

Abstract
Visual loss following head trauma is common, and the diagnosis can be challenging for the neurologist called to perform an emergency room assessment. The approach to the patient with post-traumatic visual loss is complicated by a wide range of potential ocular and brain injuries with varying pathophysiology. In addition to direct injuries of the eye and orbit, traumatic optic neuropathies, carotid cavernous fistulas, and damage to the intracranial visual pathways are classic causes of visual loss after head trauma. This review provides an update on the diagnosis and management of these conditions.
Keywords: Carotid cavernous fistula, Traumatic brain injury, Traumatic optic neuropathy, Visual field defect, Visual loss
Visual loss is common after head trauma, but its diagnosis is often delayed. The clinical assessment is complicated by the fact that trauma patients may be unconscious and unable to provide a clinical history. Examination can be limited by lack of cooperation, concomitant physical injuries, and decreased level of consciousness. Alternatively, the traumatic event may seem minimal in relation to the visual loss, and the patient may be neurologically intact. Obvious evidence of trauma in the form of ocular or periorbital hemorrhage, laceration, or ecchymosis may be absent, and the injury causing the visual loss may be concealed. A wide range of lesions can result in visual loss, and the visual system can be affected at multiple levels (Table 1). A careful examination combined with appropriate neuroimaging should elucidate the nature of the visual loss and guide initiation of optimal management.1-6 The aim of this review is to help neurologists develop a rational approach to evaluating the patient with post-traumatic visual loss.
Table 1. Causes of Post-Traumatic Acute Visual Loss.
| Refractive Error |
| Glasses or contact lenses are lost or damaged at the time of trauma |
| Ocular Injury |
| Ruptured globe (anterior with corneal laceration or posterior with scleral laceration) |
| Intraocular foreign body |
| Exposure keratopathy (secondary to proptosis, lid laceration, or 7th nerve dysfunction) |
| Corneal edema (from airbag injury) |
| Corneal abrasion |
| Hyphema (blood in anterior chamber) |
| Traumatic iritis (often delayed by approximately 24 h) |
| Traumatic mydriasis (and decreased accommodation) |
| Lens subluxation or luxation |
| Vitreous hemorrhage |
| Commotio retinae |
| Retinal detachment |
| Retinal ischemia from carotid dissection |
| Retinal fat emboli |
| Choroidal rupture |
| Optic Nerve |
| Direct traumatic optic neuropathy |
| Indirect traumatic optic neuropathy |
| Intrasheath hematoma |
| Avulsion of the optic nerve head |
| Penetrating injuries of the orbit with direct optic nerve injury |
| Intraorbital foreign body |
| Optic nerve ischemia from carotid dissection |
| Orbit |
| Orbital fracture |
| Orbital hemorrhage |
| Orbital emphysema |
| Carotid cavernous fistula |
| Subperiosteal hemorrhage |
| Intracranial Optic Pathways |
| Chiasmal or retrochiasmal direct injury |
| Chiasmal or retrochiasmal indirect injury |
| Hemorrhage or hematoma compressing the chiasm |
| Cerebral diffuse axonal injury with homonymous hemianopia |
| Intraparenchymal hemorrhage with homonymous hemianopia |
| Cerebral infarction (posterior cerebral artery) secondary to increased intracranial pressure/herniation with homonymous hemianopia or cerebral blindness |
| Cerebral infarction secondary to cervical artery dissection with homonymous hemianopia or cerebral blindness |
Approach to the Patient With Post-Traumatic Visual Loss
Unless the trauma is minimal and the visual loss is isolated, priority is given to assessment of vital functions in the emergency room. It is only once the patient is stabilized, and severe trauma to the head, face, spine, and other essential organs is ruled out, that attention is paid to visual function.1
A basic ophthalmologic evaluation should be performed systematically in the emergency room. Trauma patients, especially those with decreased consciousness, or severe pain and anxiety, may not be aware of visual changes until they are specifically questioned. Examination should include external inspection of the eyes and periorbital region, measurement of visual acuity, pupillary examination, testing of extraocular movements, and funduscopic examination. Any abnormality should prompt an emergent ophthalmologic consultation.
In most cases, traumatic visual loss is related to direct ocular injury and occurs in the setting of severe head trauma associated with loss of consciousness.3-6 Ocular trauma such as hyphema or ocular penetrating injuries and foreign body may require urgent ophthalmologic treatment (Figure 1). Less common is unilateral or bilateral visual loss with normal ocular appearance, suggesting trauma to the optic nerve or intracranial visual pathways, for which a neurologic or neurosurgical consultation is often requested.
Figure 1.
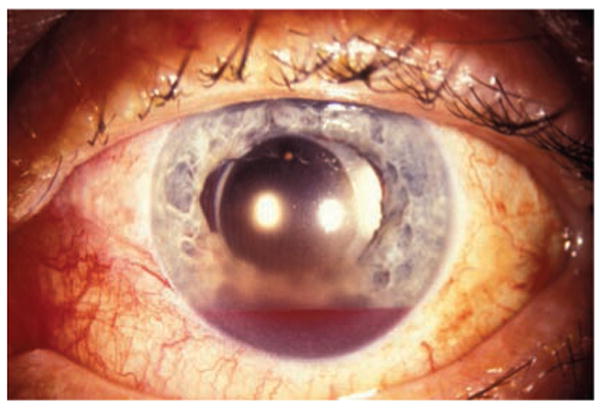
Blood in the anterior chamber (hyphema) after blunt trauma to the eye and face.
