

Abstract
Both pro- and antimitogenic activities have been ascribed to progesterone receptor (PR) agonists and antagonists in breast cancer cells; however, the transcriptional responses that underlie these paradoxical functions are not apparent. Using nontransformed, normal human mammary epithelial cells engineered to express PR and standard microarray technology, we defined 2370 genes that were significantly regulated by the PR agonist R5020. Gene ontology (GO) analysis revealed that GO terms involved in inflammation and nuclear factor-κB (NF-κB) signaling were among the most significantly regulated. Interestingly, on those NF-κB responsive genes that were inhibited by agonist-activated PR, antagonists either 1) mimicked the actions of agonists or 2) reversed the inhibitory actions of agonists. This difference in pharmacological response could be attributed to the fact that although agonist- and antagonist-activated PR is recruited to NF-κB-responsive promoters, the physical presence of PR tethered to the promoter of some genes is sufficient for transcriptional inhibition, whereas on others, an agonist-activated PR conformation is required for inhibition of NF-κB signaling. Importantly, the actions of PR on the latter class of genes were reversed by an activation function-2-inhibiting, LXXLL-containing peptide. Consideration of the relative activities of these distinct antiinflammatory pathways in breast cancer may be instructive with respect to the likely therapeutic activity of PR agonists or antagonists in the treatment of breast cancer.
The progesterone receptor can function as a passive or an active inhibitor of NFkb target gene transcription in cellular models of breast cancer.
Whereas the physiological actions of progestins acting through their cognate receptors in reproductive function are well understood, the role of this signaling axis in disease is poorly defined. In breast cancer, for instance, the majority of estrogen receptor (ER)-positive tumors express the progesterone receptor (PR), although it is unclear whether this receptor plays any role in the pathogenesis of the disease or whether it is merely a surrogate marker of estrogen responsiveness. In this regard, it is of interest that high-dose progestins are currently used as third-line endocrine therapies for breast cancer, although the mechanism by which this intervention impacts disease progression is unclear. Paradoxically, antiprogestins were also shown, albeit in a limited number of small clinical trials, to improve outcome in metastatic breast cancer patients (1,2). Further complicating our understanding of progestin/antiprogestin action, however, is the observation in the Women’s Health Initiative study that breast cancer incidence was slightly increased in women assigned to the estrogen plus progestin arm, as opposed to those who were taking estrogens alone (3). Taken together, these findings highlight the need to define roles of progestins and their cognate receptor(s) in breast cancer, a first step in the development of strategies to optimally exploit this signaling axis for the identification of clinically useful pharmaceuticals.
The observation that progestins can have both positive and negative effects on breast cancer progression is recapitulated in cellular and animal models of this disease. In T47D cells, for instance, progestins induce the expression of E2F1 and cyclin D1, facilitate hyperphosphorylation of Rb, and initiate one round of cell replication (4,5). When the same cells are implanted in athymic nude mice, robust progestin-dependent tumor growth is observed (6). In other cell lines, such as MCF-7, it can be shown that progestins efficiently inhibit estrogen-dependent cell proliferation in vitro (7), an activity that has generally been considered to underlie their efficacy as breast cancer therapeutics. However, of particular relevance to breast cancer are data that suggest that PR also functions as an efficient inhibitor of nuclear factor-κB (NF-κB)-dependent transcription through its interaction with the p65 subunit (8). The functional importance of this regulatory axis was first described in studies performed in the uterus, where it was shown that activation of NF-κB leads to the expression of proteins, such as cyclooxygenase 2 (COX-2), that facilitate uterine contractility and that this process could be inhibited by progestins (9,10). Likewise, it has recently been shown that COX-2 expression is also negatively regulated by progestins in cellular models of breast cancer (11). These latter findings are important in light of accumulating data that suggest that NF-κB plays an important role in mammary gland development and in the etiology of breast cancer (12). Whereas it is apparent that the divergent actions of PR are likely due to its ability to interact in a differential manner with functionally distinct transcriptional coregulators and transcription factors, this study was undertaken with the specific goal of understanding how agonist and/or antagonist-activated PR interferes with NF-κB signaling in breast cancer cells, a question of both pathological and pharmacological importance.
Results
PR negatively regulates NF-κB target gene transcription
Although both pro- and antimitogenic activities have been ascribed to PR ligands in different experimental settings, the transcriptional responses that underlie these paradoxical functions are not apparent in gene expression data derived from cultured breast cancer cells. Consequently, we have developed a model using nontransformed, normal human mammary epithelial cells (hMECs) in which hPR-B is transiently expressed. Transient expression of PR in these cells was required because the endogenous expression of the receptor alone was not sufficient to support robust progestin-dependent transcriptional activity. Using standard microarray technology, we identified 3847 probe sets, corresponding to 2370 unique previously characterized transcripts that were significantly regulated in cells treated for 16 h with the PR-specific agonist R5020 (Fig. 1A). Further analysis revealed that genes involved in the regulation of the IκB-NF-κB pathway were significantly overrepresented in the R5020-regulated genes identified (GO:0007249 corrected P = 0.0015). A total of 33 genes, belonging to the IκB-NF-κB gene ontology (GO) node, were identified (Supplemental Table 1, published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). The GO analysis also led to the identification of an additional 62 PR-regulated genes that were noted as being involved in inflammatory processes (GO:0006954, Supplemental Table 2). At the end of this analysis, we had in hand a set of 88 unique genes (seven genes displayed overlap between the two GO nodes) involved in NF-κB action and inflammatory response whose expression in hMECs were regulated by PR/R5020. Significantly, none of these genes were identified as PR responsive in a previously published microarray of T47D cells treated with R5020 (Fig. 1B). However, quantitative PCR (qPCR) analysis revealed that 66 of these 88 genes were expressed at some level in PR-expressing T47D-A18 cells and that the expression of most of these genes was dramatically up-regulated upon treatment with either IL-1β or TNFα, classical inducers of the NF-κB pathway. Notably, of the 66 genes of interest, 22 were induced more than 2.5-fold by IL-1β in this cell system. Interestingly, under these conditions, the inhibitory actions of progestin-activated PR were apparent, resembling what we observed in the hMEC model system (Supplemental Table 3).
