

Abstract
The tumor suppressor, phosphatase, and tensin homologue deleted on chromosome 10 (PTEN), is a phosphoinositide (PI) phosphatase specific for the 3-position of the inositol ring. PTEN has been implicated in autism for a subset of patients with macrocephaly. Various studies identified patients in this subclass with one normal and one mutated PTEN gene. We characterize the binding, structural properties, activity, and subcellular localization of one of these autism-related mutants, H93R PTEN. Even though this mutation is located at the phosphatase active site, we find that it affects the functions of neighboring domains. H93R PTEN binding to phosphatidylserine-bearing model membranes is 5.6-fold enhanced in comparison to wild-type PTEN. In contrast, we find that binding to phosphatidylinositol-4,5-bisphosphate (PI(4,5)P2) model membranes is 2.5-fold decreased for the mutant PTEN in comparison to wild-type PTEN. The structural change previously found for wild-type PTEN upon interaction with PI(4,5)P2, is absent for H93R PTEN. Consistent with the increased binding to phosphatidylserine, we find enhanced plasma membrane association of PTEN-GFP in U87MG cells. However, this enhanced plasma membrane association does not translate into increased PI(3,4,5)P3 turnover, since in vivo studies show a reduced activity of the H93R PTEN-GFP mutant. Because the interaction of PI(4,5)P2 with PTEN's N-terminal domain is diminished by this mutation, we hypothesize that the interaction of PTEN's N-terminal domain with the phosphatase domain is impacted by the H93R mutation, preventing PI(4,5)P2 from inducing the conformational change that activates phosphatase activity.
Keywords: lipid phosphatase, phosphatidylinositol, conformational change, infrared spectroscopy, fluorescence quenching, confocal microscopy
Introduction
A tumor suppressor on human chromosome 10 plays a major role in the development of gliomas. This tumor suppressor was cloned and is now known as phosphatase and tensin homologue deleted on chromosome 10 (PTEN).1–3 Deletions or mutations have been detected in many tumor types, identifying PTEN as the second most commonly mutated tumor suppressor in human cancer.4–6 The PTEN protein is a phosphatidylinositol phosphate phosphatase specific for the 3-position of the inositol ring.7 PTEN and phosphoinositide 3-kinase (PI3K) have opposing effects on phosphatidylinositol (3,4,5) trisphosphate (PI(3,4,5)P3) levels and, consequently, on cell proliferation and survival.8,9 PI(3,4,5)P3 mediates these processes by inducing phosphorylation and activation of the Akt kinase.10
PTEN's structure consists of a short N-terminal PI(4,5)P2-binding domain, a phosphatase domain rich in α-helix, a C2 domain dominated by β-sheet, and a C-terminal tail with several phosphorylation sites.11,12 The C2 domain stabilizes the phosphatase domain through interdomain contacts and aids the targeting of the protein to the bilayer-water interface by Ca2+-independent binding to phospholipids, including phosphatidylserine (PS).12,13 The tail plays a regulatory role.14 When phosphorylated, the tail inhibits phosphatase activity, probably by binding to the active site by a pseudo substrate mechanism.15,16
Recently, we and others have proposed a model for regulation of PTEN by PI(4,5)P2.17–19 Efficient action of PTEN requires three steps. First, PTEN binds to PI(4,5)P2 and PS in the membrane. Second, PI(4,5)P2 induces a conformational change that increases the α-helical content and activates the phosphatase domain.19 Binding of the C2 domain to PS is associated with a structural change that leads to a slightly increased β-sheet content.19 Although important for binding to the membrane, this structural change appears to have no effect on phosphatase activity. Third, PTEN diffuses on the surface of the membrane and hydrolyzes PI(3,4,5)P3. Two approaches have demonstrated the consequences of the PI(4,5)P2-induced conformational change. First, PI(4,5)P2 enhances PTEN phosphatase activity in in vitro assays.20,21 Furthermore, mutations of the N-terminus (including single point mutations, such as K13E) that eliminate PI(4,5)P2 binding also decrease phosphatase activity.19,20,22 Second, the PI(4,5)P2-binding domain is required for membrane binding and biological function in cells.16,23
Autism spectrum disorder is a family of diseases with impaired communication and social interactions. Although environmental factors or autoimmune responses may be partially responsible in some cases, the origin is thought to be largely genetic, even though no single gene is likely sufficient to cause autism. Instead, mutations in multiple genes contribute to this disorder.24 Mutations in the PTEN gene were implicated in a subset of autistic patients with macrocephaly.25–32 The 10 known autism-associated point mutations occur in the phosphatase domain, the C2 domain, and the phosphatase/C2 boundary. None of these autism-associated mutations are located in the PI(4,5)P2-binding domain or the C-terminal regulatory tail. The R173H mutation decreased phosphatase activity,33 but there has been little characterization of the other autism-associated mutant PTEN proteins.
Loss of both PTEN genes in neural precursor cells leads to mice with enlarged brains and disorganized laminar patterning.34 Deletion of PTEN also leads to abnormal dendritic spines and weakened synaptic transmission and plasticity.35 Bear and coworkers suggested that mutations of PTEN and other autism-related genes affect synaptic plasticity by misregulation of synaptic protein synthesis.36,37 Alternatively, formation of synapses may be faulty because of the rapid growth of head and brain.38
Given the proposed role of PTEN, a natural question is whether other proteins in the PI(3,4,5)P3 pathway are associated with autism. Tuberous sclerosis complex (TSC) is a disease clearly associated with autism.39 A fraction of TSC patients show macrocephaly40 and autistic behavior.39 In the PI(3,4,5)P3 pathway, the Akt kinase inhibits the activity of the TSC complex (also known as hamartin and tuberin).41 These results strongly implicate the PI(3,4,5)P3 pathway in the etiology of autism.