Assessment of Visual Function in the Emergency Room
A simple, systematic approach allows neurologists to evaluate the visual function of trauma patients.
External Inspection
External inspection should always be performed to detect signs of globe laceration or intraocular foreign body. The presence of lid laceration, subconjunctival hemorrhage, corneal laceration, collapsed globe, irregularity or displacement of the pupil, and blood in the anterior chamber (hyphema) should prompt an emergent ophthalmologic consultation. The eye should not be touched and should be protected by a shield (or a plastic cup) taped over the eye.4 If the eye appears intact, palpating the orbital rim can identify fractures. Periorbital swelling can conceal enophthalmos or proptosis. Auscultation of the head and orbit checking for an orbital bruit (suggesting a carotid cavernous fistula) should be performed in any trauma patient with periorbital ecchymosis, proptosis, or ophthalmoplegia.
Visual Acuity
Visual acuity can easily be checked at the bedside with a hand-held near card, newspaper, or magazine. It should be kept in mind that injured patients often lose their glasses at the time of trauma and may not be alert enough to mention this in the emergency room. For patients older than 50 years, a pinhole card, reading glasses, or a +3.00 lens are useful at the bedside to correct a refractive error.
Confrontation Visual Fields
Confrontation visual fields can be tested by checking for the ability to count fingers in all 4 quadrants. This is helpful in the emergency room; however, it can only be performed in alert and cooperative patients. Formal visual field testing can be obtained later, once the patient is able to sit upright.
Pupils
Pupils can easily be assessed in both responsive and unresponsive trauma patients. In cases of unilateral visual loss, a relative afferent pupillary defect indicates an optic neuropathy or a large retinal lesion (such as retinal detachment or retinal artery occlusion). In cases of bilateral optic neuropathies, there is no relative afferent pupillary defect, but the pupils are sluggish in response to light (as compared with cerebral blindness, in which the pupils are normal). The pupils may also be asymmetric (anisocoria) as a result of a Horner syndrome (from carotid dissection), a third nerve palsy, or from direct ocular trauma (traumatic mydriasis or ocular perforation). Anisocoria or a relative afferent pupillary defect should prompt an emergent ocular examination.
Inspection of Extraocular Movements
This is part of the emergency room examination for patients with head trauma. Conscious patients with abnormal eye movements and good vision may complain of diplopia. Abnormal eye movements may result from brainstem or cranial nerve injuries. Fourth nerve palsy is most common with mild to moderate closed head injury, followed by sixth nerve palsy, and then by third nerve palsy with relatively more severe closed head injury.2 Third nerve palsy may indicate herniation and is always of concern in the acute trauma patient who is not awake and alert. Ophthalmoplegia is also common in carotid cavernous fistula and with any orbital syndrome or orbital fracture. In comatose patients, inspection of eye position may disclose ocular deviation, suggesting ophthalmoplegia. In the acute trauma patient, the oculocephalic responses with the doll's eye maneuver should not be tested as this requires active mobilization of the neck.
Funduscopic Examination
Funduscopic examination should be attempted in the emergency room in all trauma patients. This should be done without pharmacologic dilation. Visualizing the optic nerve head confirms that the eye is clear of hemorrhage and that the ocular media is clear. There is no need to systematically perform a dilated funduscopic examination acutely unless the patient complains of visual loss. In these cases, the pupils should be pharmacologically dilated only by the consulted ophthalmologist, who will first verify the absence of ocular injury or globe perforation. If there is a globe perforation, then further examination is performed in the operating room by the ophthalmologist.
Neuroimaging
The presence of a neuroimaging abnormality, particularly intracranial hemorrhage, is significantly associated with specific neuro-ophthalmic deficits. Neuroimaging abnormalities may be a more reliable predictor of specific neuro-ophthalmic outcomes than loss of consciousness.6 Brain CT without contrast is still the best imaging modality when assessing acute head trauma. When visual loss is suspected, both axial and coronal orbital views should be included. CT is the neuroimaging study of choice for visualizing the bony anatomy of the optic canals and the paranasal and frontal sinuses, to rule out an intraocular or orbital foreign body, and to look for acute orbital or intracranial hemorrhage.7 MRI is the study of choice for visualizing soft tissue, but it is not usually performed acutely in head trauma patients, who are often unstable and poorly cooperative. When brain ischemia is suspected, MRI with diffusion-weighted images and magnetic resonance angiogram can be extremely helpful.8 Some centers systematically recommend a CT angiogram of the aortic arch, cervical vessels, and head in cases of severe head or neck trauma to investigate the possibility of associated arterial dissection or carotid cavernous fistula.9
Clinical Syndromes of Post-Traumatic Visual Loss
Traumatic Optic Neuropathy
Traumatic optic neuropathy has traditionally been separated into direct and indirect traumatic optic neuropathy. 1,9-30 In direct traumatic optic neuropathy, there is direct injury of the optic nerve causing functional and anatomic disruption of the optic nerve. Indirect traumatic optic neuropathy refers to the transmission of force distant from the optic nerve, preserving ocular and cerebral tissue plane integrity but indirectly disrupting the functional and anatomic integrity of the optic nerve. Clinically, traumatic injury to the orbital, intracanalicular, or intracranial segment of the optic nerve is presumed when the neuro-ophthalmologic examination shows unilateral decreased visual acuity and color vision, an ipsilateral relative afferent pupillary defect, a normal appearing fundus or mild optic nerve edema acutely, and no apparent intraocular pathology. The affected optic nerve becomes pale only 4 to 6 weeks after injury. Finding optic nerve pallor at the time of trauma suggests pre-existing optic neuropathy. The diagnosis of traumatic optic neuropathy is clinical and neuroimaging is only useful to rule out compression of the optic nerve by a bone fragment or orbital damage such as fracture or hemorrhage.