Figure 1.

Induction profiles of genes with putative NF-κB1/NF-κB-binding sites by R5020. A, Genes significantly regulated by R5020 in hMECs are analyzed for the presence of putative NF-κB1/NF-κB-binding elements in their regulatory sequences as described in Supplemental Data. Five hundred twenty-two genes were identified. Their regulation in hMECs transiently overexpressing hPR-B was assessed by two-way plotting of induction/repression levels vs. −log10 (P value) for corresponding probesets (A). B, Similar plot for T47D cells (primary expression data courtesy of Dean Edwards, Baylor College of Medicine). Vertical dashed lines represent 1.5-fold induction/repression boundaries. Horizontal dashed line in A represents Holm-adjusted statistical significance cutoff (6.02). None of the genes expressed in T47D cells passed the significance cutoff (α = 0.05) when adjusted for multiple comparisons. C, Dendogram showing the expression profile of 22 NF-κB and inflammatory genes in cells treated with vehicle (dimethylsulfoxide), a NF-κB inhibitor (Bay11-7082), or a JNK inhibitor (SP600125) with and without IL-β. The profiles were analyzed with the Ward hierarchical clustering algorithm using standardized data.
NF-κB and activator protein 1 (AP-1) are essential regulators of genes involved in inflammation, and both are activated in response to IL-1β and TNFα. As a first step in our mechanistic studies, we probed whether NF-κB and/or c-Jun/AP-1 were the primary effectors of IL-1β on the 22 target genes identified above. To this end, their expression in T47D-A18 cells was assessed after IL-1β treatment in the presence or absence of the NF-κB (Bay11-7082) or the c-Jun N-terminal kinase (JNK) (SP600125) inhibitor. The results of this analysis (Fig. 1C) reveals that the induction of all of these genes was significantly inhibited by the NF-κB inhibitor, although a subset was also inhibited after treatment with the JNK inhibitor. These data suggest that progestins may have a more global effect on inflammatory responses and NF-κB pathways in breast cancer cells than previously anticipated. Therefore, in this study, we focused on defining the mechanism(s) by which progestins inhibit IL-1β- (or TNFα)-stimulated NF-κB-dependent target gene transcription in T47D cells.
hPR-A and hPR-B have equivalent activity as inhibitors of IL-1β-induced mRNA expression
Given the distinct activities of hPR-A and hPR-B as inducers of transcription and as repressors of estrogen-mediated transcription, we assessed the ability of each isoform to repress IL-1β-induced expression of IL-8 and CCL20 mRNAs. These genes were chosen for this initial analysis because they are among the best characterized NF-κB target genes in the set of 22 targets we analyzed above. For this analysis, expression was assessed in T47D-C42 cells (a variant T47D cell line that does not express hPR-A or hPR-B) that were engineered to stably express LacZ (control), hPR-A, or hPR-B. We determined that IL-1β-induced expression of both IL-8 and CCL20 mRNAs were quantitatively inhibited by agonist-activated hPR-A or hPR-B, with further inhibition observed in T47D-A18 cells in which both receptor isoforms were expressed (Fig. 2A). Importantly, the expression levels of PR were equivalent in the cell lines used (Fig. 2B).
Figure 2.
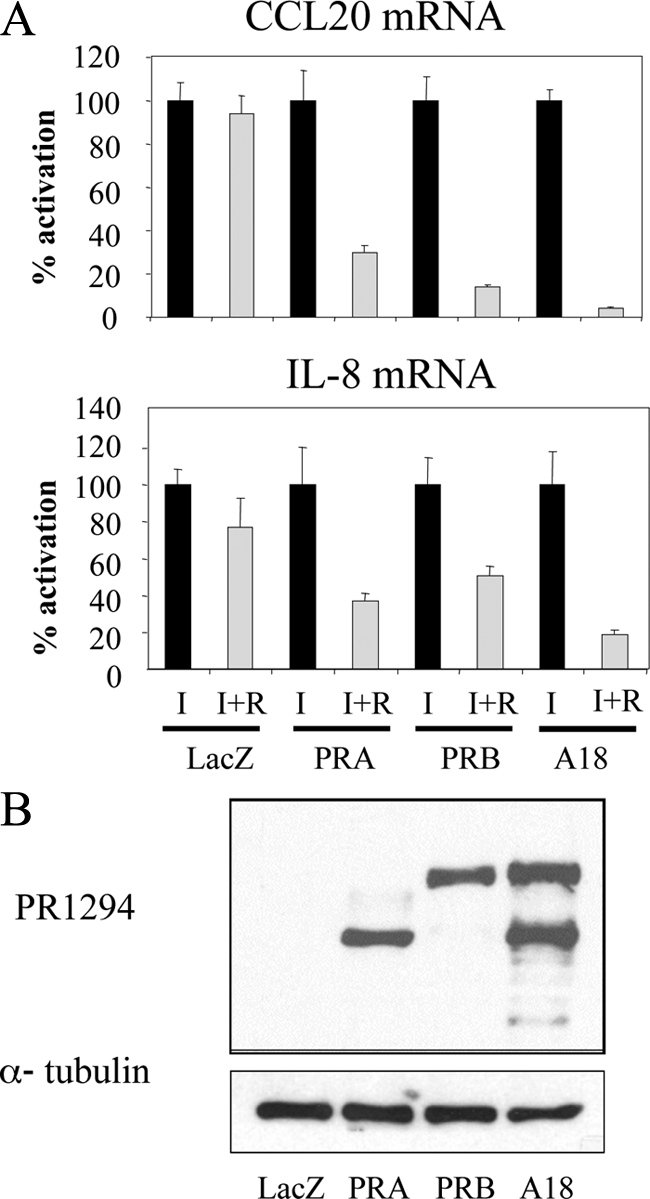
NF-κB target genes are repressed by both hPR-A and hPR-B. A, T47D-C42 cells, stably expressing lacZ, hPR-A, or hPR-B, or T47D-A18 cells were treated with vehicle or 10 nm R5020 (R) in the presence of 10 ng/ml IL-1β (I) for 6 h, followed by qPCR analysis of mRNA levels. Results are expressed as percent activation compared with IL-1β- and vehicle-treated cells. B, Immunoblotting of the protein levels of PR in whole-cell extracts from T47D-C42 cells or T47D-A18 cells with α-tubulin as a loading control.
