

Abstract
BACKGROUND AND PURPOSE
Pharmacological preconditioning (PPC) with mitochondrial ATP-sensitive K+ (mitoKATP) channel openers such as diazoxide, leads to cardioprotection against ischaemia. However, effects on Ca2+ homeostasis during PPC, particularly changes in Ca2+ channel activity, are poorly understood. We investigated the effects of PPC on cardiac L-type Ca2+ channels.
EXPERIMENTAL APPROACH
PPC was induced in isolated hearts and enzymatically dissociated cardiomyocytes from adult rats by preincubation with diazoxide. We measured reactive oxygen species (ROS) production and Ca2+ signals associated with action potentials using fluorescent probes, and L-type currents using a whole-cell patch-clamp technique. Levels of the α1c subunit of L-type channels in the cellular membrane were measured by Western blot.
KEY RESULTS
PPC was accompanied by a 50% reduction in α1c subunit levels, and by a reversible fall in L-type current amplitude and Ca2+ transients. These effects were prevented by the ROS scavenger N-acetyl-L-cysteine (NAC), or by the mitoKATP channel blocker 5-hydroxydecanoate (5-HD). PPC signficantly reduced infarct size, an effect blocked by NAC and 5-HD. Nifedipine also conferred protection against infarction when applied during the reperfusion period. Downregulation of the α1c subunit and Ca2+ channel function were prevented in part by the protease inhibitor leupeptin.
CONCLUSIONS AND IMPLICATIONS
PPC downregulated the α1c subunit, possibly through ROS. Downregulation involved increased degradation of the Ca2+ channel, which in turn reduced Ca2+ influx, which may attenuate Ca2+ overload during reperfusion.
Keywords: calcium channel, preconditioning, L-type channels, diazoxide, cardiac muscle, α1c subunit, mitoKATP channels, channel expression, reactive oxygen species, infarction
Introduction
Heart muscle can be protected from injury by severe and prolonged ischaemia by pre-treatment with ischaemic periods of a few minutes (Murry et al., 1986). This ischaemic preconditioning (IPC) can be mimicked by pharmacological agents like diazoxide, which, among other activities, opens mitochondrial ATP-sensitive K+ (mitoKATP) channels (Garlid et al., 1997; Pain et al., 2000; channel and receptor nomenclature follow Alexander et al., 2009). Furthermore, both IPC and pharmacological preconditioning (PPC) can be antagonized by mitoKATP channel blockers (Ardehali and O'Rourke, 2005; Halestrap et al., 2007). Based on this pharmacological evidence, mitoKATP channels are proposed to be central in protection of the heart muscle by IPC and PPC (Ardehali and O'Rourke, 2005; Halestrap et al., 2007). However, the possible involvement in IPC and PPC of other ion channels, such as the L-type Ca2+ channel, a key element in excitation–contraction coupling and Ca2+ homeostasis in the heart and other tissues (Bers, 2002), is largely unexplored. In smooth muscle, L-type channels are involved in refilling intracellular stores, and this is inhibited by high concentrations of diazoxide (Dessy and Godfraind, 1996). Recent evidence suggests that L-type channels may contribute to pacing-induced protection against anoxia-reoxygenation in the embryonic heart (Bruchez et al., 2008).
In this study, we tested the hypothesis that L-type Ca2+ channels are regulated during PPC. We found that preincubation with diazoxide drastically reduced the magnitude of L-type Ca2+ currents and intracellular Ca2+ transients. PPC also downregulated α1c, the principal subunit of the cardiac L-type Ca2+ channel (Catterall, 2000; Lacinová, 2005). We propose that PPC produced by diazoxide involves an increase in reactive oxygen species (ROS) production that is involved in downregulation of the α1c subunit via a protease-dependent process.
Methods
Preparation of hearts
All animal care and experimental procedures conformed to protocols approved by the Division of Laboratory Animal Units, Cinvestav-IPN, in compliance with federal law, federal statute and Consejo Nacional de Ciencia y Tecnología (CONACYT) regulations. Male Wistar rats (300–350 g) were anaesthetized with 50 mg·kg−1 of pentobarbital sodium injected intraperitoneally. A 500 U·kg−1 heparin sodium were also administered intraperitoneally. Hearts (1.5–1.7 g) were rapidly excised, arrested in modified Krebs-Henseleit buffer (in mM): 117.8 NaCl, 1.2 NaH2PO4, 6.0 KCl, 24.3 NaHCO3, 1.2 MgSO4, 0.027 EDTA, 5.1 glucose and 1.6 CaCl2, gassed with 95% O2, 5% CO2 at pH 7.4), and perfused in a Langendorff apparatus with an aortic cannula delivering 37 ± 0.5°C buffer at 66 mmHg. Care was taken to avoid damage to the aorta. The whole procedure took ∼ 30 s. Left ventricle (LV) pressure was continuously monitored and recorded by Axotape software (Axon Instruments, Foster City, CA, USA). The following parameters of cardiac function were assessed: Left ventricle developed pressure (LVDP) calculated as the difference between the systolic and diastolic pressure of the LV, LV end-diastolic pressure (LVEDP), and the maximal (dP/dtmax) and minimal (dP/dtmin) value of the first derivative of LV pressure. LVDP was monitored with a water-filled balloon inserted into the left ventricle, set to deliver diastolic pressure of 5–10 mmHg. The resulting coronary flow was in the range of 8.7–14 ml·min−1·g−1. Diazoxide was added from a 1 M stock solution in dimethyl sulphoxide (DMSO) to a final DMSO concentration of less than 0.01%.
Isolation of ventricular myocytes
Ventricular myocytes were isolated as described previously (Sánchez et al., 2001), with slight modifications. In brief, hearts were perfused for 5 min at 37°C with Ca2+-free Tyrode solution containing (in mM): 136 NaCl, 5.4 KCl, 1 MgCl2, 10 HEPES, and 11 glucose plus heparin (10 U·ml−1, Sigma). Hearts were recirculated for ∼30 min with Tyrode solution with 20–40 µM CaCl2, 0.1% bovine serum albumin (BSA), collagenase (Worthington, Lakewood, NJ, USA, type II, 70 U·ml−1), and protease type XIV (Sigma, 0.5 mg/100 ml). Ventricles were minced and shaken 2–3 times at 45 r.p.m. for 7 min in the same solution. Cells were filtered through a cell strainer (100 µm nylon BD Falcon) and centrifuged at 700×g for 2 min. The pellet was resuspended for 90 min in Tyrode solution containing (in mM): 136 NaCl, 5.4 KCl, 1 CaCl2, 2 MgCl2, 10 HEPES, and 11 glucose, plus 1% BSA in control experiments, or in an identical solution containing diazoxide (100 µM), 5-hydroxydecanoate (5-HD; 100 µM), N-acetyl-L-cysteine (NAC; 2 or 4 mM) or leupeptin (100 µM) for the indicated time periods. Myocytes were used immediately.
Electrophysiological methods
L-type currents were recorded in dissociated adult ventricular myocytes using a whole-cell patch-clamp technique as described elsewhere (Sánchez et al., 2001). Currents were recorded with an Axopatch 200-A amplifier (Axon Instruments) using the pulse protocol shown in Figure 1A. Two consecutive −10 mV hyperpolarizing pulses, lasting 150 ms, were followed by a 150 ms depolarizing pulse of preselected amplitude. The sequence was repeated 10 times, increasing the amplitude of the depolarizing pulse by +10 mV steps from a holding potential of −40 mV. Hyperpolarizing currents were used to subtract linear currents, and to measure membrane capacitance. Current records were digitized with a Digidata interface (Axon Instruments, Foster City, CA, USA) at 16 bit resolution. Data were analysed using pCLAMP 8.0 (Axon Instruments) and in-house software.
Figure 1.
