

Abstract
BACKGROUND AND PURPOSE
The present work aimed to investigate whether and through which mechanisms selective α7 and α4β2 nicotinic receptor (nAChR) agonists stimulate endogenous glutamate (GLU) and aspartate (ASP) release in rat hippocampus.
EXPERIMENTAL APPROACH
Rat hippocampal synaptosomes were purified on Percoll gradients and superfused in vitro to study endogenous GLU and ASP release. The synaptosomes were superfused with selective α7 and α4β2 nAChR agonists and antagonists. The excitatory amino acid (EAA) content of the samples of superfusate was determined by HPLC after pre-column derivatization and separation on a chromatographic column coupled with fluorimetric detection.
KEY RESULTS
Choline (Ch), a selective α7 receptor agonist, elicited a significant release of both GLU and ASP which was blocked by the α7 receptor antagonist methyllycaconitine (MLA), but was unaltered by the α4β2 receptor antagonist dihydro-β-erythroidine (DHβE). The stimulant effect of Ch was strongly reduced in a Ca2+-free medium, was not inhibited by Cd2+ and tetrodotoxin (TTX), but was antagonized by dantrolene, xestospongin C and thapsigargin. 5-Iodo-A-85380 dihydrochloride (5IA85380), a selective α4β2 receptor agonist, elicited EAA release in a DHβE-sensitive, MLA-insensitive fashion. The 5IA85380-evoked release was dependent on extracellular Ca2+, blocked by Cd2+ and TTX, but unaffected by dantrolene.
CONCLUSIONS AND IMPLICATIONS
Our study shows for the first time that rat hippocampal synaptosomes possess α7 and α4β2 nAChR subtypes, which can enhance the release of endogenous GLU and ASP via two distinct mechanisms of action. These results extend our knowledge of the nicotinic modulation of excitatory synaptic transmission in the hippocampus.
Keywords: nicotinic receptor subtypes, endogenous glutamate, endogenous aspartate, synaptosomes, rat hippocampus, neurotransmitter release
Introduction
Several lines of evidence have pointed to the fundamental role of ACh in the attentional functions and capacities, as well as in cognitive processes at the hippocampal level (Clementi et al., 2000; Gold, 2003; McKay et al., 2007). Additionally, nicotinic acetylcholine receptor (nAChR) agonists are already being used in clinical trials for Alzheimer's disease and for other cognitive disorders. It has been suggested that nicotinic receptors play a pivotal role in memory and cognition possibly through a facilitating action on excitatory synaptic transmission (McGehee et al., 1995; Gray et al., 1996; Radcliffe et al., 1999). This is most likely to occur because nicotine can evoke the release of glutamate (GLU), as demonstrated both in vivo (Toth et al., 1992; 1993; Fedele et al., 1996; Toth, 1996) and in vitro, using [3H]-d-aspartate ([3H]-d-ASP) (Marchi et al., 2002; Wang et al., 2006; Dickinson et al., 2007). Moreover, chronically administered nicotine can also regulate glutamatergic neurotransmission functions by modulating the sensitivity of excitatory amino acid (EAA) receptors (Risso et al., 2004a,b; Parodi et al., 2006; Grilli et al., 2009).
It is well known that, in the CNS, nicotine can act on several nAChR subtypes, which differ not only in their sensitivity to agonists and antagonists, but also in their kinetics of activation and inactivation, and Ca2+ permeability. As for their functional diversity, recent evidence supports the hypothesis that different nAChR subtypes trigger GLU release in the rat prefrontal cortex and hippocampus, both in vivo and in vitro, via different mechanisms (Dickinson et al., 2007; 2008; Bancila et al., 2009; Konradsson-Geuken et al., 2009). However, the nicotinic modulation of EAA release may be more complex because, in different brain areas, the evoked overflow might be also Ca2+ independent and partially mediated by excitatory amino acid transporters (EAATs; Reid et al., 2000). As to the targets of the released amino acid in the CNS, GLU is known to activate all the ionotropic and metabotropic EAA receptors. However, the natural neurotransmitter ASP is known to mimic the action of GLU, but acts selectively on NMDA receptors with a low, if any, affinity for AMPA receptors (Patneau and Mayer, 1990; Curras and Dingledine, 1992; Fleck et al., 1993). Interestingly, ASP is co-localized with GLU in the same nerve terminals of hippocampal neurones accumulated into a common vesicular pool by different transporter (Fleck et al., 2001a,b;) and is released upon depolarization by Ca2+-dependent, Clostridium toxin-sensitive exocytosis (Gundersen et al., 1998; Fleck et al., 2001a,b; Bradford and Nadler, 2004; Wang and Nadler, 2007). However, its role in the physiology and pathology of excitatory neurotransmission is still unclear. As a matter of fact, in vivo microdialysis studies have shown that nicotine is able to increase ASP and GLU extracellular levels in the hippocampus, striatum and frontal cortex (Toth et al., 1993; Fedele et al., 1996; Toth, 1996). However, to the best of our knowledge, no studies have investigated and fully characterized the possible role of nAChR subtypes on endogenous ASP and GLU release from isolated nerve endings in the rat hippocampus.
In the present study, we have therefore evaluated the effects of different agonists selective for α7 and α4β2 nAChR subtypes (Bencherif et al., 1998; Alkondon et al., 1999; Mukhin et al., 2000; Mogg et al., 2004; Wishka et al., 2006) to comparatively assess whether, to what extent and through which mechanisms they stimulate endogenous GLU and ASP release from purified hippocampal synaptosomes in superfusion. Our results demonstrate that, in the rat hippocampus, nAChR of the α7 and α4β2 subtypes are present on EAA nerve endings, and stimulate the release of both endogenous EAAs through two different molecular pathways.
Methods
Animals and brain tissue preparation
Adult male Sprague–Dawley rats (200–250 g) were housed at constant temperature (22 ± 1°C) and relative humidity (50%) under a regular light–dark schedule (light 7–19 h). Food and water were freely available. The animals were killed by decapitation and the hippocampus rapidly removed at 0–4°C. The experimental procedures were approved by the Ethical Committee of the Pharmacology and Toxicology Section, Department of Experimental Medicine, in accordance with the European legislation (European Communities Council Directive of 24 November 1986, 86/609/ EEC). All efforts were made to minimize animal suffering and to use a minimum number of animals necessary to produce reliable results.
The drug/molecular target nomenclature reported in the text conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2009).