To understand the molecular basis for the mutant PTEN proteins in autism, we characterized the autism-related mutation, H93R, which is located near the phosphatase active site.12 We found that the H93R mutation affects multiple steps of PTEN action. First, the mutation alters binding of PTEN to membranes bearing negatively charged lipids. Binding to PS-bearing membranes was increased 5.6-fold for H93R PTEN relative to wild-type PTEN, while binding to membranes that contain PI(4,5)P2, but not PS, was reduced 2.5-fold in comparison to wild-type PTEN. Second, the phosphatase activity was decreased for H93R PTEN. Third, a PTEN-GFP fusion protein showed increased localization at the plasma membrane of U87MG glioblastoma cells. This localization was further enhanced by mutations of the phosphorylation sites in the C-terminal tail.
Results
We expressed the H93R PTEN protein in E. coli with a yield indistinguishable from wild-type PTEN (1–2 mg/L culture). The circular dichroism spectrum for H93R protein was indistinguishable from that of wild type, and based on dynamic light scattering, the H93R PTEN was not aggregated (data not shown). We next assessed phosphatase activity using a colorimetric assay and soluble C8 PI(3,4,5)P3 as substrate.20 H93R PTEN was ∼15% as active as wild-type PTEN [Fig. 1(A)]. A negative control, C124S PTEN, showed no phosphatase activity. We also assessed PTEN activity by cotransfecting PTEN expression plasmids with an activated PI3K plasmid to drive PI(3,4,5)P3 production and a HA-Akt plasmid to assess PI(3,4,5)P3 levels. U87MG cells are ideal for this assay because they are efficiently transfected and lack functional PTEN. We measured phosphorylation of immunoprecipitated HA-Akt by Western blotting with anti-phospho-Akt antibody [Fig. 1(B)]. Active PTEN decreases Akt phosphorylation. The H93R mutant showed reduced activity.
Figure 1.
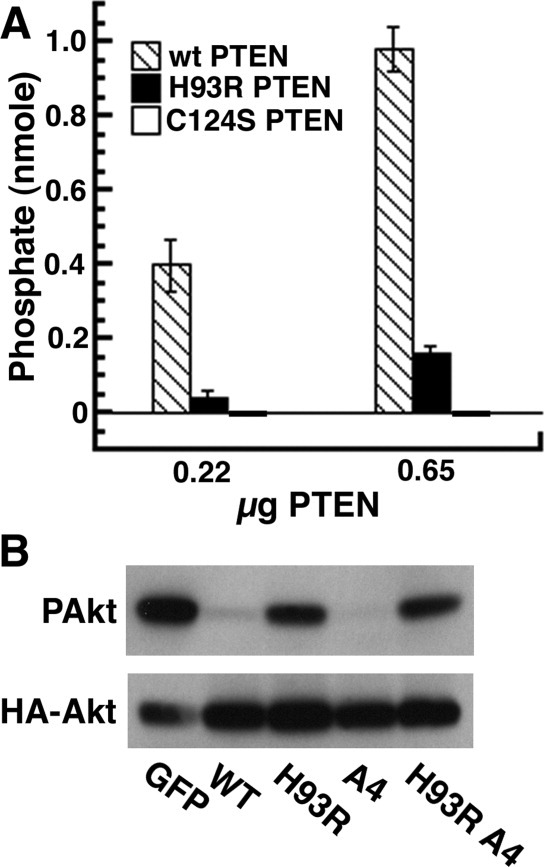
PTEN phosphatase assays for wild type (WT), H93R-PTEN, and C124S-PTEN. For both recombinant protein and PTEN expressed in U87MG cells, H93R had reduced phosphatase activity. A: PTEN protein was expressed in bacteria and phosphatase activity was assessed using short chain PI(3,4,5)P3. These results include three independent experiments, each carried out in triplicate. The results are presented as means ± SEM. There are no error bars for C124S-PTEN because every assay with C124S-PTEN gave zero phosphate release. B: U87MG cells were transfected with expression vectors for GFP, PTEN-GFP, or H93R-PTEN-GFP, HA-Akt and a constitutively activated PI3K. The HA-Akt protein was immunoprecipitated with anti-HA antibodies and phosphorylation of Akt was assessed by Western blotting. WT and A4 PTEN proteins were more active than H93R or H93R A4 mutants. These results are representative of three independent experiments.
Using our multistep model for PTEN action, we analyzed which step(s) is affected by the H93R mutation. Because binding to negatively charged lipids is the first step for membrane localization and PTEN activation, we assessed binding of wild-type and H93R PTEN to lipid vesicles doped with negatively charged lipids and pyrene-labeled phosphatidylethanolamine (PE). Binding of PTEN to these vesicles quenches tryptophan fluorescence. The Kd was calculated using the total lipid concentration. Also, for samples in which the binding was dominated by the anionic lipid (PI(4,5)P2 or PS), we calculated the binding constants for just the anionic lipid(s), which we call KPI(4,5)P2 or KPS. We previously demonstrated that PTEN binds more strongly to PI(4,5)P2 bearing membranes (Kd = 163±6 μM; KPI(4,5)P2 = 8.1±0.3 μM) than PS-membranes (Kd = 508±13 μM; KPS = 22.0±0.5 μM).19 In contrast, H93R PTEN binds more strongly to PS-membranes (Kd = 91±7 μM; KPS = 4.5±0.3 μM) than PI(4,5)P2 (Kd = 408±20 μM; KPI(4,5)P2 = 20.4±1.0 μM) (Fig. 2). Because H93R PTEN binds more strongly to PS and less strongly to PI(4,5)P2, we predicted that wild-type and H93R PTEN proteins would have similar affinities for membranes with both PS and PI(4,5)P2. Indeed, this turned out to be the case. Binding to PC/PS/PI(4,5)P2 membranes yielded similar binding constants for wild-type PTEN (Kd = 41±5 μM; KPS/PI(4,5)P2 = 2.1±0.2) and H93R PTEN (Kd = 45±6.0 μM; KPS/PI(4,5)P2 = 2.3±0.3 μM).