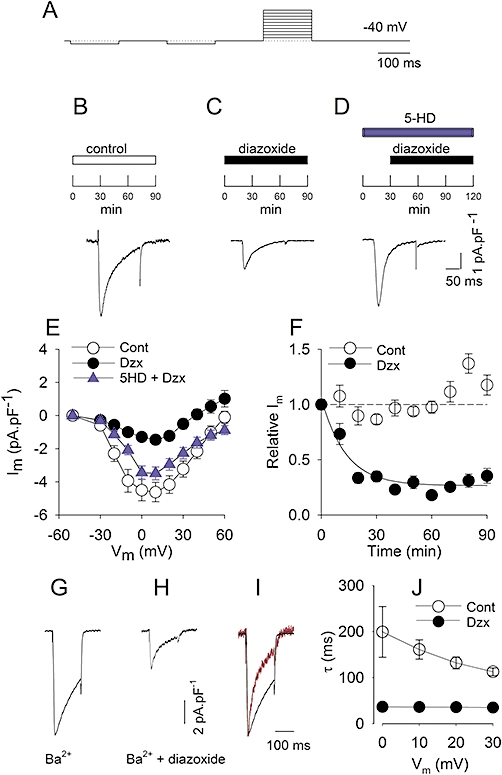
PPC reduces L-type currents. A, pulse protocol. B–D, experimental protocol and representative recordings of L-type Ca2+ currents at 0 mV. B, representative L-type Ca2+ current after 90 min in control solution. C, myocyte after 90 min in diazoxide (100 µM). D, myocyte preincubated for 30 min in 5-HD (100 µM), then 90 min in 5-HD plus diazoxide (100 µM). E and subsequent panels show mean ± SE. E, I-V relationship of peak L-type currents. At the indicated potentials, peak currents are shown from controls (Cont; n = 38); from diazoxide-incubated myocytes (Dzx; n = 29) and from 5-HD plus diazoxide pretreated myocytes (Dzx + 5HD; n = 14). Differences between diazoxide only and control data sets were statistically significant between −20 mV and +30 mV, and at +50 mV (P < 0.05). F: relationship between incubation time and peak L-type Ca2+ currents (○): peak L-type currents as a function of time in control solution (n = 15–38). Corresponding mean values from diazoxide-treated myocytes (n = 10–14); data were significantly different from control data for t >10 min. Dotted line: mean amplitude of control L-type Ca2+ currents at t = 0. Solid line: best fit of a single exponential for diazoxide-treated myocytes with τ= 13.2 min. G, representative recording of L-type Ba2+ currents at 0 mV after 90 min in control solution. H, myocyte after 90 min in diazoxide (100 µM). I, normalized recordings from G (black) and H (red). J, relationship between the time constant of inactivation of L-type Ba2+ currents and voltage under control (Cont) conditions and after diazoxide (Dzx). Values were significantly different at all potentials (n = 4–8).
Peak Ca2+ channel current values were fitted to equation (1) to describe current-voltage relationships of L-type currents.
| (1) |
Gmax is the maximum conductance, Vrev is the reversal potential, Vm is the membrane potential, V is the potential for which G = 0.5 Gmax, and k is a measure of the steepness of the curve. Numerical formulae were fitted to experimental data using a non-linear, least-squares algorithm.
The pipette solution contained (in mM): 100 cesium aspartate, 20 CsCl, 20 TEACl, 2 Mg-ATP, 1.8 MgCl2, 0.05 EGTA, and 5 HEPES, pH 7.2. The standard bath saline was Tyrode solution plus 1% BSA for stability of the electrophysiological recordings. We confirmed that BSA has no significant effects on L-type currents. In Tyrode solution with no BSA, the average value of peak currents at +10 mV was −4.37 ± 0.03 pA·pF−1 (n = 7). In the presence of BSA, at the same potential, it was −4.61 ± 0.58 pA·pF−1 (n = 38). No shifts in the current-voltage (I-V) curve were observed.
To characterize the effects of PPC on L-type currents, intact cardiac myocytes were incubated in control solution or in solutions containing different drugs (indicated in the figures and legends) for the stated time period. After the incubation period was terminated, cardiomyocytes were transferred to the recording chamber where a selected myocyte was patch-clamped to record whole-cell membrane currents. Experiments were performed at room temperature (23°C).
Measurement of Ca2+ transients
Fluo-3 AM (Molecular Probes) was used to monitor the levels of intracellular Ca2+. This dye undergoes large fluorescence changes upon Ca2+ binding, has a large dynamic range and low compartmentalization (Thomas et al., 2000), and was previously used to record Ca2+ signals in ventricular myocytes (Vicencio et al., 2006). Adult cardiomyocytes were loaded with approximately 5 µM Fluo-3 AM in standard Tyrode solution for 40 min at room temperature. After loading, cells were incubated in dye-free solution for over 30 min to allow the conversion of the dye to its Ca2+-sensitive, free acid form. Dye-loaded cells were placed on a laminin-coated coverslip at the bottom of a chamber on the stage of an Optiphot microscope (Nikon, Tokyo, Japan). Fluorescence emitted by stained myocytes, illuminated episcopically with monochromatic light at 485 nm, was filtered with a high-pass barrier filter (cut-off wavelength 535 nm) and detected with a low-noise photodiode in a photovoltaic configuration. Analogue signals were digitized as described above and sampled every 60 µs for 40 s. The basal fluorescence (F) of the myocyte was recorded, and its mean value during a 300 ms interval prior to electrical stimulation was used to calculate Ca2+ signals as ΔF/F. No absolute value measurements were made for intracellular Ca2+ concentration, as this study was designed to detect temporal and relative patterns of Ca2+ transients. To induce Ca2+ transients during action potentials, myocytes were electrically stimulated with two extracellular platinum electrodes at a frequency of 0.3 Hz. Effects of PPC on Ca2+ transients were determined with an experimental procedure similar to the one used for electrophysiological experiments.
Measurement of ROS production
ROS was measured using the cell-permeant fluorescent probe, 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester (CM-H2DCFDA, Molecular Probes). Inside cells, the diacetate group of CM-H2DCFDA is cleaved by intracellular esterases, trapping the non-fluorescent product CM-H2DCFFH, which is oxidized to fluorescent CM-H2DCF upon exposure to H2O2 or ·OH (Rijstenbil et al., 2000). Intensity is dependent on the amount of generated ROS. The probe has excellent retention in live cells and is used to measure intracellular ROS in cardiomyocytes and other cells (Bodi et al., 2005). Fluorescence in arbitrary units at 470 nm excitation and 515 nm emission was measured for 200 ms in user-defined segments of cardiomyocytes. The intensity of the emitted light was detected as described above and sampled at a frequency of 17 kHz.
Cardiomyocytes were loaded with 10 µM CM-H2DCFDA by incubation in the dark for 30 min at room temperature. The dye was added from a stock solution in DMSO to result in a final DMSO concentration of less than 0.1%. At this concentration, fluorescence signals associated with ROS production are unaffected by the vehicle (Pasdois et al., 2008). Loaded myocytes were incubated in dye-free solution for over 30 min to allow conversion of the dye into its ROS-sensitive form. At the end of the experiments, dye-loaded myocytes were exposed to constant excitation for 3–6 min to induce photooxidation of CM-H2DCFDA. Fluorescence values increased with light exposure to a plateau representing the total amount of CM-H2DCFDA available for oxidation within the exposed area of the myocyte. Immediately after dye loading, ROS production was measured every 5 or 10 min for 30 min, either in myocytes in Tyrode solution, with or without diazoxide (100 µM). For normalization, ROS fluorescence data are expressed relative to mean fluorescence at the beginning of the experiment.
Isolated hearts
Hearts were subjected to the following treatments: The control group was perfused for 90 min with Krebs-Henseleit buffer. The diazoxide-treated group was perfused for 90 min in Krebs-Henseleit buffer containing 100 µM diazoxide. For the NAC plus diazoxide group, the ROS scavenger NAC was added to Krebs-Henseleit buffer at 4 mM, and hearts were perfused for 15 min, then perfused for 90 min in the same buffer with diazoxide. The NAC-only group was perfused for 90 min with Krebs-Henseleit buffer plus NAC. For the 5-HD plus diazoxide group, the mitoKATP channel inhibitor 5-HD was added to Krebs-Henseleit buffer at 100 µM, and hearts were perfused for 15 min, then perfused for 90 min in the same buffer with diazoxide. The 5-HD-only group was perfused for 90 min with Krebs-Henseleit buffer plus 5-HD.
To assess protection against ischaemia, hearts previously perfused for 90 min in Krebs-Henseleit buffer or in solutions containing diazoxide and/or the inhibitors as described above, and were subjected to global ischaemia for 30 min, followed by 2 h of reperfusion with control solution. To assess the effect of nifedipine on infarction, a group of hearts was perfused for 60 min with Krebs-Henseleit buffer, then perfused for 30 min in the same buffer with nifedipine (30 nM). This was followed by global ischaemia for 30 min, then hearts were reperfused with the same buffer for 30 min and reperfusion continued with Krebs-Henseleit buffer for 90 min. Thereafter, hearts from all groups were dyed with 2,3,5-triphenyltetrazolium chloride (TTC, at 1%) for 10 min fixed overnight with paraformaldehyde (4%), cut into 500 µm slices and photographed with a digital camera attached to a microscope (Olympus). TTC produces coloured precipitates in the presence of intact dehydrogenase enzyme systems, whereas necrotic areas lacking dehydrogenase activity or sufficient concentrations of cofactors (e.g. NADH) fail to stain (Klein et al., 1981). TTC is widely used for estimating the size of myocardial infarction areas (e.g. Garcia-Dorado et al., 1987). Infarcted areas were measured by the Analysis Soft Imaging System (Olympus).