Release experiments
Purified synaptosomes were prepared on Percoll gradients (Sigma-Aldrich, St Louis, MO, USA) essentially according to Nakamura et al. (1993) with only minor modifications. Briefly, the tissue was homogenized in 6 volumes of 0.32 M sucrose, buffered at pH 7.4 with Tris–HCl, using a glass–Teflon tissue grinder (clearance 0.25 mm, 12 up–down strokes in about 1 min). The homogenate was centrifuged (5 min, 1000×g at 4°C) to remove nuclei and debris, and the supernatant was gently stratified on a discontinuous Percoll gradient (2, 6, 10 and 20% v/v in Tris-buffered sucrose) and centrifuged at 33 500×g for 5 min at 4°C. The layer between 10 and 20% Percoll (synaptosomal fraction) was collected, washed by centrifugation and resuspended in physiological HEPES-buffered medium having the following composition (mM): 128 NaCl, 2.4 KCl, 3.2 CaCl2, 1.2 KH2PO4, 1.2 MgSO4, 25 HEPES, pH 7.5, 10 glucose, pH 7.2–7.4. Synaptosomal protein content following purification was 10–15% of that in the supernatant stratified on the Percoll gradient.
The synaptosomal suspension was layered on microporous filters at the bottom of a set of parallel superfusion chambers maintained at 37°C (Raiteri and Raiteri, 2000; Superfusion System, Ugo Basile, Comerio, Varese, Italy). Synaptosomes were superfused at 1 mL·min−1 with standard physiological medium as previously described (Grilli et al., 2009). The system was first equilibrated during 36.5 min of superfusion; subsequently, four consecutive 90 s fractions of superfusate were collected. Synaptosomes were exposed to agonists for 90 s after the first fraction had been collected (t = 38 min), while antagonists were added 8 min before. Appropriate controls were always run in parallel. The evoked overflow was calculated by subtracting the corresponding basal release from each fraction collected, and was expressed as pmol·mg−1 of synaptosomal proteins. In some experiments, synaptosomes were depolarized with KCl (9 or 15 mM) for 90 s in the absence of any drug. In this case, the overflow was calculated by subtracting the corresponding basal release from the outflow induced by KCl. We have previously demonstrated that in our superfusion system, the neurotransmitter released is immediately removed and cannot be taken up by synaptosomes (for a review, see Raiteri and Raiteri, 2000)
Endogenous amino acid determination
Endogenous amino acid content was measured by HPLC analysis following pre-column derivatization with o-phthalaldehyde and resolution through a C18 reversed-phase chromatographic column (10 × 4.6 mm, 3 µm; Chrompack, Middleburg, The Netherlands) coupled with fluorometric detection (excitation wavelength 350 nm; emission wavelength 450 nm). Homoserine was used as internal standard. Buffers and gradient program were prepared and executed as follows: solvent A, 0.1 M sodium acetate (pH 5.8)/methanol, 80:20; solvent B, 0.1 M sodium acetate (pH 5.8)/methanol, 20:80; solvent C, sodium acetate (pH 6.0)/methanol, 80:20; gradient program, 100% C for 4 min from the initiation of the program; 90% A and 10% B in 1 min; 42% A and 58% B in 14 min; 100% B in 1 min; isocratic flow 2 min; 100% C in 3 min; flow rate 0.9 mL·min−1.
Statistical analysis
All data are expressed as pmol·mg−1 protein and represent mean ± SEM of the number of experiments reported in the figure legends. Multiple comparisons were performed with one-way anova followed by an appropriate (Dunnett and Tukey–Kramer) post hoc test. Differences were considered significant for P < 0.05, at least. The EC50 has been calculated according to non-linear curve fitting algorithm of Sigma Plot 8.0 (Jandel Scientific, San Rafael, CA, USA)
Chemicals
Percoll, choline iodide, CdCl2 and dantrolene (Sigma-Aldrich); xestospongin C (Inalco, Milan, Italy); 5-iodo-A-85380 dihydrochloride (5-iodo-A-85380), RJR2429 dihydrochloride, PHA543613 hydrochloride, dl-TBOA, dihydro-(-erythroidine hydrobromide, methyllycaconitine citrate, thapsigargin, N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea (PNU 120596) (Tocris Bioscience, Bristol, UK); tetrodotoxin (TTX) (Ascent Scientific, Princeton, NJ, USA)
Results
Figure 1 illustrates the time-course of the endogenous GLU and ASP release evoked by a 90 s pulse of choline (Ch) or 5IA85380. The Ch-evoked release of GLU and ASP showed a similar pattern, reaching a maximum corresponding to min 41 of superfusion and declined to basal levels at min 42.5 (Figure 1A,B). The 5IA85380-evoked release of ASP showed a similar pattern (Figure 1), while the peak value of the 5IA85380-evoked GLU release was reached at min 39.5 (Figure 1A).
Figure 1.
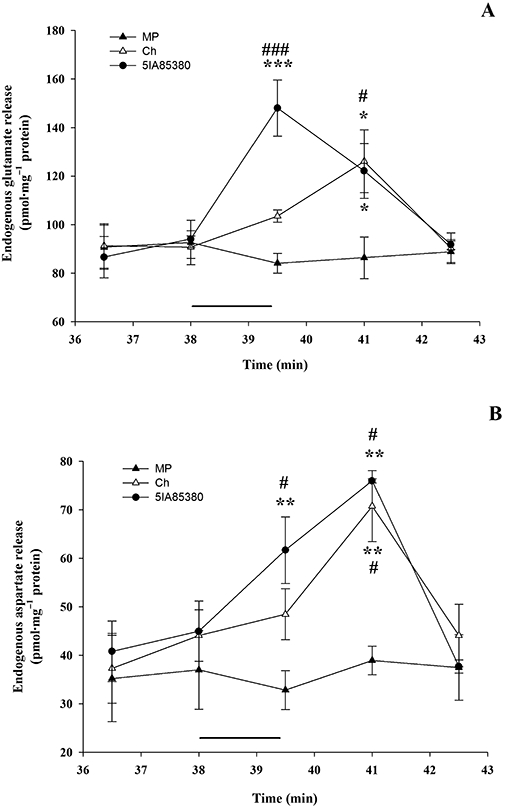
(A) Time-course of GLU release in response to stimulation with Ch (1 mM) and 5IA85380 (10 nM). Values are from two experiments and represent mean ± SEM of eight replicate superfusion chambers per condition (basal or evoked release). *P < 0.05, ***P < 0.001 versus time 36.5; #P < 0.05, ###P < 0.001 versus basal release; two-way anova followed by Tukey–Kramer post hoc test. (B) Time-course of ASP release in response to stimulation with Ch (1 mM; Δ) and 5IA85380 (10 nM). Values are from two experiments and represent mean ± SEM of eight replicate superfusion chambers per condition (basal or evoked release). **P < 0.01 versus time 36.5; #P < 0.05 versus basal release; two-way anova followed by Tukey–Kramer post hoc test.