PR does not impact the nuclear translocation of p65
It has been reported previously that both glucocorticoid receptor (GR) and PR can induce the expression of IκBα, a mediator of a negative feedback loop that attenuates NF-κB activity by interfering with the translocation of p65 NF-κB subunit from the cytoplasm to the nucleus (10,13). Thus, we evaluated the impact of R5020 on IκBα mRNA expression in T47D-A18 cells. The results of this analysis, shown in Fig. 3A, indicate that IκBα mRNA is induced by IL-1β, TNFα, or R5020. However, we observed minimal effects of these compounds, as single agents or when combined, on IκBα protein levels in either cytoplasmic or whole-cell extracts (Fig. 3A).
Figure 3.

Cytokine-mediated activation and nuclear translocation of NF-κB is not significantly influenced by PR. A, T47D-A18 cells were treated with vehicle (v) or 10 nm R5020 (R) in the presence or absence of cytokines (10 ng/ml IL-1β or 50 ng/ml TNFα) for 6 h. mRNA levels of IκBα were quantitated using qPCR. Results were expressed as fold induction over vehicle-treated cells. Whole-cell extracts (WCE) and cytoplasmic extracts (CE) were resolved by SDS-PAGE and blotted with indicated antibodies. B, T47D-A18 cells were treated as in A and harvested 2 or 6 h after treatment. Nuclear extracts (NE) and WCE were resolved by SDS-PAGE and blotted with indicated antibodies. Equal amount of protein was confirmed by using GAPDH, cytokeratin 18, and lamin A as loading controls.
We then investigated whether PR impacted the cellular compartmentalization of p65. In the absence of cytokines, R5020 treatment did not alter p65 nuclear localization by itself. Importantly, whereas both IL-1β and TNFα induce nuclear localization of p65, the addition of R5020 had no effect on this compartmentalization (Fig. 3B). Furthermore, we found that the inhibitory actions of R5020 were preserved in cells stably expressing hPR dimerization mutants, hPR-B-A604T or hPR-B-3D (hPR-B-A604T/R606D/D608C), which cannot induce IκBα (Supplemental Fig. 1). Cumulatively, these data indicate that PR must impact NF-κB signaling at a point downstream of p65 nuclear localization.
As demonstrated above, and observed by others, c-Jun/AP-1 modulates NF-κB-mediated induction of some of the genes that are down-regulated by progestins (14,15). Indeed, induction of the dual-specificity phosphatase, MKP-1, by GR inhibits p38 and JNK MAP kinase phosphorylation, downstream activation of the transcription factors c-Jun/AP-1, and the transcription of the target genes TNFα, CCL2, and COX-2 (16,17). However, only a modest induction of MKP-1 mRNA by R5020 was observed in both hMECs and T47D cells (data not shown). Furthermore, although we observed significant induction of phosphorylation of p38, JNK, MAPK kinase-1/2, and c-Jun in cells treated with either IL-1β or TNFα, this was not influenced by treatment with either the PR agonist R5020 or the antagonist RU486. Finally, phosphorylation of p65 itself was not influenced by R5020 or RU486 treatment (Supplemental Fig. 2). Given these findings, we considered it likely that the primary activity of PR on NF-κB signaling was to regulate the ability of p65 to bind its response elements in target gene promoters and/or interfere with the activity of the DNA-bound transcription factor.
PR and p65 are recruited to the enhancer/promoter regions of both progestin-regulated NF-κB target genes
We next used a series of chromatin immunoprecipitation (ChIP) assays to evaluate the colocalization of PR and p65 with the NF-κB response element within the enhancer/promoter regions of the endogenously expressed CCL20 and IL-8 genes in T47D-A18 cells. Using ChIP, a significant interaction of p65 with the NF-κB interacting regions within both the CCL20 (18) and IL-8 (19) genes were observed in IL-1β-treated cells (Fig. 4). Cotreatment with R5020 and IL-1β enhanced recruitment of p65 to the IL-8 promoter but slightly repressed recruitment to the CCL20 enhancer/promoter region. Importantly, significant recruitment of PR to the enhancers within these genes was observed under the same conditions. Pretreatment of these cells with the NF-κB inhibitor BAY11-7082 quantitatively inhibited the recruitment of p65 and PR with both genes. To determine whether PR inhibits the transcription of IL-8 and CCL20 at the initiation step, we used ChIP to determine whether RNA polymerase is recruited to the promoter/enhancer regions of these target genes. Although both total and phospho-serine-5 (S5) RNA polymerase II (Pol II) are significantly recruited to these regions upon IL-1β treatment, it was observed that treatment with R5020 disrupts both total and S5 Pol II recruitment to the enhancer/promoter region (Fig. 4B). It appears, therefore, the agonist-activated PR disrupts the expression of CCL20 and IL-8 mRNAs at the step of transcriptional initiation.
Figure 4.