Measurement of LDH
Lactate dehydrogenase (LDH), an indicator of myocardial tissue injury, was assayed in coronary effluent using a coupled spectrophotometric technique from a kit (Cayman Chemical Co., USA) according to the manufacturer's instructions. LDH activity was measured with a coupled two-step reaction, the second step of which was the reduction of a tetrazolium salt to formazan, which absorbs at 490 nm. The amount of LDH released in the first 15 min of reperfusion was calculated by integrating the area underneath individual time course curves. To compare data from different experiments, normalization used the ratio of the LDH value from PPC- or nifedipine-treated ischaemic hearts to LDH from control ischaemic hearts.
Western blotting
Membrane fractions were obtained as described (Saada et al., 2005) by Dounce homogenization of −80°C frozen pulverized rat ventricles in 10 volumes of homogenization medium containing (in mM): 250 sucrose, 50 MOPS, and 2 EGTA, at pH 7.4, to which protease inhibitors 1 mM benzamidine, 1 µM leupeptin, 0.1 µM aprotinin, 1 µM pepstatin A, 0.2 mM PMSF, and 0.4 mM pefabloc were added. Homogenates were centrifuged at 1000×g for 10 min, then at 5000×g for 10 min, and finally at 100 000×g for 1 h. The final pellet was resuspended in homogenization medium and stored at −80°C. Protein content was measured with the Bradford assay.
In some experiments, dissociated myocytes were used for Western blotting by resuspension in lysis buffer containing: 20 mM Tris (pH 7.5), 100 mM NaCl, 1% Triton X-100 and 1 mM PMSF with protease inhibitors 1 mM benzamidine, 1 µM leupeptin, 0.1 µM aprotinin, 1 µM pepstatin A, and 0.4 mM pefabloc. Lysis was achieved by continuous agitation for 60 min at 4°C followed by five cycles of sonication. Samples were centrifuged at 13 000×g for 10 min at 4°C and the soluble fraction used for Western blots. Protein content was measured with an RC DC Protein Assay kit (Bio-Rad) following the manufacturer's instructions.
Western blotting used 50 µg of membrane preparations (or 100 µg when myocytes were used) applied to 7.5% dodecyl sulphate-polyacrylamide (SDS-PAGE) gels to identify α1c subunits, and 30 µg (or 40 µg when myocytes were used) on 13.5% gel to identify α-actin. Loading buffer and gels for α1c subunit immunoblots contained 8 M urea. Proteins were transferred to nitrocellulose membranes and blocked with 4.5% nonfat dried milk in phosphate buffered saline (PBS), before probing with primary anti-α1c antibodies (ACC-003, Alomone Laboratories, Jerusalem, Israel) at 1:100 dilution. Monoclonal antibody (a gift from Dr J.M. Hernández, from the Department of Cell Biology, Cinvestav) against α-actin (42 kDa) was also used at 1:100 (or 1:200 when myocytes were used). After washing, membranes were incubated with an anti-rabbit (1:8000) or an anti-mouse (1:8000) horseradish peroxidase conjugate (Zymed) as secondary antibodies and detected by enhanced chemiluminescence using a Super Signal West Pico Chemiluminescent substrate (Pierce). To measure the densities of the α1c subunit and α-actin bands, film was scanned at 300 dpi, and images were inverted. The mean value of the intensity of each band and its corresponding area were measured with Photoshop (CS3), and absolute densities were calculated as the product of these two parameters. For normalization, the absolute density of α1c subunit bands was divided by the density for corresponding α-actin bands. Relative densities were the ratio between the density of the α1c subunit band from a treated heart, and the density of the α1c subunit band from a control heart obtained in parallel experiments.
Data analysis
Data are expressed as the mean ± standard error (s.e. mean). Analysis used independent t-tests or anova, and Tukey's or Dunnett's post-test, as appropriate. A P < 0.05 was considered to be statistically significant.
Materials
Diazoxide, NAC, 5HD, nifedipine, TTC, protease inhibitors, Tiron and other chemicals were supplied by Sigma Aldrich (USA) and leupeptin was from Roche (USA).
Results
Effect of diazoxide on L-type Ca2+ channel currents
Incubation with diazoxide led to a significant reduction in Ca2+-current amplitude. Figure 1B–D shows representative recordings during step depolarizations to 0 mV. To allow comparisons among experiments, membrane current values were normalized to unit capacitance. L-type Ca2+ currents under control conditions reached a peak of ∼−5 pA·pF−1, approximately 10 ms after pulse onset. The current was not sustained but inactivated during the pulse. These characteristics are typical of L-type Ca2+ currents of adult rat cardiac myocytes (Gomez et al., 1994; Striessnig, 1999; Bodi et al., 2005). Diazoxide produced a significant decrease in current amplitude to approximately 30% of the value observed in controls. The effects of diazoxide on L-type Ca2+ currents developed within minutes, leading to a progressive decline in current magnitude that was almost complete after 30 min (Figure 1F). Diazoxide effects were reversible after 60 min of wash-out with control solution (data not shown). At the concentration used, diazoxide is a selective opener of ATP-sensitive K+ channels in the inner mitochondrial membrane (Garlid et al., 1996), so we tested the effects of the selective mitoKATP channel blocker 5-HD (Hu et al., 1999) on the diazoxide effect. The 5-HD completely antagonized the reduction in L-type Ca2+ currents produced by diazoxide, at all potentials tested (Figure 1E).
The decrease in current amplitude might involve an increase in Ca2+-dependent inactivation of L-type channels by diazoxide. To explore this possibility, we replaced Ca2+ with an equimolar amount of Ba2+ as the charge carrier. As expected, these currents were larger than Ca2+ currents because the channels are more permeable to Ba2+ (Figure 1G). In the presence of diazoxide, Ba2+ peak currents were greatly decreased (Figure 1H), with a drop in current magnitude that was similar to that seen with Ca2+ as the charge carrier, indicating that Ca2+-dependent inactivation appears to play no role in the effects of diazoxide on the Ca2+ currents. On the other hand, Ba2+ current recordings revealed an increase in voltage-dependent inactivation of L-type currents by diazoxide. This was manifested as a significant acceleration in Ba2+ current decay, seen when current traces were normalized (Figure 1I). To estimate current time courses, the inactivation phase of the currents was fitted to a single exponential. The time constant (τ) at different potentials was distinctly smaller in diazoxide-treated myocytes (Figure 1J).
Effect of ROS scavengers on the decline of L-type Ca2+ channel currents
We explored whether diazoxide effects on L-type Ca2+ currents were mediated by ROS. Figure 2 shows results obtained with the ROS scavenger NAC. In diazoxide, 30 min led to near maximal effects on current amplitude (Figure 1). NAC largely prevented the diazoxide-induced decline in current magnitude. In its presence, diazoxide had no effect on peak current values at any potential tested, although NAC by itself produced a small decline in current amplitude (Figure 2).
Figure 2.
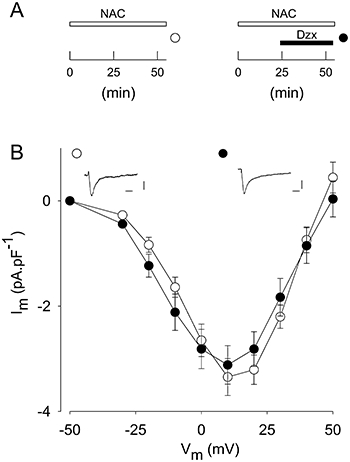
Effects of diazoxide (Dzx) on L-type Ca2+ currents are prevented by ROS scavengers. A, experimental protocol. B, I-V plots showing mean ± SE of peak L-type currents after 55 min in NAC (4 mM; n = 18) and after incubation in NAC plus diazoxide (100 µM; n = 23), as in A. Insets show representative L-type Ca2+ currents during pulses to +10 mV, under conditions as in A. Calibration bars are 20 ms and 2 pA·pF−1.