Direct traumatic optic neuropathy
Pathologic mechanisms of direct injury include optic nerve avulsion, stretch injury, shearing, contusion, laceration, and disruption. Orbital hemorrhages can create an immediate compressive optic neuropathy with a compartment syndrome. Hemorrhages within the dural sheath and interstitial optic nerve hemorrhages are often observed in autopsy studies following closed head injury.5 The optic nerve is tightly tethered in the optic canal and is subject to stretch injury, shearing, and ischemic necrosis during brain shifts. Brain shifting can also result in upward displacement of the intracranial segment of the optic nerve against the falciform dural fold, which lies above the optic nerve adjacent to the optic canal.6 The optic chiasm can also be directly injured by compression from hematomas and other direct pathologic mechanisms.
Although spontaneous visual recovery can rarely occur after direct traumatic optic neuropathy, surgical decompression of the optic nerve is usually performed when imaging confirms an orbital fracture and compression of the optic nerve by a bone fragment. Orbital hemorrhages with compartment syndrome presenting with acute proptosis, chemosis, and elevated intraocular pressure may require emergent orbital decompression, which is usually performed by doing a canthotomy and cantholysis (surgical opening of the lateral canthus) at bedside in the emergency room. Rarely, surgical orbital decompression, optic nerve sheath fenestration for intrasheath hematoma, or evacuation of subperiosteal orbital hematoma is performed.21
Indirect traumatic optic neuropathy
Indirect optic nerve injury typically arises from energy absorbed by the optic nerve within the confines of the bony optic nerve canal at the moment of impact, usually from blunt trauma to the forehead.14 The nature of the bony architecture of the orbit tends to directly transfer forces from frontal blows to the superolateral orbital rim to the intracanalicular portion of the optic nerve canal. Indirect traumatic optic neuropathy may occur even with very mild trauma. Visual loss is immediate or may be delayed over a few days, presumably because optic nerve edema may lead to progressive ischemia from compression within the confines of the bony optic canal.15 Trauma and ischemia can precipitate a destructive cascade of interrelated events that are well known to cause secondary damage within the central nervous system. These include oxidative stress, release of inflammatory mediators, breakdown of the blood-brain barrier with increased macrophage activity, and intracellular calcium influx leading to excitotoxic damage and apoptosis.31
Because of progression of vision loss, which is often delayed, some authors have suggested that a window of opportunity exists for early surgical intervention (such as optic canal decompression), or for medical intervention (steroid therapy or neuroprotection). However, no randomized study has shown that surgical decompression of the optic nerve or treatment with steroids improves the visual outcome of patients with head trauma.12,18 The International Optic Nerve Trauma study20 was a large prospective study that compared the visual outcomes of traumatic optic neuropathy treated with variable uncontrolled doses of corticosteroids, optic canal decompression, or observation alone, within 7 days of injury. No significant difference between any of the treatment options was found. The study concluded that neither corticosteroids nor optic canal decompression surgery benefited patients with traumatic optic neuropathy,20 confirming other studies.16,27
Very high-dose steroids, such as those given for the treatment of spinal cord injury, should not be prescribed for the treatment of traumatic optic neuropathy.17 Very high-dose corticosteroids (intravenous loading dose of 30 mg/kg followed by a continuous infusion of 5.4 mg/kg/h for 24 or 48 h) have been used for the treatment of acute spinal cord injury since the publication of the second and third National Acute Spinal Cord Studies (NASCIS II and III).22,23 NASCIS II22 was a multicenter, randomized, double-blinded, placebo-controlled study of acute spinal cord injury. Patients were randomized to placebo, naloxone, or high-dose methylprednisolone within 12 hours of injury. A small but significant improvement in the methylprednisolone treatment arm compared with the placebo arm was reported, but only for those treated within 8 hours after injury. Post-hoc analysis revealed that treatment past the 8-hour mark had deleterious effects.24,25 This therapeutic regimen was subsequently adopted for the treatment of traumatic optic neuropathy, although no randomized study has yet shown benefit.