PR and the p65 subunit of NF-κB colocalize to the enhancer regions of NF-κB target genes. A, Serum-starved T47D-A18 cells were pretreated with dimethylsulfoxide or 5 μm BAY11-7082 (BAY) for 30 min and then treated with vehicle (v) or 10 nm R5020 (R) in the presence or absence of 10 ng/ml IL-1β (I) for 2 h. B, Serum-starved T47D-A18 cells were treated with vehicle (v) or 10 nm R5020 (R) in the presence or absence of 10 ng/ml IL-1β (I) for 2 h. ChIP was performed with mouse IgG control (IgG), PR 1294, p65, Pol II, or S5 Pol II antibody, and qPCR analysis was performed using primers spanning a region in the NF-κB-binding region of CCL20 or IL-8. The results are presented as percent input.
Interestingly, significant R5020- and IL-1β-independent recruitment of PR to the IL-8 promoter was observed even in the presence of BAY. This particular region of the IL-8 promoter contains a putative glucocorticoid/progesterone response element (19). It was of interest, therefore, that when expressed in T47D-C42 cells, a PR DNA-binding mutant (C587A) was unable to repress IL-1β-induced expression of IL-8 mRNA in the presence of R5020. Similarly, we observed that IL-1β-mediated induction of an IL-8 promoter luciferase reporter was not inhibited by R5020 in HepG2 cells expressing the same PR DNA-binding mutant, whereas the wild-type receptor was an effective repressor (Supplemental Fig. 3A). In both the mRNA expression and luciferase reporter assays, we observed that the dimerization mutant (A604T, 3D) enabled the repressive effect of R5020 albeit to a lesser extent than the wild-type receptor (Supplemental Fig. 3B). Thus, we conclude that DNA-binding activity is necessary, whereas dimerization contributes to the ability of PR to mediate an inhibitory action of R5020 on IL-8 mRNA expression. Although not addressed in our study, it has been shown previously that PR can interact directly with the p65 subunit of NF-κB (8). Thus, it is possible that PR may interact in a direct manner with this promoter fragment and may also function as a tethering partner of p65. It is also possible that the binding of p65 to its element stabilizes PR binding to the glucocorticoid/progesterone response element and that the resulting complex is inhibitory with respect to the induction of IL-8 mRNA expression.
The differential effects of agonists and antagonists on PR-dependent inhibition of IL-1β-induced expression of inflammatory cytokines/chemokines occur by at least two distinct pathways
It has been shown that the antiprogestins mifepristone (RU486) or onapristone (ZK98299) are as effective as progesterone in inhibiting the activity of an activated NF-κB-luciferase reporter in transfected cells (8). Therefore, we performed an analogous experiment to evaluate the extent to which the pharmacology of PR, manifest on this simple reporter, was recapitulated on endogenous target genes. To this end, T47D-A18 cells were treated with IL-1β in the presence or absence of the PR agonist R5020 or the antagonists RU486 or ZK98299. Interestingly, whereas R5020 was equally effective as an inhibitor of all the genes examined, we observed that antiprogestins had marginal effects on CCL2 and CCL20 mRNA expression when administered as single agents (Fig. 5A), but they effectively reversed the inhibitory activity of R5020 on these genes (data not shown). However, all three ligands effectively inhibited IL-1β-mediated activation of transcription of both IL-8 and CCL4 genes. This pharmacological study reveals two distinct patterns of responsive genes, type I (CCL2/CCL20 like) and type II (IL-8 and CCL4 like), that likely reflect at least two different mechanisms by which PR can interface with NF-κB.
Figure 5.

Effects of progestins and antiprogestins on IL-1β-induced activation of cytokine genes. A, T47D-A18 cells were cotreated with 10 ng/ml IL-1β (I) and vehicle (v), 10 nm R5020 (R), 100 nm RU486 (RU), or 1 μm ZK98299 (ZK) for 6 h. mRNA levels of CCL2, CCL20, IL-8, and CCL4 were quantitated using qPCR. Results are expressed as relative expression over IL-1β- and vehicle-treated cells. B, Serum-starved T47D-A18 cells were treated with vehicle, 10 nm R5020, 100 nm RU486 (RU), or 1 μm ZK98299 (ZK) in the presence or absence of 10 ng/ml IL-1β for 2 h. ChIP was performed with either mouse IgG control or PR1294 antibody. qPCR analysis was performed using primers spanning a region in the NF-κB-binding region of CCL20 or IL-8. The results are presented as percent input.
The next step in our studies was to determine whether the interaction of PR with p65 at target gene promoters was sufficient to explain the inhibitory actions of both progestins and antiprogestins on NF-κB-mediated induction of the expression of type I and type II genes. To this end, we performed the ChIP experiments in the presence of different PR ligands (Fig. 5B). As expected, we did not see any recruitment of PR to the CCL20 promoter in the absence of IL-1β. However, all three PR ligands, R5020, RU486, and ZK98299, facilitated a robust, p65-dependent interaction of PR with the enhancer of this promoter. A similar result was obtained when the analogous experiment was performed using the enhancer from the CCL2 gene (data not shown). Given that RU486 and ZK98299 do not efficiently inhibit CCL20 and CCL2 mRNA expression (Fig. 5A), we conclude that the interaction of PR with p65 alone is not sufficient for inhibition of type I genes. When analyzed under similar circumstances, we observed significant interaction of PR with the IL-8 promoter in the presence of all three ligands; this binding was significantly enhanced upon the treatment of the cells with both IL-1β and each of the PR ligands (Fig. 5B). Interestingly, a more robust recruitment of PR with the IL-8 enhancer was observed in the presence of R5020, as opposed to RU486 and ZK98299, a result that reflects their relative activities as modulators of PR-mediated inhibition of IL-8 mRNA expression. Therefore, it appears that the magnitude of PR-mediated repression of IL-8 promoter activity tracks with the degree of receptor recruitment to the enhancer/promoter region.
