

Abstract
We have developed a filamentous phage display system for the detection of asparagine-linked glycoproteins in Escherichia coli that carry a plasmid encoding the protein glycosylation locus (pgl) from Campylobacter jejuni. In our assay, fusion of target glycoproteins to the minor phage coat protein g3p results in the display of glycans on phage. The glyco-epitope displayed on phage is the product of biosynthetic enzymes encoded by the C. jejuni pgl pathway and minimally requires three essential factors: a pathway for oligosaccharide biosynthesis, a functional oligosaccharyltransferase, and an acceptor protein with a D/E-X1-N-X2-S/T motif. Glycosylated phages could be recovered by lectin chromatography with enrichment factors as high as 2 × 105 per round of panning and these enriched phages retained their infectivity after panning. Using this assay, we show that desired glyco-phenotypes can be reliably selected by panning phage-displayed glycoprotein libraries on lectins that are specific for the glycan. For instance, we used our phage selection to identify permissible residues in the −2 position of the bacterial consensus acceptor site sequence. Taken together, our results demonstrate that a genotype–phenotype link can be established between the phage-associated glyco-epitope and the phagemid-encoded genes for any of the three essential components of the glycosylation process. Thus, we anticipate that our phage display system can be used to isolate interesting variants in any step of the glycosylation process, thereby making it an invaluable tool for genetic analysis of protein glycosylation and for glycoengineering in E. coli cells.
Keywords: glycosylation, glycoconjugate, lectin chromatography, N-glycan, oligosaccharyltransferase, Campylobacter jejunipgl operon
Introduction
Asparagine-linked (N-linked) protein glycosylation is an essential and conserved post-translational modification (PTM) of diverse proteins in eukaryotic organisms. It is the most abundant of all PTMs in eukaryotes with nearly 70% of all eukaryotic proteins predicted to be N-glycoproteins.1 The attachment of N-glycans serves to expand the diversity of eukaryotic secretory and membrane proteins2 and has been reported to assist protein folding and stability, oligomerization, quality control, sorting, and transport.3,4 The process of N-linked glycosylation is catalyzed in the endoplasmic reticulum (ER) and involves the assembly of glycans on a lipid carrier in the ER membrane followed by transfer to specific asparagine residues of target polypeptides.
Long considered exclusive to eukaryotes, N-linked glycoproteins have now been found in archea5 and more recently in certain bacteria such as Campylobacter jejuni.6 In C. jejuni, the genes required for N-linked glycosylation comprise a single 17-kb locus named pgl for protein glycosylation.7,8 More than 40 periplasmic and membrane glycoproteins have been identified in C. jejuni,7,9,10 and the N-linked glycan on these proteins is the heptasaccharide GalNAc5GlcBac (where Bac is bacillosamine).10 Similar to the process in eukaryotes, the bacterial glycan is synthesized by sequential addition of nucleotide-activated sugars on a lipid carrier on the cytoplasmic side of the inner membrane11 and, once assembled, is transferred across the membrane by a flippase enzyme called WlaB.12,13 Transfer of the glycan to substrate proteins occurs in the periplasm10 and is catalyzed by an oligosaccharyltransferase (OST) called PglB, a single, integral membrane protein with significant sequence similarity to the STT3 catalytic subunit of the eukaryotic OST complex.10 PglB attaches the glycan to asparagine in the motif D/E-X1-N-X2-S/T where X1 and X2 are any residues except proline, a sequon similar to (albeit more specific than) the N-X-S/T motif found in eukaryotic glycoproteins.14 A major breakthrough in our ability to study the bacterial N-glycosylation process came about when Aebi and coworkers transferred functional glycosylation machinery encoded by the pgl locus into Escherichia coli.8 These initial studies showed that AcrA, a native C. jejuni glycoprotein, could be modified with the characteristic C. jejuni heptasaccharide in E. coli cells that harbored a plasmid encoding the pgl locus. Many other diverse proteins of prokaryotic and eukaryotic origin have since been N-glycosylated in E. coli expressing the pgl genes.14–16 While these glycoengineered E. coli have great potential to expand our understanding of cellular glycosylation reactions, genetic analysis of this pathway has been slowed by the lack of a reliable screen or selection to rapidly interrogate N-linked glycosylation in these cells or to isolate mutants that confer desired N-glycosylation phenotypes.