Experimental studies have investigated the effects of high-dose steroid therapy in traumatic optic neuropathy and suggested no benefit and potentially harmful effects in induced optic nerve crush injuries.32-34 It is therefore not surprising that the “no treatment” option has been recently suggested for patients with indirect traumatic optic neuropathy. The rationale for withholding steroid treatment in indirect traumatic optic neuropathy is further supported by the results of the Corticosteroid Randomization After Significant Head Injury (CRASH) trial.35 This multicenter, randomized, placebo-controlled study randomized patients to placebo or high-dose methylprednisolone for 48 hours, within 8 hours of head injury. The study was stopped prematurely based on safety monitoring data showing a higher risk of death from all causes at 2 weeks in the methylprednisolone-treated arm.35 It was then suggested that not only were high-dose steroids not helpful after head trauma, but were potentially harmful. The same conclusions likely apply to patients with traumatic optic neuropathy who often have associated brain injuries.
The drive to treat indirect traumatic optic neuropathy may be fueled by a sense of helplessness, but there is currently no good evidence to support treatment over no treatment; therefore, no treatment is rationally and medicolegally defensible, especially when keeping in mind the ethical principle of doing no harm. Conversely, it is frustrating to offer patients no treatment when they are faced with visual loss. Experts in the field now agree that neither current medical nor surgical treatments are the standard of care in traumatic optic neuropathy.1,17 Although some authors have suggested the administration of moderate-dose methylprednisolone (250 mg intravenously 4 times daily for 24-48 h), believing that this dose might decrease intracanalicular swelling without being neurotoxic,9 there is currently no evidence that this treatment may be better than no treatment.16
Although it may be some time before a randomized controlled trial of traumatic optic neuropathy is organized, the pace of research in neuroprotection is not slow.31 There is recent experimental evidence supporting the neuroprotective and neuroregenerative effects of erythropoietin,36 minocycline,37 and progesterone38 in patients with head trauma. Transcorneal electrical stimulation for neuroprotection in the acute phase may also hold some promise.39
Carotid Cavernous Fistulas
Carotid cavernous fistulas consist of abnormally communicating blood flow from the internal carotid arterial system to the cavernous sinus venous system. Carotid cavernous fistulas can be classified etiologically as traumatic or spontaneous, hemodynamically as high flow or low flow, and anatomically as direct or dural.40-42 The classification system described by Barrow and colleagues41 includes Type A fistulas (direct shunts between the internal carotid artery and the surrounding cavernous sinus), and Types B, C, and D fistulas. The latter are low-flow, indirect dural shunts that are believed to be congenital or spontaneous arteriovenous connections; they are mostly seen in older women with hypertension, diabetes, and atherosclerosis, and rarely result from trauma. Conversely, high-flow, direct Type A carotid cavernous fistulas are a classic complication of head trauma, and they represent 70% to 90% of all carotid cavernous fistulas. They often present dramatically days or weeks after head injury with a triad of pulsating exophthalmos, conjunctival chemosis, and orbital bruit. If not treated, progressive visual loss may ensue.40
Visual loss associated with direct carotid cavernous fistula may be either immediate (most often from coincident ocular or optic nerve damage) or delayed. The threat of irreversible secondary visual loss with carotid cavernous fistula usually arises from the increased intraocular pressure that can be severe enough to damage the optic nerve or retina. There should be no delay in diagnosis because permanent visual loss may progressively develop if intraocular pressure is not reduced, and cranial nerve palsies may become permanent. Other causes of secondary delayed visual loss in carotid cavernous fistula include exposure keratopathy (secondary to proptosis), venous stasis retinopathy, central retinal vein occlusion, serous choroidal detachment, and anterior or posterior ischemic optic neuropathy. Visual loss occurs in up to 90% of patients with direct carotid cavernous fistula and is the major cause of morbidity in those whose fistulas are not complicated by intracranial bleeding.40
The ocular manifestations of carotid cavernous fistula reflect the underlying pathophysiology of overload and retrograde filling of the venous system. Arterial blood flows anteriorly into the superior or inferior ophthalmic veins, and arterial and venous stasis causes increased episcleral venous pressure, along with decreased arterial blood flow to the cranial nerves within the cavernous sinus. This situation leads to the hallmarks of carotid cavernous fistula, including arterialization of the conjunctival vessels, conjunctival chemosis (Figure 2), and the classic fundus findings of ipsilateral optic disc swelling, dilated retinal veins, and intraretinal hemorrhages. Proptosis, from congestion of orbital tissues, is often among the earliest findings, developing acutely or progressively within a few days after trauma. Pulsating exophthalmos is present in most patients with direct carotid cavernous fistula. Increased intraocular pressure from increased episcleral venous pressure and orbital congestion develops in up to half of patients with untreated direct carotid cavernous fistula. Diplopia is usually present with direct carotid cavernous fistula. Mechanisms of diplopia include the initial trauma, mechanical restriction of the extraocular muscles, cranial nerve ischemia from the fistula, and direct compression of the cranial nerves by dilated petrosal venous sinuses. The sixth cranial nerve is most commonly affected due to its free-floating location within the cavernous sinus.40
Figure 2.