Table 1 shows the effects of different nicotinic agonists, known to act selectively on the α7 (Ch and PHA543613) and on the α4β2 (RJR2429and 5IA85380) nAChR subtypes, on GLU and ASP overflow from rat hippocampal purified synaptosomes in superfusion. The GLU and ASP overflows elicited by 1 mM Ch and 100 µM PHA543613 closely resemble those elicited by the two α4β2 agonists 5IA85380 (10 nM) and RJR2429 (3 µM). When (PNU 120596) a positive allosteric modulator of α7 nAChRs was present with Ch in the superfusion fluid, the evoked EAA release was increased, although not significantly, compared to that elicited by Ch alone (Table 1). PNU 120596 did not modify, per se, the basal release of EAA (not shown). A comparison of the stimulant effects of these four agonists with classical depolarizing stimuli (KCl 9 and 15 mM) is also reported in Table 1. In general, the amounts of GLU and ASP released by all four nicotinic agonists were within the range of those released by the lower concentration of KCl (9 mM). The simultaneous presence of Ch and 5IA85380 in the superfusion fluid provoked GLU and ASP overflows that were significantly larger than those elicited by either of the nicotinic receptor agonists alone, suggesting an additive effect of these two agonists (Table 1).
Table 1.
Effects of selective nAChR subtypes on endogenous GLU and ASP overflow (pmol·mg−1 protein)
| Drugs | GLU | ASP |
|---|---|---|
| α7 nAChR subtype agonists | ||
| Ch (1 mM) | 50.49 ± 4.85### | 40.93 ± 4.71### |
| Ch (1 mM) + PNU120596 (10 µM) | 87.62 ± 14.48 | 57.95 ± 8.04 |
| PHA (100 µM) | 42.46 ± 12.02 | 30.14 ± 6.18 |
| α4β2 nAChR subtype agonists | ||
| 5IA85380 (10 nM) | 62.28 ± 6.12## | 51.66 ± 5.20## |
| RJR 2429 (3 µM) | 72.99 ± 16.83 | 42.92 ± 13.42 |
| 5IA85380 (10 nM) + Ch (1 mM) | 134.01 ± 32.04 | 96.20 ± 17.37 |
| KCl (9 mM) | 80.46 ± 19.61 | 36.61 ± 3.27 |
| KCl (15 mM) | 248.49 ± 44.10 | 95.24 ± 13.82 |
Data are means ± SEM of six experiments run in triplicate. For experimental details, see Methods.
P < 0.01
P < 0.001 versus 5IA85380 (10 nM) + Ch (1 mM).
One-way anova followed by Tukey–Kramer post hoc test.
When synaptosomes were exposed to various concentrations of Ch (0.01–1 mM) or 5IA85380 (0.1–10 nM), both nicotinic agonists were found to increase EAA overflow in a concentration-dependent manner. The potency of Ch in inducing GLU and ASP overflows was the same, the apparent EC50 values being 5.77 ± 0.59 and 11.65 ± 6.48 µM, respectively (Figure 2A). 5IA85380 was three orders of magnitude more active than Ch, but was also similarly potent at eliciting the overflow of GLU and ASP (EC50 2.81 ± 0.83 nM and 3.24 ± 1.13 nM, respectively; Figure 2B).
Figure 2.
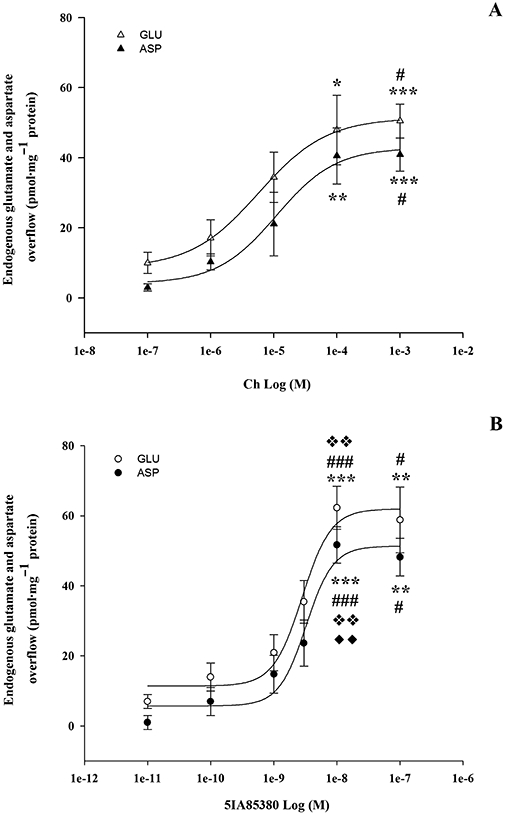
(A) Concentration-dependent effect of Ch on endogenous GLU and endogenous ASP overflow from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments for each concentration run in triplicate (three superfusion chambers for each experimental condition). *P < 0.05, **P < 0.01, ***P < 0.001 versus Ch (100 nM); #P < 0.05, versus Ch (1 µM); one-way anova followed by Tukey–Kramer post hoc test. (B) Concentration-dependent effect of 5IA85380 on endogenous GLU and endogenous ASP overflow from rat hippocampal synaptosomes. Data are mean ± SEM of three to six experiments for each concentration run in triplicate (three superfusion chambers for each experimental condition). **P < 0.01, ***P < 0.001 versus 5IA85380 (10 pM); #P < 0.05, ###P < 0.001 versus 5IA85380 (100 pM);  P < 0.01 versus 5IA85380 (1 nM); ♦♦P < 0.01 versus 5IA85380 (3 nM); one-way anova followed by Tukey–Kramer post hoc test.
P < 0.01 versus 5IA85380 (1 nM); ♦♦P < 0.01 versus 5IA85380 (3 nM); one-way anova followed by Tukey–Kramer post hoc test.
The Ch (1 mM)-evoked overflow of EAA was blocked by the selective α7 nAChR antagonist methyllycaconitine (MLA) (10 nM), but was unaffected by the selective α4β2 nAChR antagonist DHβE (1 µM), the specific voltage-dependent Na+ channel (VDSC) blocker TTX (1 µM) or the EEAT inhibitor dl-TBOA (10 µM) (Figure 3A). Figure 3B shows that the effect of 1 mM Ch on the overflow of both amino acids was totally dependent on external Ca2+, but was not altered by the non-specific voltage-dependent Ca2+ channel (VDCC) inhibitor Cd2+ (50 µM). Moreover, the Ch (1 mM)-evoked EAA overflow was markedly reduced by the ryanodine-sensitive receptor antagonist dantrolene (10 µM), by xestospongin C (1 µM) and by thapsigargin (10 µM) (Figure 3B).
Figure 3.