Genetic analysis traditionally relies on the ability to isolate interesting mutants, the properties of which can provide important information on the organization of their underlying pathways. One powerful approach for isolating peptide and protein mutants from large populations of clones is M13 phage display. Since its inception in 1985,17 phage display has been used primarily for library-based selection of ligand-binding proteins,18–21 stabilized protein variants, regulatable enzymes and protein catalysts.21–23 While it is far less common, phage display can also be used to dissect complex biological pathways that carry out specific processes. For example, the genotype–phenotype link provided by M13 phage was used to isolate mutants of E. coli disulfide isomerase DsbC that enhanced its ability to promote substrate folding in the periplasm.24
Here, we present the development of a genetic screen for glycosylation in E. coli that is based on the display of N-linked glycoproteins on M13 phage particles [Fig. 1(a)]. This system is based on genetic fusion of a target N-glycoprotein to the minor phage coat protein g3p. The fusion proteins are secreted into the periplasm by the Sec-dependent pathway and become N-glycosylated in cells carrying a functional pgl locus. Phage particles displaying these N-linked glycoproteins can then be easily recovered by lectin affinity chromatography. Importantly, a genotype–phenotype link is established between the N-glycan displayed on the phage and the phagemid inside the particle, which can be designed to carry either the gene encoding the glycoprotein or genes encoding components of the glycosylation machinery. To demonstrate the utility of this method, we show that desired glycosylation phenotypes can be reliably selected by panning phage-displayed N-glycoprotein libraries on lectins that are specific for the glycan.
Figure 1.

(a) Schematic of phage display system for N-linked glycoproteins. Plasmid encoding the entire pgl locus and phagemid encoding the acceptor-g3p fusion protein are shown. The oligosaccharide is (i) synthesized by individual glycosyltransferases and assembled on a lipid carrier, bactoprenylpyrophosphate, on the cytoplasmic side of the inner membrane, (ii) translocated across the inner membrane to the periplasmic space and (iii) transferred to specific asparagine residues of the acceptor protein-g3p fusion by the OST. After infection with helper phage VCSM13, phages that display the glycosylated acceptor protein are bound to immobilized lectin and eluted with a competitive ligand. E. coli (F+) cells are infected with eluted phages and selected for the antibiotic resistance present on the phagemid. (b) Western blot analysis of whole cell lysates from TG1 pgl or pglmut cells expressing MBPDQNAT-g3p. Lysate from TG1 cells carrying only the pgl locus is included as negative control. Blot was probed with anti-MBP antibodies. The MBPDQNAT-g3p appeared at ∼90 kDa (arrow) which is consistent with the expected molecular weight of the fusion. (c) Same samples as in (b) but probed with hR6P serum that is specific for the C. jejuni heptasaccharide. (d) Western blot analysis of whole cell lysates from TG1 pgl or pglmut cells expressing DQNAT-g3p. Lysate from TG1 cells carrying only the pgl locus is included as negative control. Blot was probed with hR6P serum. The DQNAT-g3p appeared at ∼50 kDa (arrow), consistent with the expected molecular weight of the fusion.
Results
As a first step towards engineering a phage display system for N-linked glycoproteins, we created an artificial glycosylation substrate based on the E. coli maltose binding protein (MBP). Specifically, native MBP including its N-terminal Sec export signal was modified at its C-terminus with a single glycosylation acceptor site (DQNAT) that was previously shown to be efficiently glycosylated in vitro by C. jejuni PglB.25 This MBPDQNAT construct was cloned in-frame with g3p in plasmid pBAD24, which carries an M13 phage origin of replication and thus served as the phagemid for these experiments.26 To determine if this substrate was glycosylated by PglB in the periplasm, we transformed E. coli TG1 with pBAD-MBPDQNAT-g3p and either plasmid pACYCpgl or pACYCpglmut, which encode the native (pgl) or a mutant version (pglmut) of the C. jejuni glycosylation locus, respectively.8 Whereas the native pgl locus results in transfer of C. jejuni glycan to target proteins, the pglmut locus encodes an inactive variant of PglB that is incapable of N-linked glycosylation.8 Whole cell lysates showed a major protein at a mass of ∼90 kDa corresponding to MBPDQNAT-g3p that was detected in the presence of both pgl and pglmut [Fig. 1(b)]. To determine if these proteins were glycosylated, the same fractions were probed with hR6P antiserum that is specific for the C. jejuni heptasaccharide. This serum was raised against C. jejuni whole-cell extracts and has been shown to preferentially detect C. jejuni N-glycoproteins (Dr. Markus Aebi, personal communication). Only pgl cells produced MBPDQNAT-g3p that immunoreacted specifically with hR6P antiserum [Fig. 1(c)]. In contrast, there were no specific immunoreactive bands in the lysates containing MBPDQNAT-g3p expressed in pglmut cells or the lysates from cells with only the pgl locus [Fig. 1(c)]. We also found that an MBP-g3p fusion lacking the DQNAT site was not N-glycosylated when expressed in pgl or pglmut cells (data not shown). An identical result was obtained when we used a different glycoprotein substrate where the DQNAT acceptor site was genetically fused directly to the g3p protein without the MBP domain [Fig. 1(d)]. Taken together, these results indicate that periplasmic expression of different g3p chimeras is compatible with N-linked glycosylation in E. coli.