Effect of diazoxide on Ca2+ signals
The decrease in L-type Ca2+ current magnitude illustrated in Figure 1 would be expected to reduce the amount of Ca2+ released from the sarcoplasmic reticulum through the Ca2+-induced Ca2+ release (CICR) mechanism that follows membrane depolarization. Therefore, we assessed the effect of diazoxide on intracellular Ca2+ signals. For normalization purposes, Ca2+ signal amplitude during action potentials was measured as ΔF/F. Figure 3 shows results from representative experiments. Diazoxide caused a significant decrease in ΔF/F amplitude (n = 47). Furthermore, as expected from the electrophysiological results, the inhibitory effect of diazoxide was fully prevented by 5-HD (n = 14). The ROS scavenger NAC also prevented the diazoxide-induced decline in the magnitude of Ca2+ transients, although in this case, a moderate, significant (P < 0.05) increase in the rate of Ca2+ signal decay was observed (n = 11). To estimate the rate of decay, we measured the half-width, which is the full duration of the signals at half-maximal amplitude. The half-width in control experiments averaged 282.6 ± 8.8 ms (n = 42), while in diazoxide plus NAC-treated myocytes, the half-width averaged 224.8 ± 8.0 ms (n = 11). NAC is a general antioxidant that reacts with hydroxyl radicals and with H2O2 (Aruoma et al., 1989). To determine the involvement of other ROS species, we tested the effect of Tiron (4,5-dihydroxy-1,3-benzene-disulphonic acid) which acts by scavenging superoxide (Ledenev et al., 1986; Krishna et al., 1992). In the presence of Tiron, diazoxide still significantly reduced the amplitude of Ca2+ signals, indicating that superoxide is probably not involved in its effects (n = 20). The action of diazoxide on peak ΔF/F values under different conditions is summarized in Figure 3C.
Figure 3.
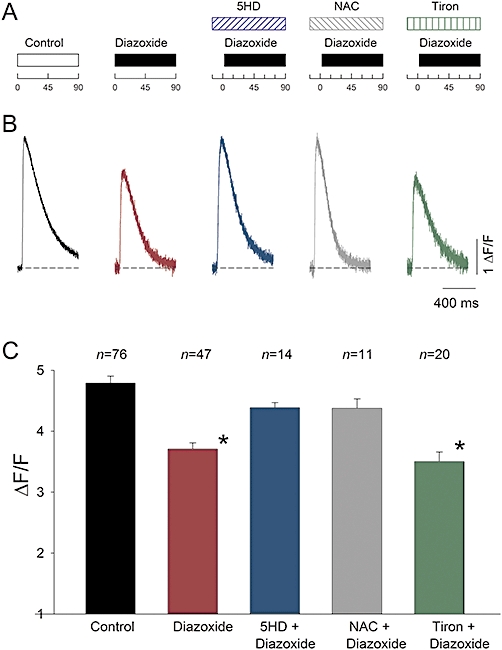
PPC reduces Ca2+ signals. A, experimental protocol. B, representative Fluo-3 Ca2+ transients recorded in Tyrode solution, expressed as ΔF/F, in response to extracellular stimulation, under conditions as indicated in A. Myocytes incubated for time (in min) indicated in A. Recordings were made immediately after incubation in control conditions, diazoxide (100 µM), diazoxide plus 5-HD (100 µM), diazoxide plus NAC (2 mM) or diazoxide plus Tiron (0.1 mM). Dotted line: basal fluorescence. C, mean peak ΔF/F values (±s.e.) as in B. Diazoxide alone and diazoxide plus Tiron significantly decreased peak ΔF/F compared with controls. Data from Tiron at 0.1 and 1 mM gave similar results and were pooled (*P < 0.05). The number of averaged experiments is indicated above each bar.
Diazoxide and ROS production
The involvement of ROS on diazoxide-induced reduction of L-type current amplitude, and on Ca2+ signals (Figures 1 and 3) was assessed further by determining if diazoxide increased ROS production. Quiescent cardiomyocytes were stained with CM-H2DCFDA, and ROS production measured during the preconditioning of myocytes with diazoxide. Figure 4 summarizes the results from several experiments, showing mean relative fluorescence values as a function of time. A significant increase in ROS production was observed in myocytes incubated in diazoxide-containing solution, compared with controls (n = 8–16). After 30 min of diazoxide incubation, ROS production was approximately 30% higher than that observed in myocytes incubated for the same time in reference solution (Figure 4). The increase occurred within a time frame consistent with the effects of diazoxide on L-type currents in Figure 1.
Figure 4.
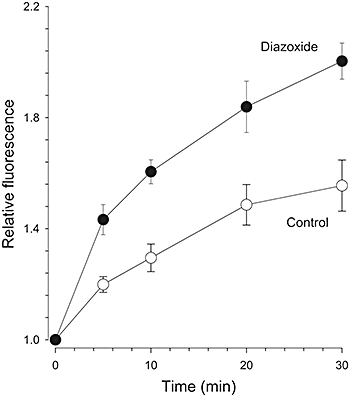
Effect of diazoxide on ROS production. Increase in relative CM-H2DCFDA fluorescence over time under control conditions (○) and in the presence of diazoxide, 100 µM (•). Data are means ± s.e. n = 8–16. The effect of diazoxide was significant for t > 0 min.
Effect of diazoxide on α1c subunit protein expression
The principal subunit of the L-type Ca2+ channel is α1c, so the PPC-induced decrease in Ca2+ current magnitude and Ca2+ transients might involve a decrease in the level of this protein at the cell membrane. Levels of α1c subunit in membrane fractions were measured by Western blot in hearts perfused with diazoxide, or under control conditions. Levels of the α1c subunit were significantly reduced by preincubation with diazoxide (Figure 5), consistent with the electrophysiological and Ca2+-transient experiments. Anti-α1c antibody recognized a single band of ∼210 kDa, the size of the cardiac α1c subunit in adult rat myocytes (Saada et al., 2005). The density of the α1c subunit band was greatly decreased in hearts after perfusion with diazoxide for 90 min. In contrast, the level of α-actin in the same membrane extracts was not significantly modified by diazoxide perfusion (Figure 5). The lack of effect of diazoxide on α-actin was observed in 14 independent experiments. Density values (in arbritary units) averaged 28.8 ± 3.6 in control experiments and 29.4 ± 4.1 in diazoxide-pretreated hearts. In contrast, the decrease in α1c subunit levels after 90 min preincubation in diazoxide was highly significant (Figure 8). Although the reduction in channel current amplitude was almost complete after 30 min in diazoxide, complete downregulation of the α1c subunit was not observed at this early time, so changes in channel function preceded changes in protein levels (Figures 1 and 8).
Figure 5.
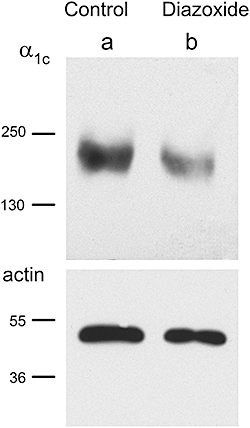
PPC downregulates the α1c subunit. Representative blots with anti-α1c antibody showing a single ∼ 210 kDa band. Band intensity was strong under control conditions (a) and weak in 90 min diazoxide-pretreated preparations (b). Corresponding blots probed with anti-α-actin are also shown.
Figure 8.
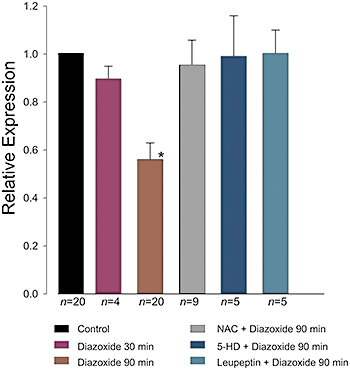
PPC and the α1c subunit. Mean of relative densities (±SE) of the α1c subunit band normalized to the density of α-actin band, under the experimental conditions indicated. Density values were significantly lower after incubation with diazoxide (100 µM) for 90 min (P < 0.05). Immunoblots with 5-HD and leupeptin were made from myocytes, all others from isolated hearts. The number of experiments in each condition is indicated below the bars.
The haemodynamic function of ischaemia-reperfused hearts with and without diazoxide was assessed in parallel experiments. After 35 min reperfusion, diazoxide-treated hearts showed significantly improved contractile function compared with control ischaemic hearts (Table 1).
Table 1.