Development and use of coactivator binding inhibitors (CBIs) to probe the role of activation function-2 (AF-2) in PR-mediated transcriptional repression
Antiprogestins function by competitively displacing agonists and facilitating a conformational change in the receptor that interferes with the presentation of the AF-2 coactivator pocket. Given the results presented above, it appears as though a functional AF-2 distinguishes the mechanisms of inhibition of the type I and type II genes. Indeed, with respect to CCL2 and CCL20, the data imply that a functional AF-2 domain is required for the inhibitory response. However, it was also possible that RU486/ZK98299-induced alterations in AF-2 architecture are merely a surrogate for another structural change occurring within PR that was required for repression. To rule out this latter possibility, we took advantage of the fact that it is possible to develop specific high-affinity LXXLL-containing peptides that function as CBIs by binding to and blocking the activity of AF-2.
Applying previously described combinatorial peptide M13 phage display methodologies (20), we were able to identify CBIs from a library of LXXLL-containing peptides that interacted with PR. A detailed characterization of one of the most interesting peptides identified, LX23 (RIHGYSPMLRALLLEEEAPK), is presented in Supplemental Fig. 4. Specifically, it was observed that this peptide interacted very well with agonist-activated hPR-A and hPR-B and inhibited progesterone-dependent activation of a transfected 2XPRE-tk-Luc reporter (Supplemental Fig. 4C). Thus, LX23 has the characteristics of a CBI that we felt would be useful in defining the role of AF-2 in PR-mediated inhibition of the expression of inflammatory cytokines. To this end, Gal4-DBD-LX23 or Gal4-DBD-LXAA (control) was overexpressed in T47D-A18 cells using an adenovirus-based expression vector, and the impact of these peptides on PR-mediated expression of endogenous target genes was evaluated. As shown in Fig. 6A, R5020-mediated repression of both CCL2 and CCL20 expression was reversed by LX23. However, this peptide was without effect on the expression of IL-8 and CCL4 mRNAs. Western immunoblot analysis confirmed that both Gal4-LX23 and the control peptide were expressed at an equivalent level in cells (Fig. 6B). Cumulatively, these results confirm that PR uses both AF-2-dependent (type I) and -independent (type II) mechanisms to repress the expression of IL-1β-induced inflammatory cytokines.
Figure 6.

Use of CBI to probe the role of AF-2 in PR-mediated transcriptional repression. A, T47D-A18 cells were infected with a Gal4-DBD-LX23 or Gal4-DBD-LXAA (control) expressing adenovirus. Viruses were removed 2 h after infection, and 42 h later, cells were treated with vehicle (v) or 10 nm R5020 (R) in the presence of 10 ng/ml IL-1β (I) for 6 h. mRNA levels of CCL2, CCL20, IL-8, and CCL4 were quantitated using qPCR. Results are expressed as relative expression compared with that observed in Gal4-LXAA-infected and IL-1β- and vehicle-treated cells. B, Western blot analysis of the expression of the Gal4-DBD fused proteins in whole-cell extracts from adenovirus-infected cells with GAPDH as a loading control.
Antiprogestins exhibit two distinct pharmacological responses on PR-regulated inflammatory response genes
The studies detailed above indicate that although the antiprogestins RU486 and ZK98299, like classical agonists, were able to inhibit IL-1β-dependent up-regulation of CCL4 and IL-8 (type II) mRNAs, they did not block induction of CCL2 or CCL20 expression. From the perspective of breast cancer therapeutics, this has significant implications because it suggests that these agents may not be as effective as agonists in inhibiting pathologically important inflammatory responses. It was important therefore to determine the extent to which this distinction between agonists and antagonists is observed on a broader set of target genes. To address this issue, we performed an analysis of the relative activity of PR agonists and antagonists on the expression of the 66 inflammatory genes described above. Briefly, T47D-A18 cells were treated with R5020, RU486, and R5020 plus RU486 together with and without IL-1β. As shown in Fig. 7, 34 of the 66 genes studied were significantly induced upon treatment with IL-1β. Of those genes that were up-regulated by IL-1β, three distinct clusters were apparent: 1) 16 genes were strongly repressed by R5020 and not affected by RU486, 2) nine genes were strongly repressed by both R5020 and RU486, and 3) nine genes were strongly repressed by R5020 and slightly repressed by RU486. Thus, all of the IL-1β up-regulated genes were repressed by R5020, but only a subset were responsive in a similar manner to the antiprogestin RU486. We performed an examination to see whether there were any correlations between elevated expression of any of the individual genes or clusters and breast cancer survival/outcome in several data sets as a means to test the significance of the mechanistic classifications noted. However, no clear patterns emerged from this study (Supplemental Table 4 and unpublished data). Thus, although antiprogestins have two distinct pharmacological responses when it comes to repression of inflammatory genes, elucidation of the significance of these differences will require further investigation.
Figure 7.

Progestins and antiprogestins differentially regulate the expression of NF-κB-activated genes. A, Dendogram showing expression profiles of endogenous NF-κB target genes upon progestin and antiprogestin treatment. A gene expression profile of 66 genes was generated using T47D-A18 cells treated with R5020, RU486 with or without IL-1β, and R5020 plus RU486. RNA was harvested for cDNA production and real-time PCR analysis. The profiles were analyzed by the Ward hierarchical clustering algorithm using standardized data. Ctl, Control.
Discussion
We were interested in defining the mechanism(s) by which PR inhibited NF-κB signaling as a first step in the rational development of drugs that may have efficacy in the treatment of breast cancer and other inflammatory conditions, such as fibroids and endometriosis, in which PR has been implicated. We determined in this study that PR has no significant effects on the action of NF-κB at any step upstream of its ability to interact with DNA. However, two distinct mechanisms were defined by which PR and progestins impact the expression of a subset of NF-κB-responsive genes. Both mechanisms involve a convergence of NF-κB and PR signaling at the level of DNA-bound p65 and the subsequent inhibition of transcription initiation. However, it was demonstrated that one class of genes (type I) requires a functional PR AF-2 domain and agonist-bound PR for repression, whereas repression of the other class of genes (type II) is AF-2 independent and is equally sensitive to both PR agonists and antagonists.