To demonstrate the potential of our method for recovering glycosylated phages, we first produced recombinant phage particles using VCSM13 helper phage. TG1 pgl cells that expressed the MBPDQNAT-g3p fusion were used for production of glycosylated phage, whereas TG1 pgl cells that expressed MBP-g3p were used for nonglycosylated phage. To quantify phage titers, fresh TG1 cells were infected with the purified phages and then selected on ampicillin to determine colony-forming units (CFUs). The resulting phage titers were ∼1 × 109 per mL of culture supernatant for TG1 pgl cells expressing MBPDQNAT-g3p or MBP-g3p. Immunoblot analysis confirmed the presence of MBP in both phage preparations [Fig. 2(a)]; however only the phage produced from TG1 pgl cells expressing MBPDQNAT-g3p immunoreacted with the hR6P antiserum [Fig. 2(a)] indicating that glycosylated MBPDQNAT-g3p was displayed on phage. We next attempted to recover phages that displayed glycosylated MBP from a background of control phages displaying nonglycosylated MBP. For this, we generated a 1:1 mixture of 1 × 109 glycosylated (MBPDQNAT-g3p) phages with 1 × 109 nonglycosylated (MBP-g3p) phages. This mixture containing ∼2 × 109 total phages [Fig. 2(b)] was then subjected to a single round of panning using agarose-bound soybean agglutinin (SBA). SBA is a lectin that binds the terminal N-acetyl-galactosamine (GalNAc) of the C. jejuni oligosaccharide.9 Unbound phages were removed by washing several times with PBS followed by washing with PBS containing 30 mM galactose [Fig. 2(b)]. Since galactose binds to SBA with a KD of 2.3 × 102 M−1, it can be used to compete with the bound oligosaccharide.27 Lastly, bound phages were eluted using 300 mM galactose in PBS and a total of ∼1 × 106 phages were recovered, which was 10 times greater than the background phages recovered when ∼2 × 109 exclusively nonglycosylated phages were applied to the SBA panning procedure [Fig. 2(b)]. To confirm that we had enriched for only glycosylated phage, we infected TG1 cells with eluted phages and selected individual colonies for PCR verification of the MBPDQNAT-g3p sequence in the corresponding phagemids. This was accomplished using primers that could amplify both the MBPDQNAT-g3p and MBP-g3p sequences, but would yield different molecular weight products that could be distinguished by gel electrophoresis. Indeed, 22/28 of the phagemids from elution fractions 1, 2, and 3 encoded MBPDQNAT-g3p [Table I and Fig. 2(c)].
Figure 2.
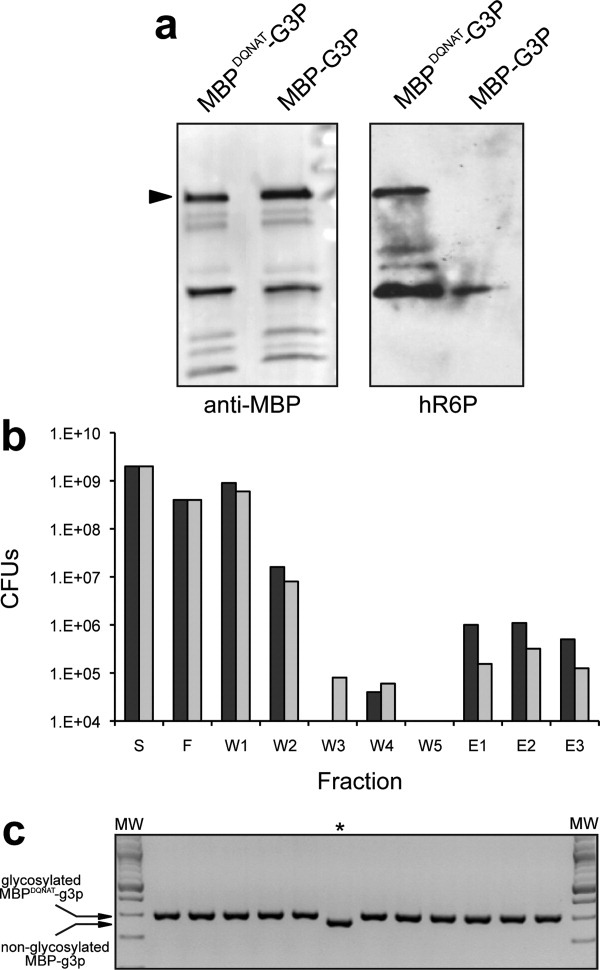
Enrichment of glycosylated phage by SBA panning. (a) Western blot analysis of phage preparations from TG1 pgl cells expressing either MBPDQNAT-g3p or MBP-g3p, as indicated. Blots were probed with anti-MBP antibodies (left panel) or hR6P serum (right panel). Arrow indicates the ∼90-kDa cross-reacting bands that corresponds to the g3p fusion proteins. (b) The total number of phages in each fraction was quantified by determining the CFUs after infection of TG1 cells and selection on ampicillin. Phages were produced from TG1 pgl cells expressing MBPDQNAT-g3p (glycosylated) and MBP-g3p (non-glycosylated) and mixed 1:1 (black bars). As a control, phages were produced exclusively from TG1 cells expressing MBP-g3p (gray bars). Solutions containing ∼2 × 109 phages were initially applied to the SBA column (S). The number of CFUs was determined during the panning procedure for each of the following fractions: F, SBA flow-through; W1 and W2, wash steps using PBS; W3–W5, wash steps using 30 mM galactose in PBS; E1–E3, elution steps using 300 mM galactose in PBS. The panning procedure was performed in triplicate and the resulting CFUs after TG1 infection varied by less than 10%. (c) Colony PCR of enriched clones from fraction E3 in (b) above. PCR products were generated using a single primer set that can amplify both MBPDQNAT-g3p and MBP-g3p, and then run in a 2% agarose gel. Only 1/12 products ran at the lower molecular weight size of 268-bp corresponding to non-glycosylated MBP-g3p (indicated by asterisk). The remaining 11 products all ran at 301-bp corresponding to glycosylated MBPDQNAT-g3p. The first and last lanes contain molecular weight ladder (MW). All PCR products were verified by DNA sequencing.