Figure 2.
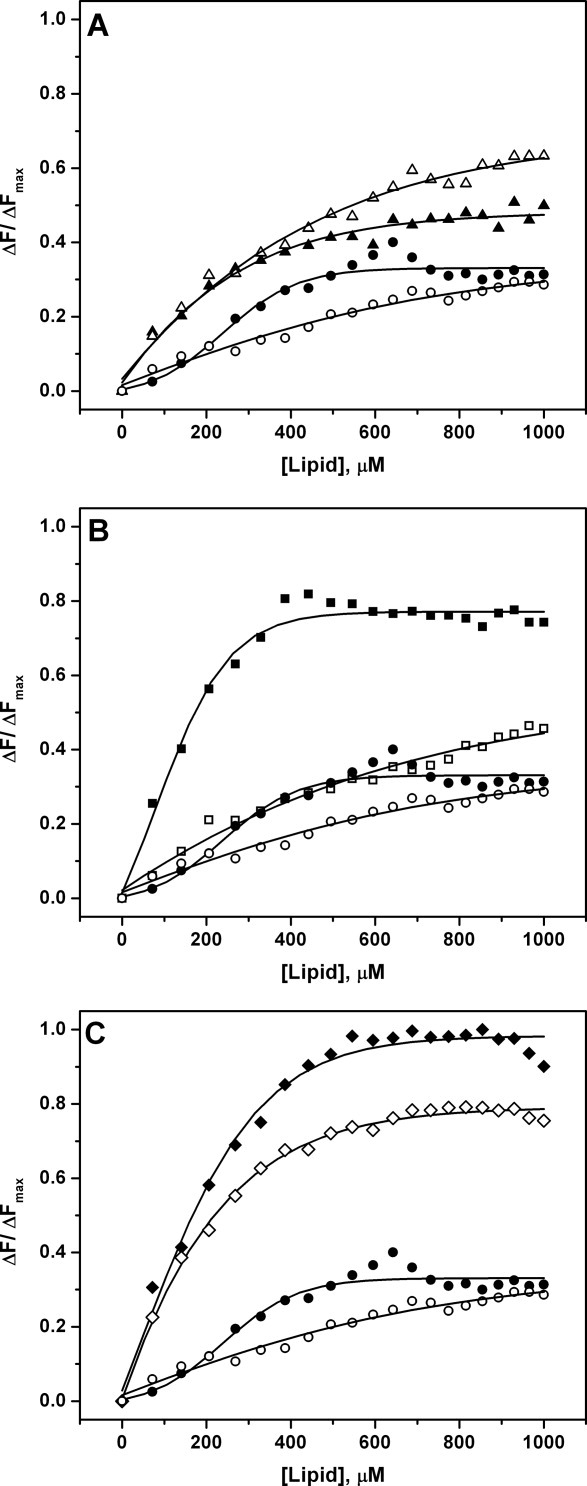
PTEN binding to lipid membranes. The vesicles contain pyrene-lipids to quench fluorescence of bound PTEN proteins. WT and H93R PTEN data points are shown in black and red, respectively. In each panel, control binding curves for vesicles lacking anionic lipids (circles) are shown. A: The H93R mutant PTEN binds PI(4,5)P2 vesicles (triangles) slightly less strongly than WT PTEN. B: In contrast, the H93R-PTEN binds to PS-bearing vesicles (squares) more strongly than WT. C: WT and H93R-PTEN bind to PI(4,5)P2 + PS vesicles (diamonds) with similar affinities. All data points represent averages of at least three repeats. The binding curves were fitted to the equation y = (K[L])/(1 + K[L]) in which [L] is the lipid concentration. The binding constants are listed in Table I.
Table I.
Binding Constants Based on Tryptophan Fluorescence Quenchinga
| PC | PC/PI(4,5)P2 | PC/PS | PC/PS/PI(4,5)P2 | |
|---|---|---|---|---|
| Wild-type PTEN, Kd (μM) | 585 ± 15 | 163 ± 6 | 508 ± 13 | 41 ± 5 |
| Wild-type PTEN, Klipid | 8.1 ± 0.3 | 22.0 ± 0.5 | 2.1 ± 0.2 | |
| H93R PTEN, Kd (μM) | 408 ± 20 | 91 ± 7 | 45 ± 6 | |
| H93R PTEN, Klipid | 20.4 ± 1.0 | 4.5 ± 0.3 | 2.3 ± 0.3 |
Binding constants were calculated from data shown in Figure 2. Kd values are based on total lipid concentrations (i.e., including PC). The Klipid values are based on the respective anionic lipid component(s) (PI(4,5)P2, PS, or PS/PI(4,5)P2).
We next assessed whether binding of H93R PTEN to negatively charged lipids induces a conformational change in the structure of PTEN. In particular, we used infrared spectroscopy to analyze the protein amide I band envelope, which is composed of several sub-bands representing the secondary structure elements of proteins. Band intensities in the 1650–1655 cm−1 region are associated with α-helical content. In contrast, band intensities in the 1630 cm−1 region are associated with antiparallel β-sheet content. The attenuated total reflectance (ATR) cell preferentially detects membrane-bound protein due to settling of the multilamellar vesicles on the surface of the ATR crystal, while unbound protein stays in solution.19 Hence, we were able to record the spectra for predominantly lipid-bound PTEN. We previously showed that binding of PTEN to PI(4,5)P2 bearing membranes increases the α-helical content19 [Fig. 3(A)]. The spectra for wild-type and H93R PTEN are identical [Fig. 3(A)], giving another indication that the H93R protein folds properly. The interaction of H93R PTEN with PI(4,5)P2- or PS-bearing membranes resulted in only a slight broadening of the amide I band at the detection limit of the experiment, indicating that the overall structure of the protein was almost unchanged. Vesicles with both PI(4,5)P2 and PS did induce a slight shift of the amide I band envelope to lower wavenumbers [Fig. 3(A–C)], indicating a slightly increased β-sheet content. Hence, the H93R mutation also affects membrane-induced conformational changes, because the PI(4,5)P2 induced change toward more α-helical structural content previously found for wild-type PTEN19 is not observed for H93R PTEN.