The effect of PPC on heart function
| Pre-ischaemic | Reperfused | |||||
|---|---|---|---|---|---|---|
| Control | Diazoxide | Nifedipine | Control | Diazoxide | Nifedipine | |
| n | 8 | 10 | 9 | 3 | 3 | 3 |
| LVEDP (mmHg) | 2.1 ± 0.6 | 3.9 ± 1.2 | 3.5 ± 6.0 | 52.5 ± 12.6* | 25.5 ± 11.5* | 24.1 ± 12.2 |
| LVDP (mmHg) | 96.6 ± 3.4 | 88.5 ± 5.3 | 78.7 ± 4.5# | 14.9 ± 5.6* | 61.2 ± 4.6* | 57.2 ± 4.9 |
| dP/dtmax (mmHg·s−1) | 2477 ± 127 | 2467 ± 132 | 1805 ± 67# | 454 ± 174* | 1720 ± 357* | 1494 ± 100 |
| dP/dtmin (mmHg·s−1) | 1532 ± 89 | 1531 ± 81 | 1305 ± 64 | 384 ± 124* | 1164 ± 182 | 1131 ± 97 |
| LDH (%) | 100 | 33 ± 8.0# | 46 ± 10.0# | |||
P < 0.05 vs. control.
P < 0.05 vs. pre-ischaemic baseline.
The decrease in α1c subunit level caused by preincubation with diazoxide depends on ROS production, as seen by Western blot. Figure 6 shows representative blots of α1c subunit and α-actin. Consistent with the results in Figure 5, diazoxide drastically reduced the α1c subunit band density and this reduction was fully prevented by NAC (n = 9). NAC by itself had no effect on the α1c subunit (n = 10) (Figure 6).
Figure 6.
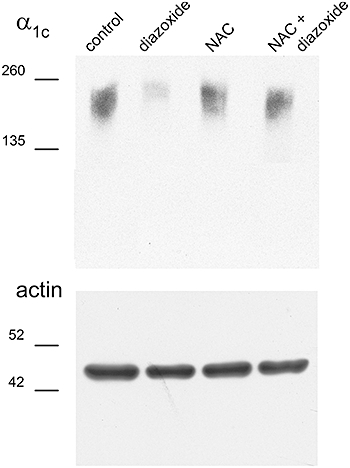
Downregulation of the α1c subunit is prevented by ROS scavengers. Representative α1c subunit and α–actin blots. From left to right: control conditions, isolated heart perfused with diazoxide (100 µM), isolated heart perfused with NAC (4 mM), and isolated heart perfused with diazoxide plus NAC.
Effect of protease inhibitors on Ca2+channel expression decline, Ca2+ currents and Ca2+ signals during PPC
The decrease in Ca2+ channel expression and activity produced by diazoxide might result from proteolytic degradation of the α1c subunit. Therefore, we tested the effect of adding the protease inhibitor leupeptin to the perfusate for Western blots, and electrophysiological and Ca2+ signal measurements. Leupeptin greatly prevented downregulation of the α1c subunit by diazoxide, as revealed by Western blot from myocytes preincubated with the protease inhibitor for 60 min (n = 3) (Figure 7A). Leupeptin also largely prevented the near maximal decrease in L-type currents by diazoxide seen when cardiomyocytes were preincubated in diazoxide for 30 min (Figure 7B, n = 22). Similar results were obtained when Ca2+ signals were recorded after 90 min in diazoxide. Leupeptin partially prevented the effects of diazoxide on Ca2+ transients (Figure 7C & D; n = 35), though by itself had no effect on Ca2+ signals (data not shown).
Figure 7.
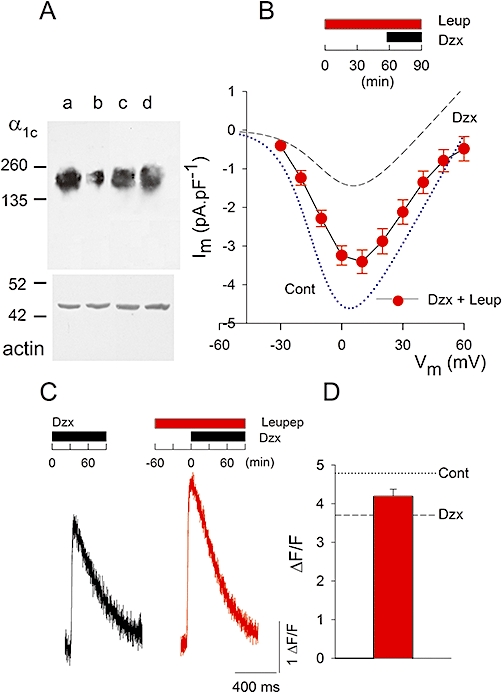
Leupeptin prevents downregulation of the α1c subunit, and reduction of L-type currents and Ca2+ signals by PPC. A, representative α1c subunit and α−actin blots. Lane a: control conditions, lane b: myocytes perfused with diazoxide (100 µM), lane c: myocytes perfused with leupeptin (100 µM), lane d: myocytes perfused with diazoxide (100 µM) plus leupeptin (100 µM). Protocol was as in panel C. B, current-voltage relationship of L-type Ca2+ channels recorded with the protocol shown above. Data shown are mean values of peak currents ± s.e. (n = 22) from diazoxide (Dzx) plus leupeptin (Leup; 100 µM)-pretreated myocytes. Dotted line: best fit of equation (1) to the current-voltage relationship of peak currents from control myocytes (Cont; from Figure 1E) with parameters: Gmax= 100 pS·pF−1, V =−11.1 mV, Vrev= 61.4 mV and k = 8.6 mV. Dashed line: corresponding fit to peak currents from diazoxide pretreated myocytes (Dzx; from Figure 1E) with parameters: Gmax= 60 pS·pF−1, V = 3.6 mV, Vrev= 40.7 mV and k = 9.8 mV. C, representative Fluo-3 Ca2+ transients, expressed as ΔF/F, in response to extracellular stimulation, under conditions shown above. D, mean value of peak ΔF/F± s.e. (n = 35) from myocytes preincubated with diazoxide plus leupeptin. Dotted line: mean of peak ΔF/F from controls (from Figure 3C). Dashed line: corresponding value from diazoxide pretreated myocytes (from Figure 3C).
Figure 8 summarizes the results from the Western blots, showing that perfusion with diazoxide for 90 min reduced the density of the α1c subunit band to approximately 50% of the control value, and this effect was fully prevented by NAC, 5-HD and leupeptin. These inhibitors by themselves had no significant effect on the density of the α1c subunit band (data not shown).
PPC and Ca2+ channels
The decrease in Ca2+ channel expression and Ca2+ channel activity caused by diazoxide might play a role in PPC, in which case, protection against ischaemia by diazoxide should be blocked if channel downregulation is prevented by NAC or 5-HD (Figures 1,3 and 5). On the other hand, protection against ischaemia would be expected if Ca2+ channels were partially blocked during reperfusion by specific drugs. Therefore, we confirmed that perfusion with diazoxide led to PPC by exposing control and diazoxide-pretreated hearts to severe ischaemia. Figure 9 A shows a cross-section of an infarcted heart perfused with control solution before ischaemia. Extensive areas of infarction are evident as light-coloured areas. When an isolated heart was perfused with diazoxide prior to ischaemia, infarcted areas were greatly reduced. Consistent with a role for channel downregulation in PPC, protection by diazoxide was abolished when NAC or the KATP channel blocker 5-HD was added to the perfusate. NAC and 5-HD by themselves had no effect on infarction areas (data not shown).
Figure 9.
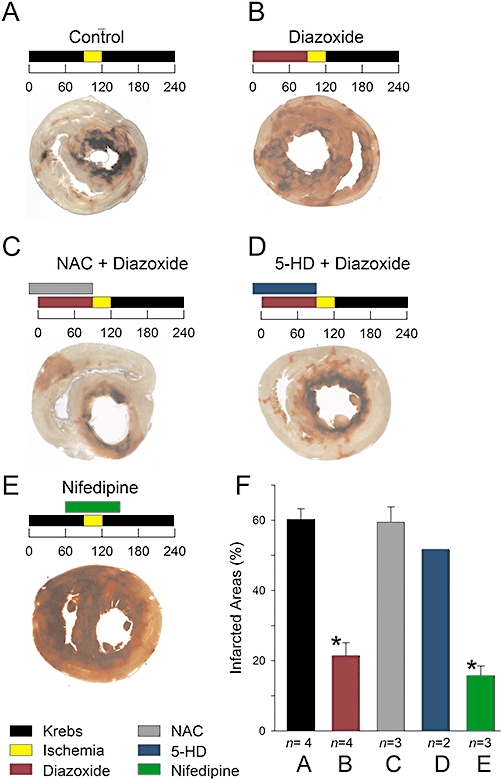
PPC and infarction areas. A–E, infarction areas (light areas) after severe ischaemia in cross-sections of isolated ventricles under conditions as indicated above. Numbers are time in minutes. Concentrations were 100 µM diazoxide, 4 mM NAC, 100 µM 5-HD, and 30 nM nifedipine. Ventricles were dyed with 2,3,5-triphenyltetrazolium chloride. F, infarction areas under indicated conditions corresponding to panels A–E. Data are the mean relative infarcted area (±s.e.) with the number of experiments indicated below. Infarcted area values from hearts perfused with diazoxide and nifedipine were significantly lower than those from control hearts (*P < 0.05).