Table I.
Enrichment of N-Glycosylated Phages Using SBA Panning
| Total number of phages in artificial library of glycosylated and nonglycosylated phages (CFUs) |
Fraction of glycosylated phages recovered from elution fractions E1–E3 |
|||
|---|---|---|---|---|
| MBPDQNAT-G3P | MBP-G3P | E1 | E2 | E3 |
| 109 | 109 | 6/8 | 5/8 | 11/12 |
| 107 | 109 | 0/8 | 2/8 | 5/8 |
| 106 | 1010 | 2/8 | 3/8 | 4/8 |
| 104 | 1010 | 1/8 | 1/8 | 3/8 |
The amount of phages in the elution fractions was ∼1 × 106, which was well below the theoretical maximum recovery one would expect with a 1:1 starting mixture. The most likely explanation for the low recovery is the generally weak binding affinity of lectins for their carbohydrate ligands, and the usual requirements of multivalency and the “cluster glycoside effect” to achieve high avidity.28 Regardless of the cause, the actual ratio of glycosylated to nonglycosylated phages in our starting mixture was probably much smaller than 1:1, indicating that our assay can recover glycosylated phages from a significant excess of control phages. Encouraged by this result, we further diluted the presumably glycosylated phage (i.e., the ratio of glycosylated to nonglycosylated phage was not quantified) in an excess (102-, 104-, and 106-fold) of nonglycosylated phages and the above procedure was repeated. In all cases, significant enrichment was observed (Table I). This was best exemplified by the recovery of 5/22 phagemids encoding MBPDQNAT-g3p from a 106-fold excess of control phages after just one round of SBA panning, equating to an enrichment factor of 2.3 × 105. Collectively, these results demonstrate that: (i) glycosylated MBPDQNAT-g3p was present on phage; (ii) glycosylated phage could be enriched by lectin chromatography and retained their infectivity; and (iii) enrichment factors of ∼105 per round of SBA panning could be achieved.
To demonstrate the ability of this method to isolate mutants that may help to reveal mechanistic details of the N-linked glycosylation process, we next used our phage selection strategy to identify permissible residues in the bacterial consensus acceptor site sequence. Specifically, a library of MBPDQNAT-g3p sequences was created by random mutagenesis of the aspartate (D) residue in the −2 position of the DQNAT acceptor site. Since previous studies showed that N-glycosylation by C. jejuni PglB requires a negatively charged side chain at the −2 position,14 we reasoned that phage selection of library clones should yield only D or glutamate (E) substitutions. Consistent with this observation, we found that MBPDQNAT-g3p with an alanine substitution in the −2 position (MBPAQNAT-g3p) was not glycosylated in TG1 pgl cells (data not shown). Using an NNK degenerate primer, a random library of MBPDQNAT-g3p was created that contained 1 × 104 members, representing a 500-fold coverage of possible variants and ensuring that every clone was represented in the library. The diversity of the library from both E. coli DH5α and TG1 pgl cells was checked by sequencing of randomly selected clones, which confirmed that the library was sufficiently random and all sequenced clones contained the XQNAT motif. A library of ∼1 × 109 phages was produced from TG1 pgl cells. These phages were subjected to a single round of SBA panning followed by four wash steps with PBS, three wash steps with 30 mM galactose in PBST and finally elution with 300 mM galactose in PBS. TG1 pgl cells were infected with the elution fraction and selected on ampicillin to obtain single colonies, from which phagemids were isolated. A total of 47 isolated phagemids were sequenced and of these, 30 encoded D at the −2 position, consistent with known acceptor site requirements.14 Importantly, this result confirms that phages displaying desired glyco-phenotypes can be specifically selected from a large background of undesired phages in even a single round of library screening.