Figure 3.
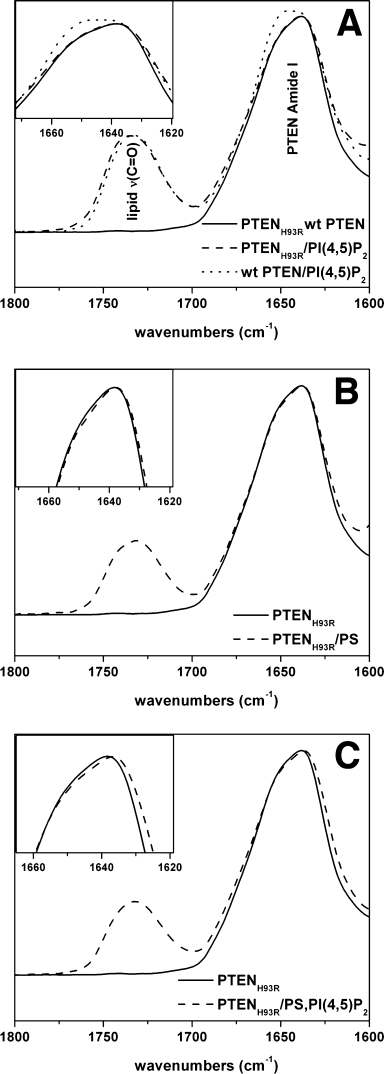
H93R-PTEN protein shows reduced membrane-induced conformational changes. A, B: Neither PI(4,5)P2 nor PS bearing vesicles lead to a shift of the absorption band at 1640 cm−1, indicating that is neither an increase in α helical nor β sheet content. The wild-type and H93R spectra in the absence of lipid are identical. The band at 1730 cm−1 is due to lipid carbonyl groups. C: Vesicles with both PI(4,5)P2 and PS induced a slight shift to lower wave numbers. All spectra are representative of at least three experiments.
We hypothesized that because H93R PTEN has an unusually strong affinity for PS, and PS is preferentially localized in the plasma membrane,42 the H93R mutant would show an increased association with the plasma membrane. Therefore, we expressed wild-type and mutant PTEN-GFP fusion proteins in U87MG cells (Fig. 4). We did not detect PTEN-GFP at the membrane [Fig. 4(A)], probably because membrane association of wild-type PTEN is transient and difficult to detect.23 There also is no obvious plasma membrane association for endogenous PTEN protein.43 In contrast, there were faint membrane patches for H93R PTEN [Fig. 4(B)]. Phosphorylation sites on PTEN are known to regulate PTEN localization to the membrane; elimination of these phosphorylation sites (S380A T382A T383A S385A = A4) enhances membrane association.14 We also tested if combining H93R and A4 would have a synergistic effect on localization. The A4-PTEN-GFP also showed very faint membrane patches [Fig. 4(C)], while H93R-A4-PTEN-GFP showed strong patches at the plasma membrane [Fig. 4(D)]. Differences in subcellular localization were not due to proteolytic cleavage of the PTEN fusion proteins; SDS-PAGE analyses showed that the PTEN-GFP fusion proteins all had the same apparent molecular weight [Fig. 4(E)]. Thus, consistent with the altered binding to lipid membranes, H93R PTEN shows enhanced binding to the PS-rich plasma membrane of U87MG cells.
Figure 4.
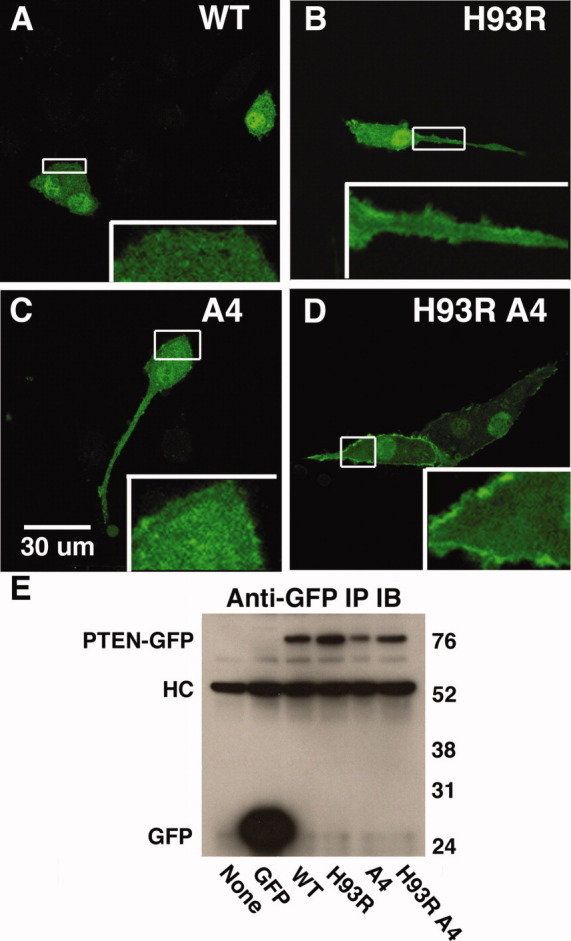
H93R-PTEN-GFP shows enhanced membrane association. U87MG cells were transfected with expression vectors, cells were fixed and subcellular localization was determined with a confocal microscope. A: WT PTEN-GFP does not show any obvious membrane association. B: H93R-PTEN-GFP shows faint localization at the plasma membrane. C: 4A-PTEN-GFP shows weak association with the plasma membrane. D: H93R-4A-PTEN-GFP showed a strong association with the plasma membrane. E: PTEN-GFP fusion proteins were visualized by an immunoprecipitation-Western blot with anti-GFP antibodies. We included the immunoprecipitation to avoid confusion with a nonspecific band that appeared in Western blots of cell extracts. HC is the immunoglobulin heavy chain from the immunoprecipitation.