Our studies have revealed that the mechanisms underlying PR-mediated inhibition of the type I genes are the most complex, the lack of understanding of which is an impediment to the development of therapeutics that target this activity. The ability to reverse the repressive activity of progestins on type I genes, using either antiprogestins or a high-affinity LXXLL-containing peptide (direct AF-2 inhibitor), suggests that the recruitment by the receptor of a coactivator is required for this aspect of PR function. Given that there are over 300 cofactors that have been implicated in nuclear receptor signaling, it will take significant effort to define the specific proteins required for PR-mediated repression of the type I genes. LXXLL motifs are abundant in different coactivators and/or corepressors. For example, the p160/steroid receptor coactivator family members and p300/CREB binding protien (CBP) coactivators contain multiple LXXLL motifs as does the ligand-dependent nuclear receptor corepressor RIP140 (21,22). Interestingly, RIP140 has also been shown to be a necessary cofactor for p65-dependent cytokine gene expression and exists in a complex with p65 and CBP in macrophages (23). It would be of interest to determine whether PR interferes with RIP140 and/or CBP recruitment to the promoter of type I genes.
The regulation of the type II genes by progestins appears to be accomplished in a much more straightforward manner and may require only the physical recruitment of ligand-activated PR to the promoter of target genes. However, it is difficult at this time to rule out the involvement of cofactors that interact with PR 1) on a surface other than AF-2 and 2) in a manner that is not significantly impacted by antiprogestins. It is interesting that GR has also been shown to inhibit IL-8 mRNA expression, although in this case there is a requirement for steroid receptor coactivator 2 (24), and the bound GR-cofactor complex is involved in actively inhibiting the activity of the positive transcription elongation factor b complex (25). However, here we demonstrate that PR inhibits both IL-8 and CCL20 gene transcription by interfering with Pol II recruitment at the point of initiation. Thus, it appears that PR- and GR-mediated repression of IL-8 promoter activity occurs by different mechanisms.
An equally important finding from this study is that although progestins caused down-regulation of NF-κB-activated genes, albeit through multiple different mechanisms, antiprogestins selectively inhibited some genes, although not affecting others. More work needs to be done to understand how genes unaffected vs. those partially and completely inhibited by antiprogestins impact the progression of breast cancer. However, the data presented here highlight important questions, the resolution of which will help to define the type of PR modulators that may be most beneficial for the treatment of this disease. It will be important, for instance, to determine whether it is appropriate to use an agent with agonist-like properties that will enable the inhibition of both type I and type II genes. The problem with this approach is that progestins also induce the expression of genes that are associated with proliferation (26) and antiapoptosis (27), thus contributing to breast cancer progression. It may be possible, however, to develop selective PR modulators that enable repression of a subset of NF-κB-responsive genes but that do not lead to the activation of other transcription factors involved in proliferation, such as AP-1, or apoptosis. Specifically, it is anticipated that a drug that allows the presentation of the AF-2 PR-coactivator binding site, but that does not enable dimerization, may have utility as an inhibitor of the expression of NF-κB-dependent inflammatory cytokines.
In summary, we have made the unexpected finding that PR uses two distinct mechanisms to inhibit NF-κB target gene transcription, both of which are engaged by classical agonists but which are differentially sensitive to antagonists. These data, although arguing against the use of classical antagonists for breast cancer treatment, provide the mechanistic information that can be used to develop selective PR modulators that exhibit selectivity appropriate for breast cancer therapy.
Materials and Methods
Biochemicals
Promegestone (R5020) was purchased from NEN Life Science Products (Boston, MA). RU486 and ZK98299 were gifts from Ligand Pharmaceuticals (San Diego, CA) and Schering Pharmaceuticals (Berlin, Germany). IL-1β and TNFα were obtained from Sigma-Aldrich (St. Louis, MO), BAY11-7082 from Calbiochem (San Diego, CA), and SP600125 from Enzo Life Sciences (Plymouth Meeting, PA).
Cell culture
The hMECs were a gift from J. Marks (Duke University, Durham, NC). The T47D-A18 cell line was kindly provided by V. Craig Jordan (Lombardi Cancer Center, Washington, DC). The PR-negative T47D-C42 cells and those stably expressing LacZ, PR-A, or PR-B were shown previously (28). hMECs were maintained in mammary epithelial cell basal media (Lonza, Basel, Switzerland) supplemented with mammary epithelial cell growth media SingleQuots (Lonza). T47D-A18 cells were maintained in DMEM supplemented with 8% fetal bovine serum (FBS) (Hyclone Laboratories, Logan, UT), 0.1 mm non-essential amino acids and 1 mm sodium pyruvate. T47D-C42 cell lines were maintained in MEM supplemented with 8% FBS, 10 mm HEPES, 25 μg/ml gentamicin, 50 U/ml penicillin/streptomycin, 0.1 mm non-essential amino acids, 60 μg/ml insulin, and for cells stably expressing LacZ, hPR-A, or hPR-B, 200 μg/ml Zeocin. When T47D-A18 cells and T47D-C42 cells were treated with ligands/cytokines, cells were seeded in phenol red-free medium containing 8% charcoal-stripped FBS (Lonza) and the appropriate supplements. Medium was replaced with fresh medium 72 h later, and cells were treated with ligands/cytokines for the indicated time periods. Unless otherwise noted, all media and supplements were purchased from Invitrogen. All cells were grown at 37 C with 5% CO2.