Finally, the specific L-type Ca2+ channel blocker nifedipine greatly protected against infarction. Nifedipine-treated hearts also showed excellent recovery of haemodynamic function after ischaemia. LVDP values recovered to 79 ± 4% (n = 3) after 35 min of reperfusion. In contrast, ischaemic control hearts recovered to only 15 ± 5% (n = 3) at a similar period of time. Likewise, dP/dtmax and dP/dtmin values recovered to 86 ± 2% and 85 ± 4% (n = 3) in hearts reperfused with nifedipine and to 18 ± 5% and 27 ± 7% (n = 3) in ischaemic control hearts. We also confirmed that LDH loss was significantly reduced in hearts subjected to PPC, compared with the loss from ischaemic control hearts. In three separate experiments, diazoxide significantly reduced (P < 0.05) LDH loss to 33 ± 8% of the value for ischaemic control hearts. Likewise, nifedipine during reperfusion reduced LDH loss to 46 ± 10% of control values (n = 3). For both LDH and infarction area measurements, 30 nM nifedipine was used to block L-type Ca2+ current in adult rat ventricular myocytes to 50% of the control value (Pignier and Potreau, 2000). Diazoxide reduced the magnitude of L-type currents and Ca2+ channel expression to approximately this level (Figures 1 and 8).
Discussion
Comparison with previous work
Diazoxide opens mitoKATP channels (Garlid et al., 1996; Liu et al., 1998), and protects against ischaemia (Wang et al., 1999; Sato et al., 2000; Wang et al., 2001; Pasdois et al., 2008; this paper; also see Ardehali and O'Rourke, 2005). MitoKATP channels play a key role in cardioprotection during IPC (Ardehali and O'Rourke, 2005; Halestrap et al., 2007). We present the novel result that downregulation of the principal subunit of the adult heart L-type Ca2+ channel is a consequence of diazoxide activation of mitoKATP channels.
PPC and Ca2+ channels
The amplitude of L-type Ca2+ currents decreased after incubation with diazoxide. A similar decrease was observed when Ba2+ was used as the charge carrier, indicating that this effect cannot be explained by changes in Ca2+-dependent inactivation, as this is a major contributor to the decline of L-type currents when Ca2+ is the permeant ion (Findlay et al., 2008). Although previous work showed that the L-type Ca2+ channel is a target for anaesthetic-induced preconditioning (Tampo et al., 2009), the mechanism of diazoxide is fundamentally different. An acceleration in the Ca2+-dependent inactivation of L-type channels was observed after isoflurane exposure, with no changes in voltage-dependent inactivation or current magnitude (Tampo et al., 2009), while diazoxide caused a large decrease in current density values and α1c subunit protein levels in our experiments. In addition, it caused an increase in voltage-dependent inactivation of the channels. Clearly, the L-type channel can be modulated in various ways by PPC.
Ca2+ entry via the L-type Ca2+ channel is the dominant Ca2+ source for the CICR of the sarcoplasmic reticulum (Bers, 2002), so downregulation of the channel protein and the associated reduction in current magnitude should decrease intracellular Ca2+ release during action potentials. Our experiments confirmed this prediction, although the decrease in Ca2+ signal amplitude was smaller than that observed for the Ca2+ current. This finding was not entirely unexpected as Altamirano and Bers (2007) have shown that when the amplitude of the macroscopic Ca2+ current decreases, the CICR amplification factor (the gain of excitation-contraction coupling) increases. The increase in gain is proposed to be due to a reduction in redundant Ca2+ channel opening, which may contribute to our findings. On the other hand, our results do not agree with Katoh et al. (2002), who described an increase in Ca2+ transient amplitude induced by diazoxide. However, their experiments involved high drug concentrations, at which toxic effects on mitochondrial function, leading to release of mitochondrial Ca2+ via a cyclosporin A sensitive pathway, are likely, as pointed out by Katoh et al. (2002).
The role of ROS
We found that diazoxide increased ROS production within minutes and that the downregulation of the α1c subunit by PPC was prevented by the ROS scavenger NAC. These observations suggest that ROS play a role in downregulation of the Ca2+ channel. Previous reports revealed that diazoxide and other mitoKATP channel openers increase ROS production (Forbes et al., 2001; Oldenburg et al., 2003; Pasdois et al., 2008), and a modest increase in ROS production is beneficial, since IPC is prevented when a ROS scavenger is present during the preconditioning phase (Forbes et al., 2001; Halestrap et al., 2007). The production of free radicals after the opening of mitoKATP channels is proposed to trigger the preconditioned state (Pain et al., 2000). However, the increase in ROS production by diazoxide is not necessarily mediated by mitoKATP channel opening, as described by Dröse et al. (2006). The targets for ROS have not been fully identified, but oxygen radicals are proposed to contribute to IPC by activating protein kinase C (Baines et al., 1997). ROS are likely to have several targets in preconditioning, including the Ca2+ channel, according to our results. In agreement, free radicals decrease the number of [H3]-nitrendipine binding sites in isolated heart membranes (Kaneko et al., 1989).
Physiological significance of channel downregulation
Long periods of ischaemia result in large increases in cytosolic Ca2+ concentration during reperfusion (Lee and Allen, 1992) and this rise is generally thought to precede muscle damage. Furthermore, reduction in this increase improves myocardial survival (see Murphy and Steenbergen, 2008). Diazoxide-induced decrease in Ca2+ influx and subsequent downregulation of the α1c subunit may, therefore, contribute to cardioprotection. Normally, Ca2+ efflux in heart cells is predominantly by the Na+/Ca2+ exchange mechanism, with a minor contribution from the sarcolemmal Ca2+ pump (Bers, 2002). During ischaemia-reperfusion, the deleterious increase in intracellular Ca2+ concentration appears to result from the reverse operation of the Na+/Ca2+ exchange mechanism associated with intracellular acidosis and membrane depolarization (Murphy and Steenbergen, 2008). Factors that diminish the increase in intracellular Ca2+ would be expected to attenuate myocardial injury, so downregulation of Ca2+ channels should be beneficial since, even if the Na+/Ca2+ exchange mechanism is not extruding Ca2+ as efficiently as under normal conditions, reduced Ca2+ channel influx would decrease Ca2+ overload. Our results agree with this as we observed protection against infarction with nifedipine, a selective blocker of L-type Ca2+ channels. Furthermore, preventing channel downregulation with NAC or 5-HD resulted in lack of protection.
The role of L-type Ca2+ channels in altering Ca2+ influx as a mechanism of infarct size reduction during preconditioning is unclear. Wallbridge et al. (1996) described no changes in infarct size when nisoldipine was administered to pigs by intravenous infusion through the ischaemic period. Likewise, LDH levels in the coronary effluent of Langendorff-perfused rat hearts subjected to global ischaemia did not change when Ca2+ channels were blocked by infusion with verapamil, with washing before the ischaemic-reperfusion period (Miyawaki et al., 1996). Mocanu et al. (1999) found that amlodipine does not reduce infarct size when administered in the perfusate until just after coronary occlusion. In all these examples, Ca2+ channel blockers were washed out before reperfusion, so Ca2+ influx, and consequently Ca2+ overload, remained unaltered during this period. In contrast, we infused nifedipine up to 30 minutes after the start of reperfusion, to reduce the influx of Ca2+ when Ca2+ overload takes place. A relevant finding is that verapamil and the Ca2+ channel blocker Ro-40-5967 significantly reduced infarct size after local ischaemia in dogs, when the drugs were administered before and during ischaemia, and through the first 25 min of reperfusion (Vander Heide et al., 1994). Finally, continuous administration of nifedipine and other Ca2+ channel blockers improved postischaemic recovery of isolated guinea pig hearts (Becker and Möbert, 1999).
MitoKATP channels and L-type Ca2+ channels
Our experiments indicated that downregulation of the Ca2+ channel followed the opening of mitoKATP channels. MitoKATP channels are suggested to be downstream regulators of myocardial protection with beneficial effects lasting into the reperfusion period (Fryer et al., 2001). After the ischaemic period, the increase in intracellular Ca2+ concentration is generally thought to be an early event in reperfusion (Murphy and Steenbergen, 2008). This does not necessarily argue against downregulation of the Ca2+ channel by diazoxide as a factor in Ca2+ overload reduction, since we found that only minutes are required for the reduction in Ca2+ channel current amplitude and channel downregulation. Therefore, a significant drop in the fraction of active channels may have already occurred at the beginning of reperfusion after PPC, thus reducing the Ca2+ overload.