Discussion
The major goal in this study was to analyze the effects of the autism-associated H93R mutation in terms of our recent model for regulation of PTEN. This mutant protein has reduced phosphatase activity but is not inactive. A similar mutation (H93A) also reduced, but did not eliminate, PTEN phosphatase activity.12 The mutation affects multiple steps for PTEN activation, including membrane binding and the PI(4,5)P2-induced conformational change. As a result, in cells, the H93R mutant protein has reduced phosphatase activity, even though there is enhanced membrane association. Since it is usually assumed that enhanced membrane association of PTEN leads to increased PI(3,4,5)P3 turnover, this is a unique and clinically important feature of this PTEN mutation.
We assessed membrane binding by two independent assays. Based on quenching of PTEN tryptophan fluorescence with pyrene-doped vesicles, the dissociation constant for binding of the H93R mutant protein to PS-bearing vesicles was 5.6-fold smaller than that for wild-type PTEN. In contrast, the dissociation constant for binding of H93R PTEN to PI(4,5)P2-bearing vesicles was 2.5-fold larger than that for wild-type PTEN. We also assessed membrane binding for H93R PTEN in biological cells. Because mammalian plasma membranes have about 15% PS and 1% PI(4,5)P2, PS is the predominant anionic lipid.44 Consistent with the observed changes in affinity, the H93R PTEN protein was preferentially associated with the PS-rich plasma membrane.42 This association was enhanced by the A4 mutation that deletes four phosphorylation sites in the C-terminal tail, opening the PTEN structure and stabilizing PTEN-membrane interactions.14,23 The C124S mutation at the active site also enhances membrane binding.23 Notably, combining the C124S mutation with the A4 mutation further enhances membrane binding. These results suggest that for a number of PTEN mutant proteins, membrane binding requires the “open” conformation, which is induced by the A4 mutation.
The H93R mutation at the active site influences both PI(4,5)P2 and PS binding. In wild-type PTEN, PI(4,5)P2 binds to the N-terminal domain and initiates a conformational change in the phosphatase domain. It is plausible that the H93R mutation perturbs the phosphatase domain and, thereby, affects the N-terminal PI(4,5)P2 binding site. A point mutation (K13E) in the PI(4,5)P2-binding domain eliminated the PI(4,5)P2-induced conformational change.19 In wild-type PTEN, PS binding is at least partly mediated by the C2 domain,12,13 and the H93R mutation might also indirectly affect the C2 domain. Indeed, it has been suggested that conformational changes in the phosphatase domain might be transmitted across the broad phosphatase-C2 interface.45 These results are consistent with the emerging model for PTEN—that it is a plastic protein with strong interdomain interactions.
How might the H93R mutation affect the bearer's health? PTEN activity affects development of the nervous system as well as synaptic plasticity in adults.35 These functions are thought to be related to autism, macrocephaly and mental retardation.36 A related question is whether autistic patients with one PTEN gene mutated will have an enhanced risk for cancer. These questions are just now being addressed.46 However, the effects of PTEN are highly quantitative so the partial activity of the H93R PTEN protein may allow it to act as a tumor suppressor and retard or prevent cancer development, whereas, a null PTEN mutant would not. Finally, an intriguing question is whether the enhanced association of H93R PTEN might have additional effects, such as the membrane association of H93R PTEN affecting complexes with other proteins.11 A question for further study is whether other autism-associated PTEN mutations also show partial phosphatase activity and altered subcellular localization.
Materials and Methods
1-Palmitoyl-2-oleoylphosphatidylcholine (POPC), 1-palmitoyl-2-oleoylphosphatidylserine (POPS), brain phosphatidylinositol (4,5) bisphosphate (PI(4,5)P2), and 1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine-N-(1-pyrenesulfonyl) (excitation = 347 nm, emission = 379 nm) (pyrene-labeled DOPE) were purchased from Avanti Polar Lipids (Alabaster, AL). Chloroform and methanol used for dissolving lipid samples were ACS grade, and water for buffers was HPLC grade (Fisher Scientific, Chicago, IL).
Preparation of PTEN protein and phosphatase assays
Human PTEN with a six-histidine tag at the C-terminus was expressed in Escherichia coli BL21(DE3) bacteria as described.19 Mutations were introduced with a Quick-Change site-directed mutagenesis kit from Stratagene. PTEN proteins were purified with a HisTrap HP kit from GE Healthcare, a Superdex 200 column and a MonoQ anion-exchange column. In vitro activity of PTEN proteins was assessed as described.20,47 In brief, release of free phosphate from 36 μM PI(3,4,5)P3 was detected using BIOMOL GREEN (BIOMOL, Plymouth Meeting, MA).