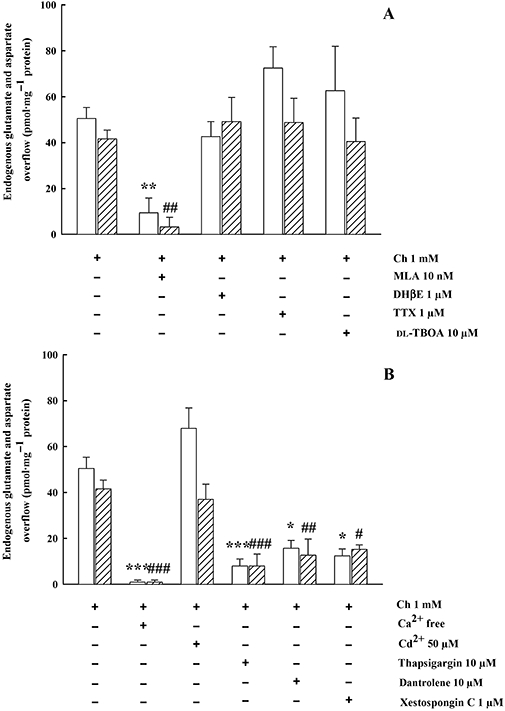
(A) Effect of MLA, DHβE, TTX and dl-TBOA on endogenous GLU (open columns) and ASP (hatched columns) overflow evoked by Ch from rat hippocampal synaptosomes. Synaptosomes were depolarized with Ch for 90 s at t = 38 min of superfusion. When appropriate, antagonists were introduced 8 min before depolarization. Data are mean ± SEM of three to six experiments run in triplicate. **P < 0.01 versus Ch evoked GLU overflow; ##P < 0.01 versus Ch evoked ASP overflow; one-way anova followed by Dunnett post hoc test. (B) Effect of Ca2+-free, Cd2+, thapsigargin, dantrolene and xestospongin C on endogenous GLU and ASP overflow evoked by Ch (1 mM) from rat hippocampal synaptosomes. When appropriate, Ca2+ was omitted 18 min before Ch. Data are mean ± SEM of three to six experiments run in triplicate. *P < 0.05, ***P < 0.001 versus Ch evoked GLU overflow; #P < 0.05, ##P < 0.01, ###P < 0.001 versus Ch evoked ASP overflow; one-way anova followed by Dunnett post hoc test.
Figure 4A shows that the 5IA85380 (10 nM)-evoked GLU/ASP overflow was blocked by DHβE (1 µM), unaffected by MLA (10 nM) or dl-TBOA (10 µM), but significantly inhibited in the presence of TTX (1 µM) (Figure 4A). The effect of 5IA85380 (10 nM) was almost totally dependent on external Ca2+ and blocked by Cd2+ (50 µM). The presence of dantrolene (10 µM) did not significantly attenuate the stimulant effect of 5IA85380 (10 nM) (Figure 4B). DHβE (1 µM), MLA (10 nM), TTX (1 µM), Cd2+ (50 µM), dantrolene (10 µM) xestospongin C (1 µM) and thapsigargin (10 µM) did not produce on their own any significant effect on basal EAA release.
Figure 4.
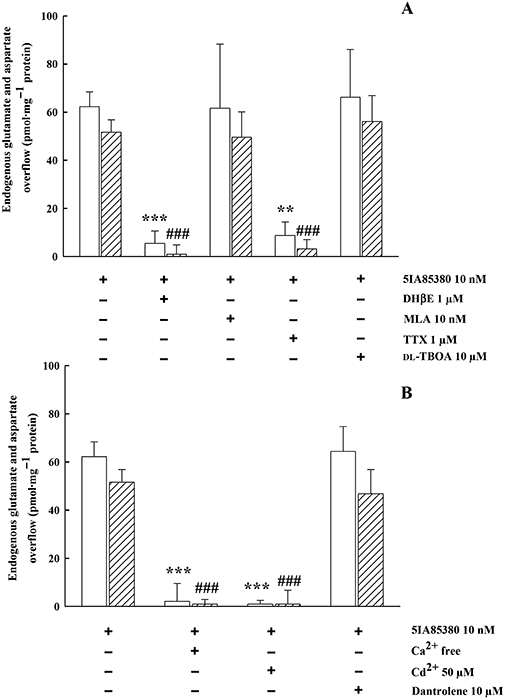
(A) Effect of DHβE, MLA, TTX and dl-TBOA on endogenous GLU (open columns) and ASP (hatched columns) overflow evoked by 5IA85380 from rat hippocampal synaptosomes. Synaptosomes were depolarized with 5IA85380 for 90 s at t = 38 min of superfusion. When appropriate, antagonists were introduced 8 min before depolarization. Data are mean ± SEM of three to six experiments run in triplicate. **P < 0.01, ***P < 0.001 versus 5IA85380 evoked GLU overflow; ###P < 0.001 versus 5IA85380 evoked ASP overflow; one-way anova followed by Dunnett post hoc test. (B) Effect of Ca2+-free, Cd2+ and dantrolene on endogenous GLU and ASP overflow evoked by 5IA85380 from rat hippocampal synaptosomes. When appropriate, antagonists were introduced 8 min before 5IA85380. Data are mean ± SEM of three to six experiments run in triplicate. ***P < 0.001 versus 5IA85380 evoked GLU overflow; ###P < 0.001 versus 5IA85380 evoked ASP overflow; one-way anova followed by Dunnett post hoc test.
Discussion
The present study is the first to demonstrate that nicotinic stimulation of endogenous GLU and ASP release occurs at the level of hippocampal nerve endings through the activation of different nAChR subtypes, which trigger distinct Ca2+-dependent molecular events.
Although it has been known since the 1960s that ASP has excitatory effects on neurones (Curtis et al., 1960), it took almost 40 years for this amino acid to be considered a classical neurotransmitter. A large body of evidence has indeed accumulated, indicating that ASP shares the role of excitatory neurotransmitter of the CNS with GLU. In fact, neurochemical, as well as immunohistochemical, studies have shown that ASP can be actively taken up into nerve terminals by EAATs, concentrated into synaptic vesicles and released upon depolarization of nerve terminals in a Ca2+-dependent, Clostridium toxin-sensitive fashion, thus fulfilling the criteria of canonical exocytosis (Gundersen and Storm-Mathisen, 2000; Fleck et al., 2001a,b; Raiteri et al., 2007; Cavallero et al., 2009 and references therein). Interestingly, electron microscopy studies have shown that ASP is co-localized with GLU in the same hippocampal nerve terminals (Gundersen et al., 1998), although it is not known whether the two amino acids are present in the same synaptic vesicles (Fleck et al., 2001a,b;) or whether they are stored in different vesicular pools (Bradford and Nadler, 2004).
However, it has been postulated that ASP release is only apparently mediated by Ca2+-dependent exocytosis, and the amino acid is released by carrier-mediated heteroexchange with GLU. That is, endogenous GLU, released by canonical exocytosis, would be taken up into nerve endings through EAATs and exchanged for intracellular cytosolic ASP. This does not occur in our case, because the release of ASP (and GLU) evoked by nicotinic agonists was unaffected by the EAAT blocker TBOA, clearly demonstrating the true exocytotic nature of the phenomenon.