Discussion
In traditional phage display applications, expression of a peptide or protein on the surface of a phage particle produces a physical link between the phenotype (e.g., antigen binding) and the genotype of the expressed protein (e.g., antibody fragment).17–23 Phage-displayed proteins with desirable traits can be selectively enriched by panning over an immobilized ligand or substrate, and the identity of the selected protein can be determined by sequencing the encoding phagemid. In our approach, we have extended the principle of phage display to the post-translational modification of N-linked glycosylation. This is not the first application of phage display for genetic analysis of post-translational modifications as Georgiou and coworkers have reported a phage display system for disulfide bond formation in E. coli.24 However, to the best of our knowledge, ours is the first use of phage display for N-linked glycoproteins and one of the first reports of a high-throughput genetic assay for N-linked protein glycosylation in any host. Specifically, we show that a fusion protein comprised of MBPDQNAT and the minor phage coat protein g3p is glycosylated in E. coli carrying the entire C. jejuni pgl locus. Importantly, this glycosylated fusion protein is assembled on phage particles, thereby permitting affinity capture of glycosylated phages using proteins, such as lectins, that specifically recognize the N-linked glycan. Isolation of glycosylated phages appears to be a robust process, as enrichment factors as high as ∼105 were achieved following just a single round of panning on the immobilized lectin SBA. As a result, we were able to use this method to screen a library of MBPDQNAT variants in which the acceptor site residue in the −2 position was randomly mutated. Following one round of library screening by panning on immobilized SBA, we show that recovered phages were greatly enriched for D in the −2 position, which is consistent with the known substrate specificity of the C. jejuni OST.14 Interestingly, none of the clones encoded E at the −2 position, which may reflect the naturally occurring abundance of D over E in the −2 position of C. jejuni glycoproteins.14
Our ability to isolate mutants from an acceptor site library suggests that this new phage display method can be used to dissect or engineer other aspects of the acceptor protein. Along similar lines, Yamamoto et al.29 used phage display to isolate proteins with affinity towards immobilized carbohydrates. However, this method differs fundamentally from ours in that the displayed protein was a lectin called galectin-3 and the immobilized capture agent was blood group-specific oligosaccharides. Thus, while this earlier approach might permit genetic manipulation of the binding protein (i.e., for improved carbohydrate affinity or binding to a nonnative carbohydrate), it does not allow genetic manipulation of the enzymes involved in biosynthesis or transfer of the oligosaccharides. In contrast, it is conceivable that our system could be easily extended to genetic dissection of any of the enzymes in the glycosylation pathway. This could be accomplished by encoding any of the pgl enzymes (e.g., the PglB OST), either alone or as a bicistronic construct with the acceptor-g3p fusion, in the phagemid with the M13 ori. The resulting phage particles would permit physical linkage between a glycosylated acceptor protein and the genetic sequence of the pathway enzyme underlying the glyco-phenotype. In this manner, one could envision screening libraries of pathway enzymes for (i) revealing the molecular determinants of glycosylation enzyme function or (ii) directed evolution of glycosylation enzymes with altered or novel activities.
One drawback at the moment of our phage display system is the relatively low recovery rate achieved with the lectin capture method. While the cause is not currently known, we believe that this low recovery results from the relatively weak affinity of lectins for their carbohydrate lectins. One possible strategy to circumvent this issue would be to oxidize glycans and capture the larger glycoconjugates using hydrazide resins.30 Such a covalent capture method could increase the recovery of the putatively glycosylated phages and dramatically eliminate the phage present in the flow-through and washes shown in Figure 2(b). Another possible explanation for the low recovery rate that cannot be entirely ruled out is that a subpopulation of phages produced in pgl cells may not have glycans owing to: (i) inefficiency of PglB-mediated glycosylation of unnatural substrates14,15 like MBPDQNAT-g3p; and (ii) competition for assembly into phage particles between engineered g3p fusions and wild-type g3p encoded on the helper phage chromosome.31 Our own recent studies indicate that the efficiency of PglB is not an issue as a fairly high percentage (∼40–50%) of MBP with a C-terminal DQNAT glycosylation locus is N-glycosylated (Fisher and DeLisa, unpublished observations). Nonetheless, increasing the efficiency of phage glycosylation could have a net positive effect on our method. One possible improvement would be to use an E. coli strain lacking enzymes that are known to compete with the C. jejuni N-linked glycosylation process. For instance, Feldman et al. showed that the E. coli WaaL ligase transfers lipid-linked N-glycans onto lipid A and may deplete the pool of available substrates for PglB.11 These same authors found that strains lacking WaaL (e.g., E. coli CLM24) exhibited more efficient N-glycosylation of a native C. jejuni glycoprotein by PglB. Another improvement would be to utilize helper phage that lack a copy of the native g3p protein, so-called hyperphage,32 thereby permitting more efficient incorporation of the engineered MBPDQNAT-g3p fusion into phage particles. Alternatively, the “helperless” phage system developed by Bradbury and coworkers could offer a similar improvement.31 A final strategy that may improve phage display of glycoproteins would be to use the cotranslational signal recognition particle (SRP) pathway, which is the primary export route used by eurkaryotic N-glycoproteins. The use of the SRP pathway was also recently demonstrated to enhance the display levels of designed ankyrin-repeat proteins (DARPins) by 700-fold compared to post-translational Sec export.33
In conclusion, our phage display system provides a novel genetic assay for bacterial N-glycosylation in a genetically tractable host and thus provides a unique opportunity to render glycosylation an experimentally tunable mechanism. We anticipate that the reduced complexity of bacteria will allow for detailed genetic characterization of this important mechanism, which is currently very challenging to study in eukaryotes owing to the essential nature of glycosylation in these cells. Yet, because of the striking similarities between the prokaryotic and eukaryotic N-glycosylation pathways,6,34 lessons learned in glycosylation-competent E. coli may help to shed light on similar steps in the eukaryotic process. Moreover, the combination of a phage display system for N-linked glycosylation with the extensive toolkit available for metabolic pathway engineering and protein engineering promises to make E. coli one of the premier hosts for glycoengineering.