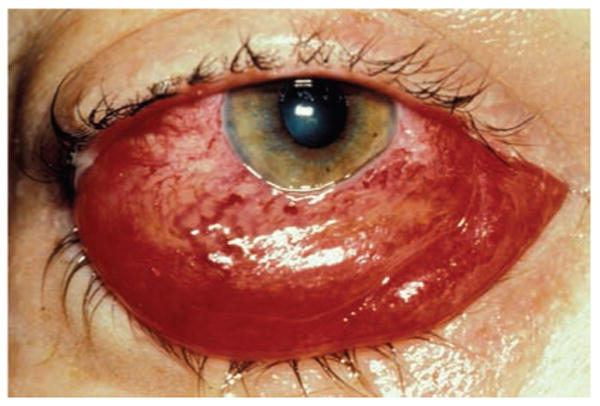
Conjunctival chemosis secondary to orbital congestion in a patient with a direct traumatic carotid cavernous fistula.
The diagnosis of direct carotid cavernous fistula is usually made with noninvasive imaging such as CT and MRI. Findings of a distended superior or inferior orbital vein and diffuse enlargement of the extraocular muscles are highly suspicious for carotid cavernous fistula in the context of trauma (Figures 3, 4, and 5). Catheter angiography showing filling of the cavernous sinus during the arterial phase is still the gold standard to visualize the complex anatomy of the cavernous sinus and the exact localization or nature of a fistula, and is usually performed at the time of treatment.
Figure 3.
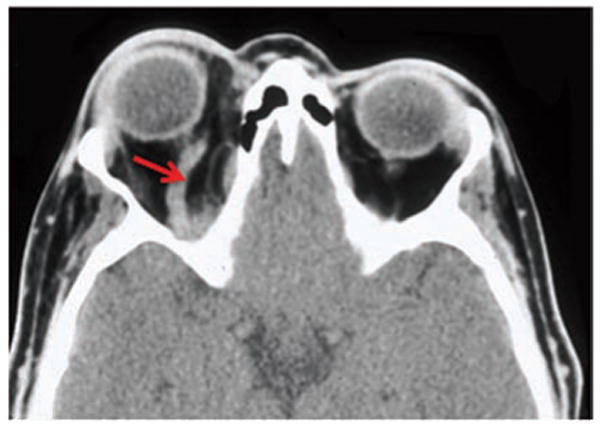
Axial CT with contrast showing a dilated right superior ophthalmic vein (arrow) secondary to a direct traumatic carotid cavernous fistula.
Figure 4.
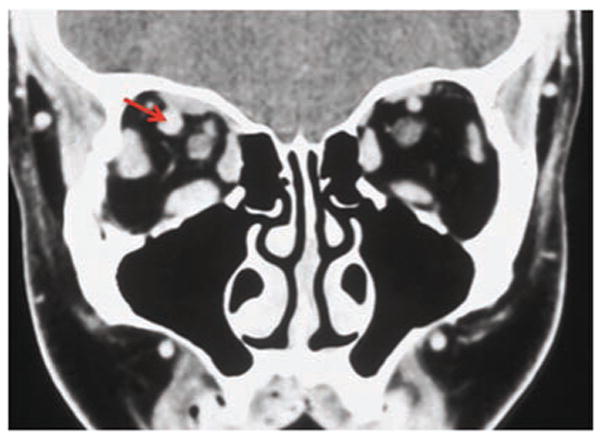
Coronal CT showing the dilated right superior ophthalmic vein (arrow), along with diffuse enlargement of the extraocular muscles.
Figure 5.
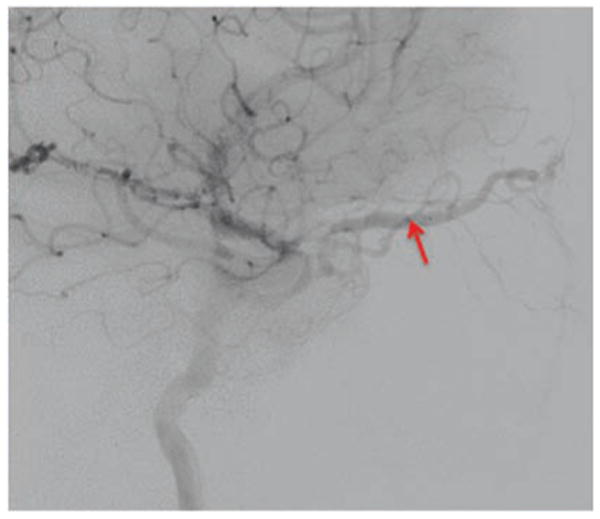
Catheter angiography (lateral view) showing a very dilated superior ophthalmic vein (arrow).
Urgent treatment is usually required for high-flow Type A fistulas, especially when visual loss is present. The intraocular pressure can be reduced medically, but only treatment of the fistula (most often with intravascular approaches using selective embolization and detachable balloon occlusion) prevents visual loss.42
Optic Pathway Trauma
Optic pathway trauma consisting of damage to the chiasm or the retrochiasmal visual pathways is common in head trauma.43-49 Chiasmal damage is second to intracanalicular optic nerve segment damage in frequency,11 and may be a result of indirect or penetrating injury. Visual field testing demonstrating bitemporal hemianopia, in combination with brain MRI, is usually diagnostic; however, little is known about optimal management, and most patients are treated empirically with corticosteroids or observation.