The ability of nicotinic agonists to release both amino acids from hippocampal nerve endings therefore suggests that part of the in vivo effects produced by the drugs may be due not only to GLU, but also to ASP. It is well known that while GLU can activate all the EAA receptors, ASP selectively stimulates only NMDA receptors (Patneau and Mayer, 1990; Curras and Dingledine, 1992; Fleck et al., 1993). Nevertheless, because ASP is a less potent agonist than GLU at EAA ionotropic receptors, a physiological role of ASP in neurotransmission has for long been a matter of debate. However, an elegant study by Fleck et al. (1993) has demonstrated that this amino acid is able to mediate excitatory synaptic transmission in the hippocampal CA1 region. In fact, confirming previous studies (Szerb and O'Regan, 1987; Szerb, 1988), they showed that a low extracellular glucose concentration was able to dramatically decrease GLU release and to favour an increase in the release of ASP in hippocampal slices. These effects were paralleled by the almost complete abrogation of GLU-evoked AMPA-mediated pEPSPs (7% of controls) at Schaffer collateral–CA1 synapses, whereas NMDA-mediated pEPSPs were maintained at 27% of their initial amplitude, thus demonstrating that ASP acts, at least in this synaptic pathway, as an excitatory neurotransmitter (Fleck et al., 1993). The effects of nicotinic agonists add further support to the function of this amino acid as a neurotransmitter and to its possible role in the physiology of hippocampal excitatory neurotransmission. In the presence of normal glucose, it seems that activation of nAChRs increases ASP more than GLU release in comparison with high KCl depolarization. In fact, the GLU/ASP release ratio with KCl amounted to 2.1–2.6 (see Table 1), whereas the corresponding ratios with nicotinic receptor agonists were in the range of 1.2–1.7. Whether this preferential effect on ASP release has a functional impact on NMDA-mediated transmission in the hippocampus remains to be established.
Our results with the specific α7 receptor agonist Ch show that this endogenous compound is able to elicit EAA release from hippocampal synaptosomes. The Ch-evoked EAA release was blocked by MLA, but not by DHβE, thus confirming the involvement of an α7 nAChR. The effect was also dependent on calcium and largely sensitive to dantrolene, xestospongin C and thapsigargin, but was not altered in the presence of TTX or Cd2+, indicating that it is not due to classical membrane depolarization, triggered by VDSCs or the opening of VDCCs. It seems therefore that Ch can increase Ca2+ influx directly through the α7 nAChR channel that generates Ca2+-induced calcium release (CICR) from endoplasmic reticulum stores, which ultimately leads to the exocytosis of EAA. It is worth noting that in prefontal cortex synaptosomes and hippocampal mossy fibre terminals, activation of α7 nAChRs by nicotine has been shown to enhance [3H]-d-ASP and endogenous GLU release by a similar CICR-mediated mechanism (Dickinson et al., 2008; Bancila et al., 2009). Moreover, this type of nicotinic modulation seems to be of physiological relevance in regulating excitatory synaptic transmission in the hippocampus, as the α7 nAChR-mediated, CICR-induced release of GLU from mossy fibre terminals is capable of evoking high-frequency bursts of mEPSCs in post-synaptic CA3 pyramidal neurones (Sharma and Vijayaraghavan, 2003). Because our preparation of purified EAA synaptosomes lacks mossy fibre nerve terminals, it remains to be established whether this phenomenon also impacts on excitatory synaptic transmission in other terminal regions of the hippocampus (i.e. CA1 and dentate gyrus).
The effect of Ch on EAA release occurred at rather low concentrations (EC50s= 5–11 µM), a result that is at variance with the potency of this agonist (EC50= 1.6 mM) at stimulating GABA release from cultured hippocampal neurones through α7 nAChRs (Alkondon et al., 1997; 1999;). Apart from obvious differences between the two experimental models, it should be noted that diverse nicotinic responses can be evoked by Ch according to the concentrations used (Alkondon et al., 1999). In fact, Ch was found to cause mild stimulation of α7 nAChRs and to induce a cascade of Ca2+-dependent intracellular events at concentrations between 50 and 300 µM, but, on the other hand, these concentrations did not induce excitation of hippocampal interneurones (Alkondon et al., 2000). The observation that the α7 nAChRs described in this study are sensitive to low concentration of Ch also opens up the possibility that they might be activated by volume transmission in a non-synaptic manner by diffusing Ch, which derives from ACh hydrolysis (Vizi and Lendvai, 1999; Lendvai and Vizi, 2008)
In addition to α7 nAChRs, our findings demonstrate that endogenous EAA release from hippocampal synaptosomes is also modulated by α4β2 nicotinic receptors, as it was increased by the selective agonist 5IA85380 in a concentration-dependent, DHβE-sensitive and MLA-resistant manner. These findings, which question the involvement of other non-α7 nAChR subtypes, fit with previous electrophysiological data showing that α4β2 nAChRs affect excitatory neurotransmission in the prefrontal cortex (Lambe et al., 2001), and increase [3H]-d-ASP release in the same area and in rat hippocampus (Rousseau et al., 2005; Dickinson et al., 2008), but do not accord with the findings of Yamamoto et al. (2005), which exclude the involvement of pre-synaptic α4β2 nAChRs in facilitating hippocampal GLU release. At the molecular level, the 5IA85380-evoked EAA release was TTX sensitive, dependent on external Ca2+ and completely blocked by Cd2+, while dantrolene was devoid of any effect. These results demonstrate that membrane depolarization, generated by opening of VDSCs, and the subsequent VDCC-mediated increase of synaptosolic Ca2+ are instrumental for the α4β2-evoked facilitation of EAA release, whereas ryanodine-sensitive intracellular stores seem not to be involved. This is in line with the generally accepted concept that neurotransmitter release evoked by the activation of non-α7 nAChRs is a Na+- and Ca2+-dependent process mediated by VDCCs (Mulle et al., 1992; Soliakov and Wonnacott, 1996; Léna and Changeux, 1997). Finally, the finding that EAA release evoked by a mixture of Ch and 5IA85380 was larger than that elicited by the single nicotinic agonist, suggests that the α7 and α4β2 nAChR subtypes can act in concert and are not mutually exclusive.