Materials and Methods
Bacterial strains and growth conditions
E. coli strain DH5α (F−endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG φ80lacZΔM15 Δ(lacZYA-argF)U169, hsdR17(rK− mK+), λ-) was used for cloning of phagemids and phagemid libraries, while strain TG1 (supE hsdΔ5 thi Δ(lac-proAB) F' [traD36 proAB lacI lacZΔM15]) was used for all phage production and titering experiments. E. coli strains were grown in Luria-Bertani or 2xTY medium at 30 (induction phase) or 37°C. Culture medium was supplemented with 50 mM glucose or 30 mM arabinose (induction phase), and with the appropriate antibiotics at the following concentrations: 100 μg/mL ampicillin, 20 μg/mL chloroamphenicol, and 50 μg/mL kanamycin. M9 minimal medium was used to select for the presence of F' plasmid when needed. VCSM13 (Stratagene) was used as the helper phage.
Phagemid and phagemid library construction
All phagemids constructed in this study were based on the phagemid pBAD2426 and were made using standard protocols.35 Initially, pBAD-g3p was constructed by amplifying the g3p cDNA from VCSM13 helper phage, using the primers g3pSalI (5'- ATA TAGGTCGACGCTGAAACTGTTGAAAGTTGTTTAGC -3') and g3pHindIII (5'-GCGATGAAGCTTTTATTAA GACTCCTTATTACGCAGTATGTTAG -3'), and ligated with SalI-HindIII digested pBAD24. Native MBP including its N-terminal Sec export signal was amplified from E. coli genomic DNA with primers MBPE coRI (5'-CACCGAATTCATGAAAATAAAAACAGGTG CACG-3') and MBPSalI (5'-TGCGTCGACTGTCGCA TTCTGATCGCTACCGCCGCCCTCGAGCTTGGTGA TACGAGTCTG -3') that introduced a single C-terminal glycosylation acceptor site (DQNAT).25 The resulting PCR product was ligated with EcoRI-SalI digested pBAD-g3p, resulting in the phagemid pBAD-MBPDQNAT-g3p. The phagemids pBAD-MBP-g3p, pBAD-DQNATg3p (including the signal sequence of MBP), pBAD-MBPAQNAT-g3p, and the phagemid library pBAD-MBPXQNAT-g3p were created similarly, using the same restriction sites, where the inserts were amplified using relevant primer pairs. All phagemids were confirmed by DNA sequencing. For the phagemid library, an NNK degenerate primer was used (Integrated DNA Technologies), where N represents any nucleotide and K represents guanine or thymine. Library construction and diversity was confirmed by DNA sequencing of phagemids that had been purified from randomly selected E. coli DH5α cells.