Changes in channel surface expression
Our results provide evidence for the involvement of a protease in downregulation of cardiac L-type channels during PPC. A single band corresponding to the intact α1c subunit decreased in density in our immunoblots, with no cleaved proteolytic products containing the epitope apparent in the membrane fraction. This does not rule out the possibility that multiple regions of the α1c subunit are cleaved by PPC. In fact, we observed changes in channel activity at times earlier than those observed by immunoblot, suggesting that the integrity of the channel is already being compromised. The particular protease involved and the exact mechanism of proteolysis remain to be determined. A possible candidate is calpain, a Ca2+-dependent protease. The 10a isoform is the most abundant calpain, and is expressed in heart (Goll et al., 2003). Calpain is fully inhibited by leupeptin at 100 µM, and is activated during inhibition of metabolism in cardiomyocytes (Atsma et al., 1995). Furthermore, calpain-dependent proteolytic events take place in rat hearts after brief ischaemic insults (Yoshida et al., 1995) and calpains are involved in degradation of ryanodine receptors during cardiac ischaemic reperfusion (Pedrozo et al., 2010).
Rapid degradation of proteins follows oxidative stress by ROS and oxidative modifications are accompanied by protein malfunction, possibly through direct modification in one of the protein domains required for function (Jung et al., 2007). Slightly oxidized proteins first show decreased activity and, after being subsequently defolded, they become ideal targets for degradation (Jung et al., 2007). The observation that the decrease in the amplitude of L-type currents preceded the decrease in α1c subunit expression is consistent with this view. The α1c subunit of the L-type Ca2+ channel contains many cysteine residues (Hool, 2008), likely to be targets of redox modification of proteins, as free thiols can easily react with ROS (Hool, 2008). Furthermore, we observed that currents not only decreased in size by diazoxide as expected by downregulation of the α1c subunit but also showed an enhanced voltage-dependent inactivation indicating alterations in channel function. Increased inactivation is expected to reduce current size which may explain the fact that the decrease in current magnitude was distinctly larger than the decrease in α1c subunit expression. The possibility that changes in channel function and in the level of the α1c subunit by PP have a common origin or alternatively, that diazoxide has additional effects on L-type currents would require further experimentation.
In conclusion, our results indicate that PPC reduces Ca2+ influx during reperfusion via increased L-type channel degradation, which protects the heart from injury caused by Ca2+ overload.
Acknowledgments
We thank J.M. Galindo for advice and O. Ramirez and E. Delgado for technical assistance. G. Gonzalez was supported by a fellowship from CONACYT, Mexico. We thank Dr Ariel Escobar for relevant discussions. This work was supported in part by CONACYT, grant number: UI60880.
Glossary
Abbreviations
- 5-HD
5-hydroxydecanoate
- IPC
ischaemic preconditioning
- mitoKATP
mitochondrial ATP-sensitive K+ channels
- NAC
N-acetyl-L-cysteine
- PPC
pharmacological preconditioning
- ROS
reactive oxygen species
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamirano J, Bers DM. Voltage dependence of cardiac excitation-contraction coupling: unitary Ca2+ current amplitude and open channel probability. Circ Res. 2007;101:590–597. doi: 10.1161/CIRCRESAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- Ardehali H, O'Rourke B. Mitochondrial KATP channels in cell survival and death. J Mol Cell Cardiol. 2005;39:7–16. doi: 10.1016/j.yjmcc.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- Atsma DE, Bastiaanse EML, Jerzewski A, Van der Valk LJM, Van der Laarse A. Role of calcium-activated neutral protease (Calpain) in cell death in cultured neonatal rat cardiomyocytes during metabolic inhibition. Circ Res. 1995;76:1071–1078. doi: 10.1161/01.res.76.6.1071. [DOI] [PubMed] [Google Scholar]
- Baines CP, Goto M, Downey JM. Oxygen radicals released during ischemic preconditioning contribute to cardioprotection in the rabbit myocardium. J Mol Cell Cardiol. 1997;29:207–216. doi: 10.1006/jmcc.1996.0265. [DOI] [PubMed] [Google Scholar]
- Becker BF, Möbert J. Low-dose calcium antagonists reduce energy demand and cellular damage of isolated hearts during both ischemia and reperfusion. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:287–294. doi: 10.1007/s002109900053. [DOI] [PubMed] [Google Scholar]
- Bers DM. Cardiac excitation–contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- Bodi I, Mikala G, Koch SE, Akhter SA, Sch A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005;115:3306–3317. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchez P, Sarre A, Kappenberger L, Raddatz E. The L-type Ca2+ and KATP channels may contribute to pacing-induced protection against anoxia-reoxygenation in the embryonic heart model. J Cardiovasc Electrophysiol. 2008;19:1196–1202. doi: 10.1111/j.1540-8167.2008.01218.x. [DOI] [PubMed] [Google Scholar]
- Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- Dessy C, Godfraind T. The effect of L-type calcium channel modulators on the mobilization of intracellular calcium stores in guinea-pig intestinal smooth muscle. Br J Pharmacol. 1996;119:142–148. doi: 10.1111/j.1476-5381.1996.tb15687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröse S, Brandt U, Hanley PJ. K+-independent actions of diazoxide question the role of inner membrane KATP channels in mitochondrial cytoprotective signaling. J Biol Chem. 2006;281:23733–23739. doi: 10.1074/jbc.M602570200. [DOI] [PubMed] [Google Scholar]
- Findlay I, Suzukib S, Murakami S, Kurachi Y. Physiological modulation of voltage-dependent inactivation in the cardiac muscle L-type calcium channel: A modelling study. Prog Biophys Mol Biol. 2008;96:482–498. doi: 10.1016/j.pbiomolbio.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Forbes RA, Steenbergen C, Murphy E. Diazoxide-induced cardioprotection requires signaling through a redox-sensitive mechanism. Circ Res. 2001;88:802–809. doi: 10.1161/hh0801.089342. [DOI] [PubMed] [Google Scholar]
- Fryer RM, Hsu AK, Gross GJ. Mitochondrial KATP channel opening is important during index ischemia and following myocardial reperfusion in ischemic preconditioned rat hearts. J Mol Cell Cardiol. 2001;33:831–834. doi: 10.1006/jmcc.2001.1350. [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D, Theroux P, Elizaga J, Galinanes M, Solares J, Riesgo M, et al. Myocardial reperfusion in the pig heart model: infarct size and duration and coronary occlusion. Cardiovasc Res. 1987;21:537–544. doi: 10.1093/cvr/21.7.537. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as receptor for potassium channel openers. J Biol Chem. 1996;271:8796–8799. doi: 10.1074/jbc.271.15.8796. [DOI] [PubMed] [Google Scholar]
- Garlid KD, Paucek P, Yarov-Yarovoy V, Murray HN, Darbenzio RB, D'Alonzo AJ, et al. Cardioprotective effect of diazoxide and its interaction with mitochondrial ATP-sensitive K+ channels. Circ Res. 1997;81:1072–1082. doi: 10.1161/01.res.81.6.1072. [DOI] [PubMed] [Google Scholar]
- Goll DE, Thompson VF, Li H, Wei W. The Calpain System. Physiol Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- Gomez JP, Potreau D, Branka JE, Raymond G. Developmental changes in Ca2+ currents from newborn rat cardiomyocytes in primary culture. Pflugers Arch. 1994;428:241–249. doi: 10.1007/BF00724503. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Clarke SJ, Khaliulin I. The role of mitochondria in protection of the heart by preconditioning. Biochim Biophys Acta. 2007;1767:1007–1031. doi: 10.1016/j.bbabio.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC. Evidence for the regulation of L-type Ca2+ channels in the heart by reactive oxygen species: mechanism for mediating pathology. Clin Exp Pharmacol Physiol. 2008;35:229–234. doi: 10.1111/j.1440-1681.2007.04727.x. [DOI] [PubMed] [Google Scholar]