Whether the two nAChRs co-exist on the same EAA nerve endings or are present on different subsets of nerve terminals is difficult to say. The possibility that these receptors are localized at the preterminal level (Léna et al., 1993; McMahon et al., 1994) is unlikely due to our experimental approach of synaptosomes in superfusion. However, the nAChR subtypes could also be differently located on the membrane of the nerve endings and subserve diverse pre-synaptic functions. Accordingly, as previously reported (Wonnacott et al., 1996; Wonnacott, 1997), the sensitivity or insensitivity to TTX might be related to the relative proximity of nAChR to the synapse and the exocytotic machinery. This observation is compatible with the existence of extrasynaptic nAChR and other forms of interaction among neurones. Evidence from neurochemical and morphological studies has demonstrated that non-synaptic communication is a fundamental element in the chemical transmission between neurones, and between neurones and non-neuronal cells (for a review, see Vizi et al., 2010). The understanding of the physiological role of these nAChRs, as well as the determination of their location, provides the bases for possible selective pharmacological strategies to treat neuronal disorders involving the disruption of the normal function of the hippocampal cholinergic system.
Another important consequence of the existence, at the pre-synaptic level, of two different nAChR subtypes modulating EAA release is that the nicotinic responses can undergo significant changes depending both on the nAChR subtypes involved and on the modification in their number and/or in their functions. Indeed, the Ca2+ permeability of a given nAChR subtype, and the time-course of the nAChR activation and desensitization are expected to influence the extent and duration of pre-synaptic facilitation induced by nicotinic agonists. Therefore, the nicotinic modulatory effect of EAA release might result in a different effect according to which of the specific nAChRs is preferentially activated. Moreover, functional changes of these nAChRs may also occur under chronic drug treatment (including the chronic consumption of nicotine in smokers or in the case of the nicotinic replacement therapy for smoking cessation). Studies have consistently indicated that long-term nicotine exposure differentially affects the function of pre-synaptic nAChR subtypes which modulate the release of NA, DA, ACh and GLU (Lapchak et al., 1989; Marshall et al., 1997; Sershen et al., 1997; Salminen et al., 2004; Grilli et al., 2005), as well as the activity of non-nicotinic receptors such as glutamatergic NMDA and AMPA receptors (Risso et al., 2004b; Grilli et al., 2009). The differential changes in the responses of α7 and α4β2 nAChR subtypes, following chronic nicotine treatment, may therefore produce significant modifications in the nicotinic modulation of EAA release and also in the physiological responses to ACh.
Finally, drugs targeting nAChRs, in particular at the hippocampal level, may have the potential to alleviate numerous disorders, including cognitive diseases. In fact, behavioural studies with selective nicotinic agonists and antagonists, or with knockout animals have shown that both α7 and α4β2 nAChR subtypes play crucial roles in memory functions; moreover, their role in the mechanism of addiction and smoking cessation is of course crucial (Levin et al., 2009 and references therein). Our results showing that endogenous GLU and ASP release are increased by nAChR agonists extend our knowledge on the cholinergic modulation of hippocampal excitatory transmission, and provide the rationale for the use of selective nicotinic agonists to treat cognitive disorders.
Acknowledgments
This work was supported by the Italian Ministero Università Ricerca to M.M. (prot. N°20072BTSR2-002), and by Compagnia di San Paolo, University of Genoa ‘Progetto Ricerca Ateneo’ and project AROMA (ALCOTRA 2007-13). We wish to thank Maura Agate for editorial assistance, and Silvia E. Smith (University of Utah) for reviewing the manuscript.
Glossary
Abbreviations
- AMPA
α-amino-3-idrossi-5-metil-4-isoxazolone propionate
- ASP
aspartate
- Ch
choline
- CICR
Ca2+-induced calcium release
- DHβE
dihydro-β-erythroidine
- dl-TBOA
dl-threo-b-benzyloxyaspartic acid
- EAA
excitatory amino acid
- GLU
glutamate
- 5IA85380
5-iodo-A-85380 dihydrochloride
- MLA
methyllycaconitine
- nAChR
nicotinic acetylcholine receptor
- PHA543613
PHA543613 hydrochloride
- PNU 120596
N-(5-chloro-2,4-dimethoxyphenyl)-N′-(5-methyl-3-isoxazolyl)-urea
- RJR2429
RJR2429 dihydrochloride
- TTX
tetrodotoxin
- VDSC
voltage-dependent sodium channel
- VDCC
voltage-dependent Ca2+ channel
Conflict of interest
The authors state no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) 4th edn. Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Barbosa CTF, Albuquerque EX. Neuronal nicotinic acetylcholine receptor activation modulates γ-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther. 1997;283:1396–1411. [PubMed] [Google Scholar]
- Alkondon M, Pereira EFR, Eisenberg HM, Albuquerque EX. Choline and selective antagonists identify two subtypes of nicotinic acetylcholine receptors that modulate GABA release from CA1 interneurons in rat hippocampal slices. J Neurosci. 1999;19:2693–2705. doi: 10.1523/JNEUROSCI.19-07-02693.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Braga MFM, Pereira EFR, Maelicke A, Albuquerque EX. α7 Nicotinic acetylcholine receptors and modulation of GABAergic synaptic transmission in the hippocampus. Eur J Pharmacol. 2000;393:59–67. doi: 10.1016/s0014-2999(00)00006-6. [DOI] [PubMed] [Google Scholar]
- Bancila V, Cordeiro M, Bloc A, Dunant Y. Nicotine-induced and depolarisation-induced glutamate release from hippocampus mossy fibres synaptosomes: two distinct mechanisms. J Neurochem. 2009;110:570–580. doi: 10.1111/j.1471-4159.2009.06169.x. [DOI] [PubMed] [Google Scholar]
- Bencherif M, Schmitt JD, Bhatti BS, Crooks P, Caldwell WS, Lovette ME, et al. The heterocyclic substituted pyridine derivative (+/−)-2-(-3-pyridinyl)-1-azabicyclo[2.2.2]octane (RJR-2429): a selective ligand at nicotinic acetylcholine receptors. J Pharmacol Exp Ther. 1998;284:886–894. [PubMed] [Google Scholar]
- Bradford SE, Nadler JV. Aspartate release from rat hippocampal synaptosomes. Neuroscience. 2004;128:751–765. doi: 10.1016/j.neuroscience.2004.06.065. [DOI] [PubMed] [Google Scholar]