Expression of g3p fusion proteins and purification of phage
20-mL cultures of E. coli TG1 cells carrying a phagemid expressing g3p fusion protein and either pACYCpgl or pACYCpglmut plasmid (kindly provided by Dr. Brendan Wren and Dr. Markus Aebi) were superinfected with VCSM13 helper phage at OD600 of 0.5–0.6, and incubated at 37°C for 30 min without shaking. Infected cells were grown in 100 mL 2xTY medium supplemented with ampicillin, chloroamphenicol, kanamycin, and arabinose, for 16 h at 30°C. Phage purification using PEG/NaCl, and phage and helper phage titer determinations with 100 μL total assay volume were performed identically as described elsewhere.36
Biopanning on immobilized SBA
Phage particles (109 CFU in PBS buffer supplemented with 1 mM CaCl2, 1 mM MnCl2 and 1 mM MgCl2) were mixed with 1 mL of agarose-bound soybean agglutinin (SBA, Vector Laboratories) and incubated for 1 h at 25°C. Following incubation, mixtures were subjected to four wash steps with 10 mL of PBS, followed by three wash steps with 10 mL of 30 mM galactose in PBS or PBST (PBS with 0.1% Tween20). The wash buffers were decanted after centrifugation at 100 g for 5 min. The glycosylated phages were eluted in three steps using 1 mL of 300 mM galactose in PBS. Eluted phages were used directly to infect E. coli TG1 cells to quantify phage titer or stored at −20°C. The infected cells were selected on ampicillin, and the resulting colonies were used to determine the number of CFU per mL of culture supernatant and/or were characterized by colony PCR. In the case of colony PCR, a single primer set was developed that yielded a 301-bp product for MBPDQNAT-g3p and a 268-bp product for MBP-g3p, thereby allowing facile discrimination between glycosylated and nonglycosylated phages, respectively. All colony PCR reactions were separated on 2% agarose gels and also confirmed by DNA sequencing.
Western blot analysis
Proteins were separated by 10% SDS-polyacrylamide gels (Bio-Rad), and Western blotting was performed as described previously.37 Blots were probed with either anti-MBP antibodies conjugated with horseradish peroxidase (HRP) (New England Biolabs) or hR6P antiserum that is specific for the C. jejuni heptasaccharide (kindly provided by Dr. Brendan Wren and Dr. Markus Aebi). In the case of hR6P, anti-rabbit IgG-HRP (Promega) was used as the secondary antibody.
Acknowledgments
We thank Brendan Wren for plasmids and Markus Aebi for antiserum used in this work. This work was supported by a Corning, Inc. Foundation Science Fellowship (to T.J.M.).
References
- 1.Apweiler R, Hermjakob H, Sharon N. On the frequency of protein glycosylation, as deduced from analysis of the SWISS-PROT database. Biochim Biophys Acta. 1999;1473:4–8. doi: 10.1016/s0304-4165(99)00165-8. [DOI] [PubMed] [Google Scholar]
- 2.Turnbull JE, Field RA. Emerging glycomics technologies. Nat Chem Biol. 2007;3:74–77. doi: 10.1038/nchembio0207-74. [DOI] [PubMed] [Google Scholar]
- 3.Helenius A, Aebi M. Intracellular functions of N-linked glycans. Science. 2001;291:2364–2369. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]
- 4.Helenius A, Aebi M. Roles of N-linked glycans in the endoplasmic reticulum. Annu Rev Biochem. 2004;73:1019–1049. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 5.Yurist-Doutsch S, Chaban B, VanDyke DJ, Jarrell KF, Eichler J. Sweet to the extreme: protein glycosylation in Archaea. Mol Microbiol. 2008;68:1079–1084. doi: 10.1111/j.1365-2958.2008.06224.x. [DOI] [PubMed] [Google Scholar]
- 6.Szymanski CM, Wren BW. Protein glycosylation in bacterial mucosal pathogens. Nat Rev Microbiol. 2005;3:225–237. doi: 10.1038/nrmicro1100. [DOI] [PubMed] [Google Scholar]
- 7.Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P. Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol. 1999;32:1022–1030. doi: 10.1046/j.1365-2958.1999.01415.x. [DOI] [PubMed] [Google Scholar]
- 8.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science. 2002;298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 9.Linton D, Allan E, Karlyshev AV, Cronshaw AD, Wren BW. Identification of N-acetylgalactosamine-containing glycoproteins PEB3 and CgpA in Campylobacter jejuni. Mol Microbiol. 2002;43:497–508. doi: 10.1046/j.1365-2958.2002.02762.x. [DOI] [PubMed] [Google Scholar]
- 10.Young NM, Brisson JR, Kelly J, Watson DC, Tessier L, Lanthier PH, Jarrell HC, Cadotte N, St Michael F, Aberg E, Szymanski CM. Structure of the N-linked glycan present on multiple glycoproteins in the gram-negative bacterium, Campylobacter jejuni. J Biol Chem. 2002;277:42530–42539. doi: 10.1074/jbc.M206114200. [DOI] [PubMed] [Google Scholar]