Phosphatase activity of PTEN in mammalian cells and confocal microscopy for subcellular localization
U87MG glioblastoma cells obtained from the ATCC were plated in 35-mm cell culture dishes and subjected to transfection with 1.0 μg pcDNA3 PTEN-GFP plasmid (a gift from Drs. Radhar and Devreotes),48 0.4 μg HA-Akt plasmid (HA, hemagglutinin epitope), 0.4 μg PI3K plasmid with a CAAX sequence to induce prenylation, and 0.2 μg pcDNA3 GFP plasmid per dish. After 48 h, the cells were lysed with 0.5 mL of buffer per dish (1% NP-40, 10% (v/v) glycerol, 100 mM NaCl, 1 mM EDTA, 2 μL/mL PIC protease inhibitor mix (Sigma, St. Louis, MO), 2 mg/mL β-glycerophosphate, 2 mg/mL sodium fluoride, 50 mM Tris, pH 7.5). Cell debris was eliminated by centrifugation (17,800g) for 10 min at 4°C. To prepare immunoprecipitates, we incubated 1 μg of anti-HA antibody (monoclonal HA.11 from Covance, Princeton, NJ) with 450 μL of cell extract for 1 h at 4°C. The antibody complexes were collected with Protein G Sepharose beads (Sigma) and analyzed by Western blotting with rabbit anti-phospho-Akt antibody (Cell Signaling, Beverly, MA) and anti-HA antibody.
To assess subcellular localization, U87MG cells were grown on glass coverslips and transfected with PTEN-GFP expression plasmids, as described above. After 24 h, the cells were fixed with 4% paraformaldehyde for 10 min at 4°C, and the coverslips, mounted in Vectashield (Vector Laboratories, Burlingame, CA). The samples were examined with a Leica SP1 confocal microscope with a 40× objective.
In some cases, cells were transfected with the PTEN-GFP expression vectors (1 μg/35-mm dish).16 After 24 h, these cells were extracted and clarified as described above. The extract (0.5 mL) as mixed with 5 μg of anti-GFP rabbit polyclonal antibody (ab290, Abcam, Cambridge, MA) for 1 h at 4°C. The immunoprecipitates were captured with protein G beads, and GFP fusion proteins were detected by Western blotting with mouse monoclonal anti-GFP antibody (0.5 μg/mL, MMS-118P, Covance, Richmond, CA).
Preparation of lipid samples
Lipid stock solutions were dissolved in mixtures of chloroform and methanol (3:1 v/v) and stored in amber glass vials to protect the lipids from UV light. Large unilamellar vesicles were prepared by drying lipid solutions at about 50°C under a stream of nitrogen, producing a lipid film. The samples were kept overnight at about 45°C in high vacuum to remove residual solvent. The lipid films were resuspended in buffer solution and vortexed for 60 s three times, waiting ∼ 5 min between vortexing periods. The resulting multilamellar vesicles were extruded through a polycarbonate film (100-nm pore size; Avestin, Ottawa, ON). The unilamellar vesicles were characterized by dynamic light scattering (HPPS and Nanosizer Malvern Instruments, Southborough, MA) before and after experiments. Typically, these vesicles exhibited a narrow size distribution centered at about 110-nm diameter.
Tryptophan quenching experiments
Using a Varian Eclipse spectrometer, tryptophan fluorescence spectra were recorded at 20°C, using an excitation wavelength of 290 nm and scanning the emission wavelength from 300 to 380 nm. Measurements were made using 1.0 μM protein solutions in pH 7.0 HEPES buffer (10 mM HEPES, 100 mM NaCl, and 0.1 mM EDTA). The maximum value of the tryptophan spectrum was recorded, and unilamellar vesicles were titrated into the protein solution in 20 steps, resulting in a final 1 mM lipid concentration. The compositions of the lipid vesicles included: 93 mol% POPC and 5 mol% brain PI(4,5)P2, 93 mol% POPC and 5 mol% POPS; 88 mol% POPC and 5 mol% brain PI(4,5)P2 with 5 mol% POPS. All lipid mixtures also contained 2 mol% pyrene-labeled DOPE as a quencher for tryptophan fluorescence. The maximum intensity of each tryptophan fluorescence spectrum was corrected for dilution as well as fluorescence changes due to an altered tryptophan environment upon binding (e.g., as a result of protein structure changes and the placement of the protein at the membrane). The latter correction was made using vesicles lacking pyrene-labeled DOPE.19 The results were then normalized to the initial value, which was set at 100%.
Infrared spectroscopy
FTIR experiments were carried out using a Bruker Tensor Spectrometer (Billerica, MA) equipped with a narrow band MCT detector and a Bruker BioATRII unit. Interferograms were collected at 2 cm−1 resolution (512 scans, 20°C), apodized with a Blackman-Harris function, and Fourier transformed with one level of zero-filling to yield spectra encoded at 1 cm−1 intervals. Protein samples were concentrated using 10,000 MWCO Centricon tubes obtained from Fisher Scientific (Chicago, IL), reaching a concentration of 8–9 mg/mL. Protein samples were exchanged against D2O buffer as described above. Dialysis against buffer was done in three steps, each 20 min in length. Then 125 μg of the D2O-exchanged protein was placed in the ATR unit and analyzed with the Bruker OPUS software. Mixed multlamellar lipid vesicle/protein samples were obtained by resuspending the appropriate lipid mixtures in protein solutions. The multilamellar vesicles were formed with protein present, so that all aqueous compartments included protein. Lipid samples varied but followed the form 95 mol% POPC, 5 mol% PI(4,5)P2 or 5 mol% PS as well as 90 mol% POPC, 5 mol% POPS, and 5 mol% PI(4,5)P2. The protein-bearing samples included 1 mole of protein per 8 moles of phosphoinositide or PS lipid. After adding the protein solution to the dried lipids, they were vortexed for 60 s, three times, waiting ∼5 min between vortexing cycles. The resulting protein/vesicle solutions were placed in the BioATR II unit and analyzed with the Bruker OPUS software. The vesicles did not form an anisotropic, ordered film on the ATR crystal but remained isotropic, as checked by IR measurements using polarized radiation. We obtained buffer spectra using the buffers against which the proteins were exchanged. The D2O buffer samples were adjusted with respect to their H2O (HOD) content so that the intensities of the H2O and HOD bands matched between the respective protein and buffer solutions. Subsequently, the buffer spectra were subtracted from the protein/lipid samples to yield a flat baseline between 1600 and 1900 cm−1. All subtraction values were 1.0000 ± 0.0005. The resulting spectra were exported to Origin software, where the lipid/protein spectra were normalized to the protein only spectrum, using the maximum value of the amide I band (∼1638 cm−1).