- Cavallero A, Marte A, Fedele E. l-Aspartate as an amino acid neurotransmitter: mechanisms of the depolarization-induced release from cerebrocortical synaptosomes. J Neurochem. 2009;110:924–934. doi: 10.1111/j.1471-4159.2009.06187.x. [DOI] [PubMed] [Google Scholar]
- Clementi F, Fornasari D, Gotti C. Neuronal nicotinic receptors, important new players in brain function. Eur J Pharmacol. 2000;393:3–10. doi: 10.1016/s0014-2999(00)00066-2. [DOI] [PubMed] [Google Scholar]
- Curras MC, Dingledine R. Selectivity of amino acid transmitters acting at N-methyl-d-aspartate and amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptors. Mol Pharmacol. 1992;41:520–526. [PubMed] [Google Scholar]
- Curtis DR, Phillis JW, Watkins JC. The chemical excitation of spinal neurones by certain acidic amino acids. J Physiol. 1960;150:656–682. doi: 10.1113/jphysiol.1960.sp006410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JA, Hanrott KE, Mok MH, Kew JN, Wonnacott S. Differential coupling of alpha7 and non-alpha7 nicotinic acetylcholine receptors to calcium-induced calcium release and voltage-operated calcium channels in PC12 cells. J Neurochem. 2007;100:1089–1096. doi: 10.1111/j.1471-4159.2006.04273.x. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Fedele E, Jin Y, Varnier G, Raiteri M. In vivo microdialysis study of a specific inhibitor of soluble guanylyl cyclase on the glutamate receptor/nitric oxide/cyclic GMP pathway. Br J Pharmacol. 1996;119:590–594. doi: 10.1111/j.1476-5381.1996.tb15713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck MW, Henze DA, Barrionuevo G, Palmer AM. Aspartate and glutamate mediate excitatory synaptic transmission in area CA1 of the hippocampus. J Neurosci. 1993;13:3944–3955. doi: 10.1523/JNEUROSCI.13-09-03944.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleck MW, Barrionuevo G, Palmer AM. Release of d,l-threo-beta-hydroxyaspartate as a false transmitter from excitatory amino acid-releasing nerve terminals. Neurochem Int. 2001a;39:75–81. doi: 10.1016/s0197-0186(00)00111-x. [DOI] [PubMed] [Google Scholar]
- Fleck MW, Barrionuevo G, Palmer AM. Synaptosomal and vesicular accumulation of l-glutamate, l-aspartate and d-aspartate. Neurochem Int. 2001b;39:217–225. doi: 10.1016/s0197-0186(01)00018-3. [DOI] [PubMed] [Google Scholar]
- Gold PE. Acetylcholine modulation of neural systems involved in learning and memory. Neurobiol Learn Mem. 2003;80:194–210. doi: 10.1016/j.nlm.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA. Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature. 1996;383:713–716. doi: 10.1038/383713a0. [DOI] [PubMed] [Google Scholar]
- Grilli M, Parodi M, Raiteri M, Marchi M. Chronic nicotine differentially affects the function of nicotinic receptor subtypes regulating neurotransmitter release. J Neurochem. 2005;93:1353–1360. doi: 10.1111/j.1471-4159.2005.03126.x. [DOI] [PubMed] [Google Scholar]
- Grilli M, Pittaluga A, Merlo-Pich E, Marchi M. NMDA-mediated modulation of dopamine release is modified in rat prefrontal cortex and nucleus accumbens after chronic nicotine treatment. J Neurochem. 2009;108:408–416. doi: 10.1111/j.1471-4159.2008.05792.x. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Chaudry FA, Bjaalie JG, Fonnum F, Ottersen OP, Storm-Mathisen J. Synaptic vesicular localization and exocytosis of l-aspartate in excitatory nerve terminals: a quantitative immunogold analysis in rat hippocampus. J Neurosci. 1998;18:6059–6070. doi: 10.1523/JNEUROSCI.18-16-06059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen V, Storm-Mathisen J. Aspartate-neurochemical evidence for a transmitter role. In: Ottersen OP, Storm-Mathisen J, editors. Handbook of Chemical Neuroanatomy. Vol. 18. Amsterdam, The Netherlands: Glutamate. Elsevier Science BV; 2000. pp. 45–62. [Google Scholar]
- Konradsson-Geuken A, Gash CR, Alexander K, Pomerleau F, Huettl P, Gerhardt GA, et al. Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats. Synapse. 2009;63:1069–1082. doi: 10.1002/syn.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2001;28:216–225. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- Lapchak PA, Araujo DM, Quirion R, Collier B. Effect of chronic nicotine treatment on nicotinic autoreceptor function and N-[3H]methylcarbamylcholine binding sites in the rat brain. J Neurochem. 1989;52:483–491. doi: 10.1111/j.1471-4159.1989.tb09146.x. [DOI] [PubMed] [Google Scholar]
- Léna C, Changeux JP. Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci. 1997;17:576–585. doi: 10.1523/JNEUROSCI.17-02-00576.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Léna C, Changeux JP, Mulle C. Evidence for ‘preterminal’ nicotinic receptors on GABAergic axons in the rat interpeduncular nucleus. J Neurosci. 1993;13:2680–2688. doi: 10.1523/JNEUROSCI.13-06-02680.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendvai B, Vizi ES. Nonsynaptic chemical transmission through nicotinic acetylcholine receptors. Physiol Rev. 2008;88:333–349. doi: 10.1152/physrev.00040.2006. [DOI] [PubMed] [Google Scholar]
- Levin DE, Petro A, Rezvani AH, Pollard N, Christopher NC, Strauss M, et al. Nicotinic α7- or β2-containing receptor knockout: effects on radial-arm maze learning and long term nicotine consumption in mice. Behav Brain Res. 2009;196:207–213. doi: 10.1016/j.bbr.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- McKay BE, Placzek AN, Dani JA. Regulation of synaptic transmission and plasticity by neuronal nicotinic acetylcholine receptors. Biochem Pharmacol. 2007;74:1120–1133. doi: 10.1016/j.bcp.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon LL, Yoon KW, Chiappinelli VA. Nicotinic receptor activation facilitates GABAergic neurotransmission in the avian lateral spiriform nucleus. Neuroscience. 1994;59:689–698. doi: 10.1016/0306-4522(94)90187-2. [DOI] [PubMed] [Google Scholar]
- Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M. Direct evidence that release-stimulating α7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem. 2002;80:1071–1078. doi: 10.1046/j.0022-3042.2002.00805.x. [DOI] [PubMed] [Google Scholar]
- Marshall DL, Redfern PH, Wonnacott S. Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J Neurochem. 1997;68:1511–1519. doi: 10.1046/j.1471-4159.1997.68041511.x. [DOI] [PubMed] [Google Scholar]
- Mogg AJ, Jones FA, Pullar IA, Sharples CG, Wonnacott S. Functional responses and subunit composition of presynaptic nicotinic receptor subtypes explored using the novel agonist 5-iodo-A-85380. Neuropharmacology. 2004;47:848–859. doi: 10.1016/j.neuropharm.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Mukhin AG, Gundisch D, Horti AG, Koren AO, Tamagnan G, Kimes AS, et al. 5-Iodo-A-85380, an alpha4beta2 subtype-selective ligand for nicotinic acetylcholine receptors. Mol Pharmacol. 2000;57:642–649. doi: 10.1124/mol.57.3.642. [DOI] [PubMed] [Google Scholar]