Adenoviral transduction
Adenoviruses overexpressing hPR-B, Gal4-LX23, Gal4-LXAA, or β-Gal were generated using the ViraPower adenoviral expression system (Invitrogen) and were amplified and purified by CsCl2 centrifugation. Cells were infected at a multiplicity of infection of 40 for 48 h (T47D-A18 cells) or an multiplicity of infection of 150 for 17.5 h (hMECs).
RNA isolation and qPCR
Total RNA was isolated by using either RNeasy kit (QIAGEN, Valencia, CA) or Aurum total RNA mini kit (Bio-Rad, Hercules, CA). cDNA was synthesized using iScript (Bio-Rad). qPCR was performed with iQ SYBR Green supermix (Bio-Rad) with 0.3 μm of each primer. All qPCR primers used in this study are listed in Supplemental Table 4. Data were analyzed by the 2−ΔΔCt method (29), normalizing expression to 36B4.
Preparation of cellular extracts and immunoblotting
Whole-cell extracts were prepared using RIPA buffer [50 mm Tris-HCl (pH 8.0), 200 mm NaCl, 1.5 mm MgCl2, 1% Triton X-100, 1 mm EDTA, 10% glycerol, and protease inhibitor cocktail (Sigma)]. Nuclear and cytoplasmic extracts were prepared as described previously (30). Protein samples were separated by 7% SDS-PAGE, transferred to a polyvinylidene fluoride membrane (Bio-Rad) and probed with the indicated antibodies. PR 1294 was described previously (31), and antibodies directed against Gal4 (DBD) (RK5C1), IκBα (C21), p65 A, cytokeratin 18 (DC-10), lamin A (H-102), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (V-18), and α-tubulin (E-19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Chromatin immunoprecipitation
T47D-A18 cells were seeded on 15-cm dishes and were grown to 80–90% confluence. After serum starvation for 24 h, cells were treated with the appropriate ligand or cytokine for the indicated time periods. Cells were subjected to ChIP analysis as previously described (32), with the following modifications. Immunoprecipitation was performed overnight at 4 C with 10 μg PR1294 antibody, 10 μg RNA Pol II (MMS-126R), 10 μg serine 5 RNA Pol II (MMS-134R; Covance, Princeton, NJ), 10 μg p65 (sc-372), 10 μg mouse IgG control (sc-2025), and 10 μg rabbit IgG control (sc-2027; Santa Cruz). After immunoprecipitation, protein A/G-PLUS agarose (70 μl, Santa Cruz) or antimouse IgM (70 μl; Sigma) beads were added and incubated for 3 h at 4 C. qPCR analysis was performed as described above (see Supplemental Table 5 for ChIP primer sequences).
Statistical analysis
qPCR and ChIP data are represented as mean ± sem. Construction of the array of NF-κB- and progestin-regulated genes was performed as previously described (33). Data shown are representative of three or more experiments with similar results.
Supplementary Material
Acknowledgments
We thank Drs. Ganesan Sathya for pcDNA3-hPR-B and Martin Tochacek for the adenoviruses expressing Gal4-DBD-LX23 and Gal4-DBD-LXAA.
Raw microarray data have been deposited to NCBI GEO, accession number GSE24468.
Footnotes
This work was supported by National Institutes of Health Grant DK048807 and HD058640 (to D.P.M.) and by Merck (Organon).
Disclosure Summary: The authors have nothing to disclose.
First Published Online October 27, 2010
Abbreviations: AF-2, Activation function-2; AP-1, activator protein 1; CBI, coactivator binding inhibitor; CBP, CREB binding protein; ChIP, chromatin immunoprecipitation; COX-2, cyclooxygenase 2; ER, estrogen receptor; FBS, fetal bovine serum; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GO, gene ontology; GR, glucocorticoid receptor; hMECs, human mammary epithelial cells; JNK, c-Jun N-terminal kinase; NF-κB, nuclear factor-κB; Pol II, polymerase II; PR, progesterone receptor; qPCR, quantitative PCR
References
- Romieu G, Maudelonde T, Ulmann A, Pujol H, Grenier J, Cavalie G, Khalaf S, Rochefort H 1987 The antiprogestin RU486 in advanced breast cancer: preliminary clinical trial. Bull Cancer 74:455–461 [PubMed] [Google Scholar]
- Robertson JF, Willsher PC, Winterbottom L, Blamey RW, Thorpe S 1999 Onapristone, a progesterone receptor antagonist, as first-line therapy in primary breast cancer. Eur J Cancer 35:214–218 [DOI] [PubMed] [Google Scholar]
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, Jackson RD, Beresford SA, Howard BV, Johnson KC, Kotchen JM, Ockene J 2002 Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 288:321–333 [DOI] [PubMed] [Google Scholar]
- Musgrove EA, Hamilton JA, Lee CS, Sweeney KJ, Watts CK, Sutherland RL 1993 Growth factor, steroid, and steroid antagonist regulation of cyclin gene expression associated with changes in T-47D human breast cancer cell cycle progression. Mol Cell Biol 13:3577–3587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade HE, Kobayashi S, Eaton ML, Jansen MS, Lobenhofer EK, Lupien M, Geistlinger TR, Zhu W, Nevins JR, Brown M, Otteson DC, McDonnell DP 2010 Multimodal Regulation of E2F1 Gene Expression by Progestins. Mol Cell Biol 30:1866–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Besch-Williford C, Brekken RA, Hyder SM 2007 Progestin-dependent progression of human breast tumor xenografts: a novel model for evaluating antitumor therapeutics. Cancer Res 67:9929–9936 [DOI] [PubMed] [Google Scholar]
- Vignon F, Bardon S, Chalbos D, Rochefort H 1983 Antiestrogenic effect of R5020, a synthetic progestin in human breast cancer cells in culture. J Clin Endocrinol Metab 56:1124–1130 [DOI] [PubMed] [Google Scholar]
- Kalkhoven E, Wissink S, van der Saag PT, van der Burg B 1996 Negative interaction between the RelA(p65) subunit of NF-κB and the progesterone receptor. J Biol Chem 271:6217–6224 [DOI] [PubMed] [Google Scholar]