Patients with head trauma often have multiple and extensive intracranial lesions that may involve the optic pathways and result in various homonymous visual field defects or bilateral visual loss (cerebral blindness).3,44 Disruption of tissue integrity secondary to trauma is the underlying etiology in most cases, but vascular causes are also common. Post-traumatic cerebral edema can cause uncal herniation to compress the ipsilateral posterior cerebral artery, resulting in unilateral or bilateral occipital lobe infarctions.45,46 Intraparenchymal hemorrhages, epidural or subdural hematoma with mass effect, and subarachnoid hemorrhage with vasospasm and resultant cerebral infarction may also produce homonymous hemianopic visual field defects. Cervical artery dissections with subsequent cerebral infarction and homonymous hemianopia are a classic cause of visual loss after trauma.47,48 Post-traumatic cerebral venous thrombosis may not only result in venous infarctions involving the occipital lobes, but may also produce visual loss from bilateral papilledema and secondary optic atrophy.49,50
Because visual loss may interfere with rehabilitation, and because many trauma patients are unaware of or unable to complain of visual loss, it is important that formal evaluation of visual function, including visual field testing, be obtained in head trauma patients during rehabilitation. Indeed, documentation of visual function is essential for proper evaluation of the ability to return to school or work, and to drive a vehicle.
Conclusions
Post-traumatic visual loss is common and can arise from a number of mechanisms. Identification of direct ocular trauma is done in the emergency room at the time of presentation. Topographic diagnosis of optic nerve lesions or lesions of the intracranial visual pathways combined with appropriate neuroimaging can help to identify neuro-ophthalmic causes of post-traumatic visual loss. Appreciation of visual field defects from intracranial lesions is often delayed; therefore, visual fields should be systematically obtained when the patient is in rehabilitation prior to returning to school, work, or driving.
Main Points.
Visual loss is common after head trauma, but its diagnosis is often delayed due to the complication that trauma patients may be unconscious and unable to provide a clinical history. Examination can be limited by lack of cooperation, concomitant physical injuries, and decreased level of consciousness.
A systematic approach allows neurologists to evaluate the visual function of trauma patients in the emergency room, and should include external inspection of the eyes and periorbital region, measurement of visual acuity, pupillary examination, testing of extraocular movements, and funduscopic examination.
CT is the neuroimaging study of choice for visualizing the bony anatomy of the optic canals and the paranasal and frontal sinuses, to rule out an intraocular or orbital foreign body, and to look for acute orbital or intracranial hemorrhage. MRI is the study of choice for visualizing soft tissue, but it is not usually performed acutely in head trauma patients, who are often unstable and poorly cooperative.
Neither corticosteroids nor optic canal decompression surgery benefited patients with indirect traumatic optic neuropathy.
Catheter angiography showing filling of the cavernous sinus during the arterial phase is still the gold standard to visualize the complex anatomy of the cavernous sinus and the exact localization or nature of a fistula, and is usually performed at the time of treatment. There should be no delay in diagnosis because permanent visual loss may progressively develop if intraocular pressure is not reduced, and cranial nerve palsies may become permanent.
Acknowledgments
This study was supported in part by a departmental grant (Department of Ophthalmology) from Research to Prevent Blindness, Inc., New York, NY, and by core grant P30-EY06360 (Department of Ophthalmology) from the National Institutes of Health, Bethesda, MD. Dr. Newman is a recipient of a Research to Prevent Blindness Lew R. Wasserman Merit Award.
References
- 1.Levin LA. Neuro-ophthalmologic diagnosis and therapy of central nervous system trauma. Ophthalmol Clin North Am. 2004;17:455–464. doi: 10.1016/j.ohc.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 2.Dhaliwal A, West AL, Trobe JD, et al. Third, fourth, and sixth cranial nerve palsies following closed head injury. J Neuroophthalmol. 2006;26:4–10. doi: 10.1097/01.wno.0000204661.48806.1d. [DOI] [PubMed] [Google Scholar]
- 3.Van Stavern GP, Biousse V, Lynn MJ, et al. Neuro-ophthalmic manifestations of head trauma. J Neuroophthalmol. 2001;21:112–117. doi: 10.1097/00041327-200106000-00012. [DOI] [PubMed] [Google Scholar]