- Hu H, Sato T, Seharaseyon J, Liu Y, Johns DC, O'Rourke B, et al. Pharmacological and histochemical distinctions between molecularly defined sarcolemmal KATP channels and native cardiac mitochondrial KATP channels. Mol Pharmacol. 1999;55:1000–1005. [PubMed] [Google Scholar]
- Jung T, Bader N, Grune T. Oxidized proteins: Intracellular distribution and recognition by the proteasome. Arch Biochem Biophys. 2007;462:231–237. doi: 10.1016/j.abb.2007.01.030. [DOI] [PubMed] [Google Scholar]
- Kaneko M, Lee SL, Wolf CM, Dhalla NS. Reduction of calcium channel antagonist binding sites by oxygen free radicals in rat heart. J Mol Cell Cardiol. 1989;21:935–943. doi: 10.1016/0022-2828(89)90761-x. [DOI] [PubMed] [Google Scholar]
- Katoh H, Nishigaki N, Hayashi H. Diazoxide opens the mitochondrial permeability transition pore and alters Ca2+ transients in rat ventricular myocytes. Circulation. 2002;105:2666–2671. doi: 10.1161/01.cir.0000016831.41648.04. [DOI] [PubMed] [Google Scholar]
- Klein HH, Puschmann S, Schaper J, Schaper W. The mechanism of the tetrazolium reaction in identifying experimental myocardiai infarction. Virchows Arch. 1981;393:287–297. [Google Scholar]
- Krishna CM, Libmann JE, Kaufman D, DeGraff W, Hahn SM, McMurry T, et al. The catecholic metal sequestering agent 1,2-dihydroxybenzene-3,5-disulfonate confers protection against oxidative cell damage. Arch Biochem Biophys. 1992;294:98–106. doi: 10.1016/0003-9861(92)90142-j. [DOI] [PubMed] [Google Scholar]
- Lacinová L. Voltage-dependent calcium channels. Gen Physiol Biophys. 2005;24(Suppl 1):1–78. [PubMed] [Google Scholar]
- Ledenev AN, Konstantinov AA, Popova E, Ruuge EK. A simple assay of the superoxide generation rate with Tiron as an EPR-visible radical scavenger. Biochem Int. 1986;13:391–396. [PubMed] [Google Scholar]
- Lee JA, Allen DG. Changes in intracellular free calcium concentration during long exposures to simulated ischemia in isolated mammalian ventricular muscle. Circ Res. 1992;71:58–69. doi: 10.1161/01.res.71.1.58. [DOI] [PubMed] [Google Scholar]
- Liu Y, Sato T, O'Rourke B, Marban E. Mitochondrial ATP-dependent potassium channels: Novel effectors of cardioprotection? Circulation. 1998;97:2463–2469. doi: 10.1161/01.cir.97.24.2463. [DOI] [PubMed] [Google Scholar]
- Miyawaki H, Zhou X, Ashraf M. Calcium preconditioning elicits strong protection against ischemic injury via protein kinase C Signaling Pathway. Circ Res. 1996;79:137–146. doi: 10.1161/01.res.79.1.137. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Gadgil S, Yellon DM, Baxter GF. Mibefradil, a T-type and L-type calcium channel blocker, limits infarct size through a Glibenclamide-sensitive mechanism. Cardiovasc Drugs Ther. 1999;13:115–122. doi: 10.1023/a:1007732025184. [DOI] [PubMed] [Google Scholar]
- Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- Oldenburg O, Yang XM, Krieg T, Garlid KD, Cohen MV, Grover GJ, et al. P1075 opens mitochondrial KATP channels and generates reactive oxygen species resulting in cardioprotection of rabbir hearts. J Mol Cell Cardiol. 2003;35:1035–1042. doi: 10.1016/s0022-2828(03)00151-2. [DOI] [PubMed] [Google Scholar]
- Pain T, Yang XM, Critz SD, Yue Y, Nakano A, Liu GS, et al. Opening of mitochondrial KATP channels triggers the preconditioned state by generating free radicals. Circ Res. 2000;87:460–466. doi: 10.1161/01.res.87.6.460. [DOI] [PubMed] [Google Scholar]
- Pasdois P, Beauvoit B, Tariosse L, Vinassa B, Bonoron-Adele S, Dos Santos P. Effect of diazoxide on flavoprotein oxidation and reactive oxygen species generation during ischemia-reperfusion: a study on Langendorff-perfused rat hearts using optic fibers. Am J Physiol Heart Circ Physiol. 2008;294:H2088–H2097. doi: 10.1152/ajpheart.01345.2007. [DOI] [PubMed] [Google Scholar]
- Pedrozo Z, Sánchez G, Torrealba N, Valenzuela R, Fernández C, Hidalgo C, et al. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of ischemic reperfusion. Biochem Biophys Acta. 2010;1802:356–362. doi: 10.1016/j.bbadis.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Pignier C, Potreau D. Characterization of nifedipine-resistant calcium current in neonatal rat ventricular cardiomyocytes. Am J Physiol Heart Circ Physiol. 2000;279:H2259–H2268. doi: 10.1152/ajpheart.2000.279.5.H2259. [DOI] [PubMed] [Google Scholar]
- Rijstenbil JW, Coelho SM, Eijsackers M. A method for the assessment of light-induced oxidative stress in embryos of fucoid algae via confocal laserscan microscopy. Mar Biol. 2000;137:763–774. [Google Scholar]
- Saada N, Carrillo E, Bosong D, Wang W, Dettbarn C, Sanchez JA, et al. Expression of multiple Cav1.2 transcripts in rat tissues mediated by different promoters. Cell Calcium. 2005;37:301–309. doi: 10.1016/j.ceca.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Sánchez JA, García MC, Sharma VK, Young KC, Matlib MA, Sheu SS. Mitochondria regulate inactivation of L-type Ca2+ channels in rat heart. J Physiol. 2001;536:387–396. doi: 10.1111/j.1469-7793.2001.0387c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Sasaki N, Seharaseyon J, O'Rourke B, Marbán E. Selective pharmacological agents implicate mitochondrial but not sarcolemmal KATP channels in ischemic cardioprotection. Circulation. 2000;101:2418–2423. doi: 10.1161/01.cir.101.20.2418. [DOI] [PubMed] [Google Scholar]
- Striessnig J. Pharmacology, structure and function of cardiac L-type Ca2+ channels. Cell Physiol Biochem. 1999;9:242–269. doi: 10.1159/000016320. [DOI] [PubMed] [Google Scholar]
- Tampo A, Hogan CS, Sedlic F, Bosnjak ZJ, Kwok WM. Accelerated inactivation of cardiac L-type calcium channels triggered by anesthetic-induced preconditioning. Br J Pharmacol. 2009;156:432–443. doi: 10.1111/j.1476-5381.2008.00026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Tovey SC, Collins TJ, Bootman MD, Berridge MJ, Lipp P. A comparison of fluorescent Ca2+ indicator properties and their use in measuring elementary and global Ca2+ signals. Cell Calcium. 2000;28:213–233. doi: 10.1054/ceca.2000.0152. [DOI] [PubMed] [Google Scholar]
- Vander Heide RS, Schwartz LM, Reimer KA. The novel calcium antagonist Ro 40-5967 limits myocardial infarct size in the dog. Cardiovasc Res. 1994;28:1526–1532. doi: 10.1093/cvr/28.10.1526. [DOI] [PubMed] [Google Scholar]
- Vicencio JM, Ibarra C, Estrada M, Chiong M, Soto D, Parra V, et al. Testosterone induces an intracellular calcium increase by a nongenomic mechanism in cultured rat cardiac myocytes. Endocrinology. 2006;147:1386–1395. doi: 10.1210/en.2005-1139. [DOI] [PubMed] [Google Scholar]
- Wallbridge DR, Schulz R, Braun C, Post H, Heusch G. No attenuation of ischemic preconditioning by the calcium antagonist nisoldipine. J Mol Cell Cardiol. 1996;28:1801–1810. doi: 10.1006/jmcc.1996.0169. [DOI] [PubMed] [Google Scholar]
- Wang S, Cone J, Liu Y. Dual roles of mitochondrial KATP channels in diazoxide-mediated protection in isolated rabbit hearts. Am J Physiol Heart Circ Physiol. 2001;280:246–255. doi: 10.1152/ajpheart.2001.280.1.H246. [DOI] [PubMed] [Google Scholar]
- Wang Y, Hirai K, Ashraf M. Activation of mitochondria ATP-sensitive K+ channels for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ Res. 1999;85:731–741. doi: 10.1161/01.res.85.8.731. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Inui M, Harada K, Saido TC, Sorimachi Y, Ishihara T, et al. Reperfusion of rat heart after brief ischemia induces proteolysis of calspectin (nonerythroid spectrin or fodrin) by Calpain. Circ Res. 1995;77:603–610. doi: 10.1161/01.res.77.3.603. [DOI] [PubMed] [Google Scholar]