- Condon JC, Hardy DB, Kovaric K, Mendelson CR 2006 Up-regulation of the progesterone receptor (PR)-C isoform in laboring myometrium by activation of nuclear factor-κB may contribute to the onset of labor through inhibition of PR function. Mol Endocrinol 20:764–775 [DOI] [PubMed] [Google Scholar]
- Hardy DB, Janowski BA, Corey DR, Mendelson CR 2006 Progesterone receptor plays a major antiinflammatory role in human myometrial cells by antagonism of nuclear factor-κB activation of cyclooxygenase 2 expression. Mol Endocrinol 20:2724–2733 [DOI] [PubMed] [Google Scholar]
- Hardy DB, Janowski BA, Chen CC, Mendelson CR 2008 Progesterone receptor inhibits aromatase and inflammatory response pathways in breast cancer cells via ligand-dependent and ligand-independent mechanisms. Mol Endocrinol 22:1812–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Karin M 2003 NF-κB in mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia 8:215–223 [DOI] [PubMed] [Google Scholar]
- Auphan N, DiDonato JA, Rosette C, Helmberg A, Karin M 1995 Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science 270:286–290 [DOI] [PubMed] [Google Scholar]
- Wolter S, Doerrie A, Weber A, Schneider H, Hoffmann E, von der Ohe J, Bakiri L, Wagner EF, Resch K, Kracht M 2008 c-Jun controls histone modifications, NF-κB recruitment, and RNA polymerase II function to activate the ccl2 gene. Mol Cell Biol 28:4407–4423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvoro A, Tzagarakis-Foster C, Tatomer D, Paruthiyil S, Fox MS, Leitman DC 2006 Distinct roles of unliganded and liganded estrogen receptors in transcriptional repression. Mol Cell 21:555–564 [DOI] [PubMed] [Google Scholar]
- Zhou Y, Ling EA, Dheen ST 2007 Dexamethasone suppresses monocyte chemoattractant protein-1 production via mitogen activated protein kinase phosphatase-1 dependent inhibition of Jun N-terminal kinase and p38 mitogen-activated protein kinase in activated rat microglia. J Neurochem 102:667–678 [DOI] [PubMed] [Google Scholar]
- Abraham SM, Lawrence T, Kleiman A, Warden P, Medghalchi M, Tuckermann J, Saklatvala J, Clark AR 2006 Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J Exp Med 203:1883–1889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harant H, Eldershaw SA, Lindley IJ 2001 Human macrophage inflammatory protein-3α/CCL20/LARC/Exodus/SCYA20 is transcriptionally upregulated by tumor necrosis factor-α via a non-standard NF-κB site. FEBS Lett 509:439–445 [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yamamoto KR 2000 The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14:2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C, Norris JD, Grøn H, Paige LA, Hamilton PT, Kenan DJ, Fowlkes D, McDonnell DP 1999 Dissection of the LXXLL nuclear receptor-coactivator interaction motif using combinatorial peptide libraries: discovery of peptide antagonists of estrogen receptors α and β. Mol Cell Biol 19:8226–8239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG 1997 A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature 387:733–736 [DOI] [PubMed] [Google Scholar]
- Heery DM, Hoare S, Hussain S, Parker MG, Sheppard H 2001 Core LXXLL motif sequences in CREB-binding protein, SRC1, and RIP140 define affinity and selectivity for steroid and retinoid receptors. J Biol Chem 276:6695–6702 [DOI] [PubMed] [Google Scholar]
- Zschiedrich I, Hardeland U, Krones-Herzig A, Berriel Diaz M, Vegiopoulos A, Müggenburg J, Sombroek D, Hofmann TG, Zawatzky R, Yu X, Gretz N, Christian M, White R, Parker MG, Herzig S 2008 Coactivator function of RIP140 for NFκB/RelA-dependent cytokine gene expression. Blood 112:264–276 [DOI] [PubMed] [Google Scholar]
- Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR 2002 Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99:16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke HF, Yamamoto KR 2005 The glucocorticoid receptor blocks P-TEFb recruitment by NFκB to effect promoter-specific transcriptional repression. Genes Dev 19:1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange CA 2008 Challenges to defining a role for progesterone in breast cancer. Steroids 73:914–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MR, Conover JL, Franks KM 2000 Progestin effects on long-term growth, death, and Bcl-xL in breast cancer cells. Biochem Biophys Res Commun 277:650–654 [DOI] [PubMed] [Google Scholar]
- Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP 2007 The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinol 21:359–375 [DOI] [PubMed] [Google Scholar]
- Bookout AL, Mangelsdorf DJ 2003 Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal 1:e012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmann BM, Sherk A, McDonnell DP 2007 Definition of functionally important mechanistic differences among selective estrogen receptor down-regulators. Cancer Res 67:9549–9560 [DOI] [PubMed] [Google Scholar]
- Clemm DL, Sherman L, Boonyaratanakornkit V, Schrader WT, Weigel NL, Edwards DP 2000 Differential hormone-dependent phosphorylation of progesterone receptor A and B forms revealed by a phosphoserine site-specific monoclonal antibody. Mol Endocrinol 14:52–65 [DOI] [PubMed] [Google Scholar]
- DuSell CD, Umetani M, Shaul PW, Mangelsdorf DJ, McDonnell DP 2008 27-hydroxycholesterol is an endogenous selective estrogen receptor modulator. Mol Endocrinol 22:65–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Chang CY, Wittmann BM, Kunder RS, Cui H, Fan D, Joseph JD, McDonnell DP 2009 The homeodomain protein HOXB13 regulates the cellular response to androgens. Mol Cell 36:405–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.