- 4.Keane JR, Baloh RW. Posttraumatic cranial neuropathies. Neurol Clin. 1992;10:849–867. [PubMed] [Google Scholar]
- 5.Crompton MR. Visual lesions in closed head injury. Brain. 1970;93:785–792. doi: 10.1093/brain/93.4.785. [DOI] [PubMed] [Google Scholar]
- 6.Gossman MD, Roberts DM, Barr CC. Ophthalmic aspects of orbital injury: a comprehensive diagnostic and management approach. Clin Plast Surg. 1992;19:71–85. [PubMed] [Google Scholar]
- 7.Seiff SR, Berger MS, Guyon J, et al. Computed tomographic evaluation of the optic canal in sudden traumatic blindness. Am J Ophthalmol. 1984;98:751–755. doi: 10.1016/0002-9394(84)90693-7. [DOI] [PubMed] [Google Scholar]
- 8.Lee AG. Imaging for neuro-ophthalmic and orbital disease. Am J Ophthalmol. 2004;138:852–862. doi: 10.1016/j.ajo.2004.06.069. [DOI] [PubMed] [Google Scholar]
- 9.Steinsapir K, Goldberg R. Traumatic optic neuropathy: a critical update. Compr Ophthalmol Update. 2005;6:11–21. [Google Scholar]
- 10.Sarkies N. Traumatic optic neuropathy. Eye. 2004;18:112–115. doi: 10.1038/sj.eye.6701571. [DOI] [PubMed] [Google Scholar]
- 11.Steinsapir KD. Traumatic optic neuropathy. Curr Opin Ophthalmol. 1999;10:340–342. doi: 10.1097/00055735-199910000-00011. [DOI] [PubMed] [Google Scholar]
- 12.Cook MW, Levin LA, Joseph MP, et al. Traumatic optic neuropathy. A meta-analysis. Arch Otolaryngol Head Neck Surg. 1996;122:389–392. doi: 10.1001/archotol.1996.01890160031006. [DOI] [PubMed] [Google Scholar]
- 13.Warner JEA, Lessell S. Traumatic optic neuropathy. Intern Ophthalmol Clin. 1995;35:57–62. doi: 10.1097/00004397-199503510-00007. [DOI] [PubMed] [Google Scholar]
- 14.Anderson RL, Panje WR, Gross CE. Optic nerve blindness following blunt forehead trauma. Ophthalmology. 1982;89:445–455. doi: 10.1016/s0161-6420(82)34769-7. [DOI] [PubMed] [Google Scholar]
- 15.Walsh FB. Pathological-clinical correlations. I. Indirect trauma to the optic nerves and chiasm. II. Certain cerebral involvements associated with defective blood supply. Invest Ophthalmol. 1966;5:433–449. [PubMed] [Google Scholar]
- 16.Entezari M, Rajavi Z, Sedighi N, et al. High-dose intravenous methylprednisolone in recent traumatic optic neuropathy; a randomized double-masked placebo-controlled clinical trial. Graefes Arch Clin Exp Ophthalmol. 2007;245(9):1267–1271. doi: 10.1007/s00417-006-0441-0. [DOI] [PubMed] [Google Scholar]
- 17.Steinsapir KD. Treatment of traumatic optic neuropathy with high dose corticosteroid. J Neuroophthalmol. 2006;26:65–67. doi: 10.1097/01.wno.0000204646.94991.68. [DOI] [PubMed] [Google Scholar]
- 18.Yu Wai, Man P. Surgery for traumatic optic neuropathy. Cochrane Database Syst Rev. 2005;4:CD005024. doi: 10.1002/14651858.CD005024.pub2. [DOI] [PubMed] [Google Scholar]
- 19.Levin LA, Baker RS. Management of traumatic optic neuropathy. J Neuroophthalmol. 2003;23:72–75. doi: 10.1097/00041327-200303000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Levin LA, Beck RW, Joseph MP, et al. The treatment of traumatic optic neuropathy: the International Optic Nerve Trauma Study. Ophthalmology. 1999;106:1268–1277. doi: 10.1016/s0161-6420(99)00707-1. [DOI] [PubMed] [Google Scholar]
- 21.Spoor TC, McHenry JG. Management of traumatic optic neuropathy. J Craniomaxillofac Trauma. 1996;2:14–26. [PubMed] [Google Scholar]
- 22.Bracken MB, Shepard MJ, Collins WF, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- 23.Bracken MB, Shepard MJ, Holford TR, et al. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury. Results of the Third National Acute Spinal Cord Injury Randomized Controlled Trial. National Acute Spinal Cord Injury Study. J Am Med Assoc. 1997;277:1597–1604. [PubMed] [Google Scholar]
- 24.Bracken MB, Holford TR. Effects of timing of methylprednisolone or naloxone administration on recovery of segmental and long tract neurologic function in NASCIS 2. J Neurosurg. 1993;79:500–507. doi: 10.3171/jns.1993.79.4.0500. [DOI] [PubMed] [Google Scholar]
- 25.Bracken MB. Steroids for acute spinal cord injury. Cochrane Database Syst Rev. 2002;22:CD001046. doi: 10.1002/14651858.CD001046. [DOI] [PubMed] [Google Scholar]
- 26.Levin LA, Beck RW, Joseph MP, et al. The treatment of traumatic optic neuropathy: the International Optic Nerve Trauma Study. Ophthalmology. 1999;106:1268–1277. doi: 10.1016/s0161-6420(99)00707-1. [DOI] [PubMed] [Google Scholar]
- 27.Chen CT, Huang F, Tsay PK, et al. Endoscopically assisted transconjunctival decompression of traumatic optic neuropathy. J Craniofacial Surg. 2007;18:19–26. doi: 10.1097/01.scs.0000248654.15287.89. [DOI] [PubMed] [Google Scholar]