Acknowledgments
The authors thank Drs. Radher and Devreotes for the PTEN-GFP expression vectors. They also thank Melonnie Furgason for her help with these experiments. The confocal microscopy was carried out with the help of Dr. Hong Cao.
Glossary
Abbreviations:
- ATR
attenuated total reflectance
- HA
hemagglutinin epitope
- PE
phosphatidylethanolamine
- PI(3,4,5)P3
phosphatidylinositol (3,4,5) trisphosphate
- PI(4,5)P2
phosphatidylinositol (4,5) bisphosphate
- PI
phosphoinositide
- PI3K
phosphoinositide 3-kinase
- POPC
1-palmitoyl-2-oleoylphosphatidylcholine
- POPS
palmitoyl-2-oleoylphosphatidylserine
- PS
phosphatidylserine
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- pyrene-labeled DOPE
1,2-dioleoyl-sn-glycerol-3-phosphoethanolamine-N-(1-pyrenesulfonyl)
References
- 1.Li D-M, Sun H. TEP1, encoded by a candidate tumor suppressor locus, is a novel protein tyrosine phosphatase regulated by transforming growth factor β. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 2.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 3.Steck PA, Pershouse MA, Jasser SA, Yung WKA, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DHF, Tavtigian SV. Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 4.Li L, Ross AH. Why is PTEN an important tumor suppressor? J Cell Biochem. 2007;102:1368–1374. doi: 10.1002/jcb.21593. [DOI] [PubMed] [Google Scholar]
- 5.Sansal I, Sellers WR. The biology and clinical relevance of the PTEN tumor suppressor pathway. J Clin Oncol. 2004;22:2954–2963. doi: 10.1200/JCO.2004.02.141. [DOI] [PubMed] [Google Scholar]
- 6.Sulis ML, Parsons R. PTEN: from pathology to biology. Trends Cell Biol. 2003;13:478–483. doi: 10.1016/s0962-8924(03)00175-2. [DOI] [PubMed] [Google Scholar]
- 7.Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-triphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 8.Furnari FB, Lin H, Huang H-JS, Cavenee WK. Growth suppression of glioma cells by PTEN requires a functional phosphatase catalytic domain. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myers MP, Pass I, Batty IH, van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK. The lipid phosphatase activity of PTEN is critical for its tumor suppressor function. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E. Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science. 2002;295:2088–2091. doi: 10.1126/science.1068094. [DOI] [PubMed] [Google Scholar]
- 11.Gericke A, Munson M, Ross AH. Regulation of the PTEN phosphatase. Gene. 2006;374:1–9. doi: 10.1016/j.gene.2006.02.024. [DOI] [PubMed] [Google Scholar]
- 12.Lee J-O, Yang H, Georgescu M-M, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 13.Das S, Dixon JE, Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci USA. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol Cell Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- 16.Radhar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN. Phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN. Proc Natl Acad Sci USA. 2009;106:480–485. doi: 10.1073/pnas.0811212106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN. Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis. J Biol Chem. 2004;279:16606–16613. doi: 10.1074/jbc.M312098200. [DOI] [PubMed] [Google Scholar]
- 18.Leslie NR, Batty IH, Maccario H, Davidson L, Downes CP. Understanding PTEN regulation: PIP2, polarity and protein stability. Oncogene. 2008;27:5464–5476. doi: 10.1038/onc.2008.243. [DOI] [PubMed] [Google Scholar]
- 19.Redfern RE, Redfern D, Furgason ML, Munson M, Ross AH, Gericke A. PTEN phosphatase selectively binds phosphoinositides and undergoes structural changes. Biochemistry. 2008;47:2162–2171. doi: 10.1021/bi702114w. [DOI] [PubMed] [Google Scholar]
- 20.Campbell RB, Liu F, Ross AH. Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J Biol Chem. 2003;278:33617–33620. doi: 10.1074/jbc.C300296200. [DOI] [PubMed] [Google Scholar]
- 21.McConnachie G, Pass I, Walker SM, Downes CP. Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids. Biochem J. 2003;371:947–955. doi: 10.1042/BJ20021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walker SM, Leslie NR, Perera NM, Batty IH, Downes CP. The tumour-suppressor function of PTEN requires an N-terminal lipid-binding motif. Biochem J. 2004;379:301–307. doi: 10.1042/BJ20031839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazquez F, Matsuoka S, Sellers WR, Yanagida T, Ueda M, Devreotes PN. Tumor suppressor PTEN acts through dynamic interaction with the plasma membrane. Proc Natl Acad Sci USA. 2006;103:3633–3638. doi: 10.1073/pnas.0510570103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geschwind DH. Autism: many genes, common pathways? Cell. 2008;135:391–395. doi: 10.1016/j.cell.2008.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boccone L, Dessi V, Zappu A, Piga S, Piludu MB, Rais M, Massidda C, de Virgiliis S, Cao A, Loudianos G. Bannayan-Riley-Ruvalcaba syndrome with reactive nodular lymphoid hyperplasia and autism and a PTEN mutation. Am J Med Genet A. 2006;140:1965–1969. doi: 10.1002/ajmg.a.31396. [DOI] [PubMed] [Google Scholar]
