. [DOI] [PubMed] [Google Scholar]
- Zheng S, Chen A. Activation of PPARgamma is required for curcumin to induce apoptosis and to inhibit the expression of extracellular matrix genes in hepatic stellate cells in vitro. Biochem J. 2004;384:149–157. doi: 10.1042/BJ20040928. [DOI] [PMC free article] [PubMed] [Google Scholar]