- Mulle C, Choquet D, Korn H, Changeux JP. Calcium influx through nicotinic receptor in rat central neurons: its relevance to cellular regulation. Neuron. 1992;8:135–143. doi: 10.1016/0896-6273(92)90115-t. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Iga K, Shibata T, Shudo M, Kataoka K. Glial plasmalemmal vesicles: a subcellular fraction from rat hippocampal homogenate distinct from synaptosomes. Glia. 1993;9:48–56. doi: 10.1002/glia.440090107. [DOI] [PubMed] [Google Scholar]
- Parodi M, Patti L, Grilli M, Raiteri M, Marchi M. Nicotine has permissive role on the activation of metabotropic glutamate 5 receptor coexisting with nicotinic receptors on rat hippocampal noradrenergic nerve terminals. Neurochem Int. 2006;48:138–143. doi: 10.1016/j.neuint.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Structure–activity relationships for amino acid transmitter candidates acting at N-methyl-d-aspartate and quisqualate receptors. J Neurosci. 1990;10:2385–2399. doi: 10.1523/JNEUROSCI.10-07-02385.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA. Nicotine modulation of glutamate and GABA synaptic transmission of hippocampal neurons. Ann N Y Acad Sci. 1999;868:591–610. doi: 10.1111/j.1749-6632.1999.tb11332.x. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of superfusion. Neurochem Res. 2000;25:1265–1274. doi: 10.1023/a:1007648229795. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Zappettini S, Milanese M, Fedele E, Raiteri M, Bonanno G. Mechanisms of glutamate release elicited in rat cerebrocortical nerve endings by ‘pathologically’ elevated extraterminal K+ concentrations. J Neurochem. 2007;103:952–961. doi: 10.1111/j.1471-4159.2007.04784.x. [DOI] [PubMed] [Google Scholar]
- Reid MS, Fox L, Ho LB, Berger SP. Nicotine stimulation of extracellular glutamate levels in the nucleus accumbens: neuropharmacological characterization. Synapse. 2000;35:129–136. doi: 10.1002/(SICI)1098-2396(200002)35:2<129::AID-SYN5>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Risso F, Parodi M, Grilli M, Molfino F, Raiteri M, Marchi M. Chronic nicotine causes functional upregulation of ionotropic glutamate receptors mediating hippocampal noradrenaline and striatal dopamine release. Neurochem Int. 2004a;44:293–301. doi: 10.1016/s0197-0186(03)00173-6. [DOI] [PubMed] [Google Scholar]
- Risso F, Grilli M, Parodi M, Bado M, Raiteri M, Marchi M. Nicotine exerts a permissive role on NMDA receptor function in hippocampal noradrenergic terminals. Neuropharmacology. 2004b;47:65–71. doi: 10.1016/j.neuropharm.2004.02.018. [DOI] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, et al. Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol. 2004;65:1526–1535. doi: 10.1124/mol.65.6.1526. [DOI] [PubMed] [Google Scholar]
- Sershen H, Balla A, Lajtha A, Vizi ES. Characterization of nicotinic receptors involved in the release of noradrenaline from the hippocampus. Neuroscience. 1997;77:121–130. doi: 10.1016/s0306-4522(96)00425-3. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Soliakov L, Wonnacott S. Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated [3H]dopamine release from rat striatal synaptosomes. J Neurochem. 1996;67:163–170. doi: 10.1046/j.1471-4159.1996.67010163.x. [DOI] [PubMed] [Google Scholar]
- Szerb JC. Changes in the relative amounts of aspartate and glutamate released and retained in hippocampal slices during stimulation. J Neurochem. 1988;50:219–224. doi: 10.1111/j.1471-4159.1988.tb13252.x. [DOI] [PubMed] [Google Scholar]
- Szerb JC, O'Regan PA. Reversible shifts in the Ca2+-dependent release of aspartate and glutamate from hippocampal slices with changing glucose concentrations. Synapse. 1987;1:265–272. doi: 10.1002/syn.890010308. [DOI] [PubMed] [Google Scholar]
- Toth E. Effect of nicotine on the level of extracellular amino acids in the hippocampus of rat. Neurochem Res. 1996;21:903–907. doi: 10.1007/BF02532339. [DOI] [PubMed] [Google Scholar]
- Toth E, Sershen H, Hashim A, Vizi ES, Lajtha A. Effect of nicotine on extracellular levels of neurotransmitters assessed by microdialysis in various brain regions: role of glutamic acid. Neurochem Res. 1992;17:265–271. doi: 10.1007/BF00966669. [DOI] [PubMed] [Google Scholar]
- Toth E, Vizi ES, Lajtha A. Effect of nicotine on levels of extracellular amino acids in regions of the rat brain in vivo. Neuropharmacology. 1993;32:827–832. doi: 10.1016/0028-3908(93)90192-6. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Lendvai B. Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res Rev. 1999;30:219–235. doi: 10.1016/s0165-0173(99)00016-8. [DOI] [PubMed] [Google Scholar]
- Vizi ES, Fekete A, Karoly R, Mike A. Non-synaptic receptors and transporters involved in brain functions and targets of drug treatment. Br J Pharmacol. 2010;160:785–809. doi: 10.1111/j.1476-5381.2009.00624.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang BW, Liao WN, Chang CT, Wang SJ. Facilitation of glutamate release by nicotine involves the activation of a Ca2+/calmodulin signaling pathway in rat prefrontal cortex nerve terminals. Synapse. 2006;59:491–501. doi: 10.1002/syn.20267. [DOI] [PubMed] [Google Scholar]
- Wang L, Nadler JV. Reduced aspartate release from rat hippocampal synaptosomes loaded with clostridial toxin light chain by electroporation: evidence for an exocytotic mechanism. Neurosci Lett. 2007;412:239–242. doi: 10.1016/j.neulet.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishka DG, Walker DP, Yates KM, Reitz SC, Jia S, Myers JK, et al. Discovery of N-[(3R)-1-azabicyclo[2.2.2]oct-3-yl]furo[2,3-c]pyridine-5-carboxamide, an agonist of the alpha7 nicotinic acetylcholine receptor, for the potential treatment of cognitive deficits in schizophrenia: synthesis and structure–activity relationship. J Med Chem. 2006;49:4425–4436. doi: 10.1021/jm0602413. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Soliakov L, Wilkie G, Redfern P, Marshall D. Presynaptic nicotinic acetylcholine receptors in the brain. Drug Dev Res. 1996;38:149–159. [Google Scholar]
- Yamamoto S, Kanno T, Nagata T, Yaguchi T, Tanaka A, Nishizaki T. The linoleic acid derivative FR236924 facilitates hippocampal synaptic transmission by enhancing activity of presynaptic alpha7 acetylcholine receptors on the glutamatergic terminals. Neuroscience. 2005;130:207–213. doi: 10.1016/j.neuroscience.2004.09.016. [DOI] [PubMed] [Google Scholar]