- 11.Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci USA. 2005;102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alaimo C, Catrein I, Morf L, Marolda CL, Callewaert N, Valvano MA, Feldman MF, Aebi M. Two distinct but interchangeable mechanisms for flipping of lipid-linked oligosaccharides. EMBO J. 2006;25:967–976. doi: 10.1038/sj.emboj.7601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelly J, Jarrell H, Millar L, Tessier L, Fiori LM, Lau PC, Allan B, Szymanski CM. Biosynthesis of the N-linked glycan in Campylobacter jejuni and addition onto protein through block transfer. J Bacteriol. 2006;188:2427–2434. doi: 10.1128/JB.188.7.2427-2434.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kowarik M, Young NM, Numao S, Schulz BL, Hug I, Callewaert N, Mills DC, Watson DC, Hernandez M, Kelly JF, Wacker M, Aebi M. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J. 2006;25:1957–1966. doi: 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwarz F, Huang W, Li C, Schulz BL, Lizak C, Palumbo A, Numao S, Neri D, Aebi M, Wang LX. A combined method for producing homogeneous glycoproteins with eukaryotic N-glycosylation. Nat Chem Biol. 2010;6:264–266. doi: 10.1038/nchembio.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kowarik M, Numao S, Feldman MF, Schulz BL, Callewaert N, Kiermaier E, Catrein I, Aebi M. N-linked glycosylation of folded proteins by the bacterial oligosaccharyltransferase. Science. 2006;314:1148–1150. doi: 10.1126/science.1134351. [DOI] [PubMed] [Google Scholar]
- 17.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 18.Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- 19.Rader C, Barbas CF., III Phage display of combinatorial antibody libraries. Curr Opin Biotechnol. 1997;8:503–508. doi: 10.1016/s0958-1669(97)80075-4. [DOI] [PubMed] [Google Scholar]
- 20.Smith GP. Surface presentation of protein epitopes using bacteriophage expression systems. Curr Opin Biotechnol. 1991;2:668–673. doi: 10.1016/0958-1669(91)90032-z. [DOI] [PubMed] [Google Scholar]
- 21.Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Methods Enzymol. 2000;326:480–505. doi: 10.1016/s0076-6879(00)26071-0. [DOI] [PubMed] [Google Scholar]
- 22.Watters AL, Baker D. Searching for folded proteins in vitro and in silico. Eur J Biochem. 2004;271:1615–1622. doi: 10.1111/j.1432-1033.2004.04072.x. [DOI] [PubMed] [Google Scholar]
- 23.Forrer P, Jung S, Pluckthun A. Beyond binding: using phage display to select for structure, folding and enzymatic activity in proteins. Curr Opin Struct Biol. 1999;9:514–520. doi: 10.1016/S0959-440X(99)80073-6. [DOI] [PubMed] [Google Scholar]
- 24.Lafond R, Zhan X, Georgiou G. Screening and selection strategies for disulfide isomerase activity. Methods Mol Biol. 2003;230:239–257. doi: 10.1385/1-59259-396-8:239. [DOI] [PubMed] [Google Scholar]
- 25.Chen MM, Glover KJ, Imperiali B. From peptide to protecomparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry. 2007;46:5579–5585. doi: 10.1021/bi602633n. [DOI] [PubMed] [Google Scholar]
- 26.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swamy MJ, Krishna Sastry MV, Khan MI, Surolia A. Thermodynamic and kinetic studies on saccharide binding to soya-bean agglutinin. Biochem J. 1986;234:515–522. doi: 10.1042/bj2340515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monsigny M, Mayer R, Roche AC. Sugar-lectin interactions: sugar clusters, lectin multivalency and avidity. Carbohydr Lett. 2000;4:35–52. [PubMed] [Google Scholar]
- 29.Yamamoto M, Kominato Y, Yamamoto F. Phage display cDNA cloning of protein with carbohydrate affinity. Biochem Biophys Res Commun. 1999;255:194–199. doi: 10.1006/bbrc.1999.0175. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Li XJ, Martin DB, Aebersold R. Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat Biotechnol. 2003;21:660–666. doi: 10.1038/nbt827. [DOI] [PubMed] [Google Scholar]
- 31.Chasteen L, Ayriss J, Pavlik P, Bradbury AR. Eliminating helper phage from phage display. Nucleic Acids Res. 2006;34:e145. doi: 10.1093/nar/gkl772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rondot S, Koch J, Breitling F, Dubel S. A helper phage to improve single-chain antibody presentation in phage display. Nat Biotechnol. 2001;19:75–78. doi: 10.1038/83567. [DOI] [PubMed] [Google Scholar]
- 33.Steiner D, Forrer P, Stumpp MT, Pluckthun A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat Biotechnol. 2006;24:823–831. doi: 10.1038/nbt1218. [DOI] [PubMed] [Google Scholar]
- 34.Weerapana E, Imperiali B. Asparagine-linked protein glycosylation: from eukaryotic to prokaryotic systems. Glycobiology. 2006;16:91R–101R. doi: 10.1093/glycob/cwj099. [DOI] [PubMed] [Google Scholar]
- 35.Sambrook JF, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- 36.Hertveldt K, Belien T, Volckaert G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol Biol. 2009;502:321–339. doi: 10.1007/978-1-60327-565-1_19. [DOI] [PubMed] [Google Scholar]
- 37.DeLisa MP, Tullman D, Georgiou G. Folding quality control in the export of proteins by the bacterial twin-arginine translocation pathway. Proc Natl Acad Sci USA. 2003;100:6115–6120. doi: 10.1073/pnas.0937838100. [DOI] [PMC free article] [PubMed] [Google Scholar]