- 28.Yip CC, Chng NW, Eu Eong KG, et al. Low-dose intravenous methylprednisolone or conservative treatment in the management of traumatic optic neuropathy. Eur J Ophthalmol. 2002;12:309–314. doi: 10.1177/112067210201200410. [DOI] [PubMed] [Google Scholar]
- 29.Ohlsson M, Westerlund U, Langmoen IA, et al. Methylprednisolone treatment does not influence axonal regeneration or degeneration following optic nerve injury in the adult rat. J Neuroophthalmol. 2004;24:11–18. doi: 10.1097/00041327-200403000-00003. [DOI] [PubMed] [Google Scholar]
- 30.Wolin MJ, Lavin PJ. Spontaneous visual recovery from traumatic optic neuropathy after blunt head injury. Am J Ophthalmol. 1990;109:430–435. doi: 10.1016/s0002-9394(14)74609-4. [DOI] [PubMed] [Google Scholar]
- 31.Levin LA. Axonal loss and neuroprotection in optic neuropathies. Can J Ophthalmol. 2007;42:403–408. [PubMed] [Google Scholar]
- 32.Ohlsson M, Mattsson P, Svensson M. A temporal study of axonal degeneration and glial scar formation following a standardized crush injury of the optic nerve in the adult rat. Restor Neurol Neurosci. 2004;22:1–10. [PubMed] [Google Scholar]
- 33.Sheng Y, Zhu Y, Wu L. Effect of high dosage methylprednisolone on rat retinal ganglion cell apoptosis after optic nerve crush. Eye Science. 2004;20:181–186. [PubMed] [Google Scholar]
- 34.Steinsapir KD, Goldberg RA, Sinha S, et al. Methylprednisolone exacerbates axonal loss following optic nerve trauma in rats. Restor Neurol Neurosci. 2000;17:157–163. [PubMed] [Google Scholar]
- 35.Roberts I, Yates D, Sandercock P, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): randomized placebo controlled trial. Lancet. 2004;364:1321–1328. doi: 10.1016/S0140-6736(04)17188-2. [DOI] [PubMed] [Google Scholar]
- 36.King CE. Erythropoietin is both neuroprotective and neuroregenerative following optic nerve transection. Exp Neurol. 2007;205:48–55. doi: 10.1016/j.expneurol.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 37.Levkovitch-Verbin H. Minocycline delays death of retinal ganglion cells in experimental glaucoma and after optic nerve transection. Arch Ophthalmol. 2006;124:520–526. doi: 10.1001/archopht.124.4.520. [DOI] [PubMed] [Google Scholar]
- 38.Guennoun R, Meffre D, Labombarda F, et al. The membrane-associated progesterone-binding protein 25-Dx: expression, cellular localization and up-regulation after brain and spinal cord injuries. Brain Res Rev. 2008;57:493–505. doi: 10.1016/j.brainresrev.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Miyake K. Neuroprotective effect of transcorneal electrical stimulation on the acute phase of optic nerve injury. Invest Ophthalmol Vis Sci. 2007;48:2356–2361. doi: 10.1167/iovs.06-1329. [DOI] [PubMed] [Google Scholar]
- 40.Biousse V, Mendicino ME, Simon DJ, et al. The ophthalmology of intracranial vascular abnormalities. Am J Ophthalmol. 1998;125:527–544. doi: 10.1016/s0002-9394(99)80194-9. [DOI] [PubMed] [Google Scholar]
- 41.Barrow DL, Spector RH, Braun IF, et al. Classification and treatment of spontaneous carotid-cavernous sinus fistulas. J Neurosurg. 1985;62:248–256. doi: 10.3171/jns.1985.62.2.0248. [DOI] [PubMed] [Google Scholar]
- 42.Barnwell SL, O'Neill OR. Endovascular therapy of carotid-cavernous fistulas. Neurosurg Clin N Am. 1994;5:485–495. [PubMed] [Google Scholar]
- 43.Savino PJ, Glaser JS, Schatz NJ. Traumatic chiasmal syndrome. Neurology. 1980;30:963–970. doi: 10.1212/wnl.30.9.963. [DOI] [PubMed] [Google Scholar]
- 44.Bruce BB, Zhang X, Kedar S. Traumatic homonymous hemianopia. J Neurol Neurosurg Psychiatry. 2006;77:986–988. doi: 10.1136/jnnp.2006.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Server A, Dullerud R, Haakonsen M, et al. Posttraumatic cerebral infarction: neuroimaging findings, etiology and outcome. Acta Radiol. 2001;42:254–260. doi: 10.1080/028418501127346792. [DOI] [PubMed] [Google Scholar]
- 46.Stovring J. Descending tentorial herniation: findings on computed tomography. Neuroradiology. 1977;14:101–105. doi: 10.1007/BF00333050. [DOI] [PubMed] [Google Scholar]
- 47.Nedeltchev K, Baumgartner RW. Traumatic cervical artery dissection. Front Neurol Neurosci. 2005;20:54–63. doi: 10.1159/000088149. [DOI] [PubMed] [Google Scholar]
- 48.Torina PJ, Flanders AE, Carrino JA, et al. Incidence of vertebral artery thrombosis in cervical spine trauma: correlation with severity of spinal cord injury. Am J Neuroradiol. 2005;26:2645–2651. [PMC free article] [PubMed] [Google Scholar]
- 49.Abe M, Udono H, Tabuchi K, et al. Analysis of ischemic brain damage in cases of acute subdural hematomas. Surg Neurol. 2003;59:464–472. doi: 10.1016/s0090-3019(03)00078-8. [DOI] [PubMed] [Google Scholar]
- 50.Ferrera PC, Pauze DR, Chan L. Sagittal sinus thrombosis after closed head injury. Am J Emerg Med. 1998;16:382–385. doi: 10.1016/s0735-6757(98)90134-6. [DOI] [PubMed] [Google Scholar]