- 26.Butler MG, Dasouki MJ, Zhou XP, Talebizadeh Z, Brown M, Takahashi TN, Miles JH, Wang CH, Stratton R, Pilarski R, Eng C. Subset of individuals with autism spectrum disorders and extreme macrocephaly associated with germline PTEN tumour suppressor gene mutations. J Med Genet. 2005;42:318–321. doi: 10.1136/jmg.2004.024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Buxbaum JD, Cai G, Chaste P, Nygren G, Goldsmith J, Reichert J, Anckarsater H, Rastam M, Smith CJ, Silverman JM, Hollander E, Leboyer M, Gillberg C, Verloes A, Betancur C. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144:484–491. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goffin A, Hoefsloot LH, Bosgoed E, Swillen A, Fryns JP. PTEN mutation in a family with Cowden syndrome and autism. Am J Med Genet. 2001;105:521–524. doi: 10.1002/ajmg.1477. [DOI] [PubMed] [Google Scholar]
- 29.Herman GE, Butter E, Enrile B, Pastore M, Prior TW, Sommer A. Increasing knowledge of PTEN germline mutations: two additional patients with autism and macrocephaly. Am J Med Genet A. 2007;143:589–593. doi: 10.1002/ajmg.a.31619. [DOI] [PubMed] [Google Scholar]
- 30.McBride KL, Varga EA, Pastore MT, Prior TW, Manickam K, Atkin JF, Herman GE. Confirmation study of PTEN mutations among individuals with autism or developmental delays/mental retardation and macrocephaly. Autism Res. 2010;3:137–141. doi: 10.1002/aur.132. [DOI] [PubMed] [Google Scholar]
- 31.Orrico A, Galli L, Buoni S, Orsi A, Vonella G, Sorrentino V. Novel PTEN mutations in neurodevelopmental disorders and macrocephaly. Clin Genet. 2009;75:195–198. doi: 10.1111/j.1399-0004.2008.01074.x. [DOI] [PubMed] [Google Scholar]
- 32.Parisi MA, Dinulos MB, Leppig KA, Sybert VP, Eng C, Hudgins L. The spectrum and evolution of phenotypic findings in PTEN mutation positive cases of Bannayan-Riley-Ruvalcaba syndrome. J Med Genet. 2001;38:52–58. doi: 10.1136/jmg.38.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han SY, Kato H, Kato S, Suzuki T, Shibata H, Ishii S, Shiiba K, Matsuno S, Kanamaru R, Ishioka C. Functional evaluation of PTEN missense mutations using in vitro phosphoinositide phosphatase assay. Cancer Res. 2000;60:3147–3151. [PubMed] [Google Scholar]
- 34.Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H. Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science. 2001;294:2186–2189. doi: 10.1126/science.1065518. [DOI] [PubMed] [Google Scholar]
- 35.Fraser MM, Bayazitov IT, Zakharenko SS, Baker SJ. Phosphatase and tensin homolog, deleted on chromosome 10 deficiency in brain causes defects in synaptic structure, transmission and plasticity, and myelination abnormalities. Neuroscience. 2008;151:476–488. doi: 10.1016/j.neuroscience.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kelleher RJ, III, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Walsh CA, Morrow EM, Rubenstein JL. Autism and brain development. Cell. 2008;135:396–400. doi: 10.1016/j.cell.2008.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Courchesne E, Pierce K. Brain overgrowth in autism during a critical time in development: implications for frontal pyramidal neuron and interneuron development and connectivity. Int J Dev Neurosci. 2005;23:153–170. doi: 10.1016/j.ijdevneu.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 39.Curatolo P, Porfirio MC, Manzi B, Seri S. Autism in tuberous sclerosis. Eur J Paediatr Neurol. 2004;8:327–332. doi: 10.1016/j.ejpn.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 40.Fidler DJ, Bailey JN, Smalley SL. Macrocephaly in autism and other pervasive developmental disorders. Dev Med Child Neurol. 2000;42:737–740. doi: 10.1017/s0012162200001365. [DOI] [PubMed] [Google Scholar]
- 41.Bjornsti MA, Houghton PJ. The tor pathway: a target for cancer therapy. Nat Rev Cancer. 2004;4:335–348. doi: 10.1038/nrc1362. [DOI] [PubMed] [Google Scholar]
- 42.Yeung T, Heit B, Dubuisson JF, Fairn GD, Chiu B, Inman R, Kapus A, Swanson M, Grinstein S. Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. J Cell Biol. 2009;185:917–928. doi: 10.1083/jcb.200903020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lachyankar MB, Sultana N, Schonhoff CM, Mitra P, Poluha W, Lambert S, Quesenberry PJ, Litofsky NS, Recht LD, Nabi R, Miller SJ, Ohta S, Neel BG, Ross AH. A role for nuclear PTEN in neuronal differentiation. J Neurosci. 2000;20:1404–1413. doi: 10.1523/JNEUROSCI.20-04-01404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 45.Wishart MJ, Dixon JE. PTEN and myotubularin phosphatases: from 3-phosphoinositide dephosphorylation to disease. Trends Cell Biol. 2002;12:579–585. doi: 10.1016/s0962-8924(02)02412-1. [DOI] [PubMed] [Google Scholar]
- 46.Blatt J, Deal AM, Mesibov G. Autism in children and adolescents with cancer. Pediatr Blood Cancer. 2010;54:144–147. doi: 10.1002/pbc.22303. [DOI] [PubMed] [Google Scholar]
- 47.Maehama T, Taylor GS, Slama JT, Dixon JE. A sensitive assay for phosphoinositide phosphatases. Anal Biochem. 2000;279:248–250. doi: 10.1006/abio.2000.4497. [DOI] [PubMed] [Google Scholar]
- 48.Liu F, Wagner S, Campbell RB, Nickerson JA, Schiffer CA, Ross AH. PTEN enters the nucleus by diffusion. J Cell Biochem. 2005;96:221–234. doi: 10.1002/jcb.20525. [DOI] [PubMed] [Google Scholar]