

Abstract
Mitogenic and prosurvival effects underlie the tumorigenic roles of prolactin (PRL) in the pathogenesis of breast cancer. PRL signaling is mediated through its receptor (PRLr). A proteomics screen identified the pyruvate kinase M2 (PKM2), a glycolytic enzyme known to play an important role in tumorigenesis, as a protein that constitutively interacts with PRLr. Treatment of cells with PRL inhibited pyruvate kinase activity and increased the lactate content in human cells in a manner that was dependent on the abundance of PRLr, activation of Janus kinase 2, and tyrosine phosphorylation of the intracellular domain of PRLr. Knockdown of PKM2 attenuated PRL-stimulated cell proliferation. The extent of this proliferation was rescued by the knock-in of the wild-type PKM2 but not of its mutant insensitive to PRL-mediated inhibition. We discuss a hypothesis that the inhibition of PKM2 by PRL contributes to the PRL-stimulated cell proliferation.
Pyruvate kinase M2 (PKM2) is identified as a central regulator of cell metabolism and an interactor of the receptor for hormone prolactin, which signals to inhibit PKM2 in order to stimulate cell proliferation.
Prolactin (PRL) is a peptide hormone that elicits intracellular effects through engaging the homodimerized receptor, PRLr. Upon ligand binding, PRLr undergoes a conformational change which leads to activation of several kinases including protein tyrosine kinases [Janus kinase 2 (JAK2) and Src], serine/threonine kinases (such as Erk and Nek3), and lipid phosphoinositol-3 kinase. Depending on the tissue-autonomous and humoral contexts, this signal transduction program may either orchestrate normal mammary gland development and lactation or promote proliferation and increased survival of breast epithelium that contributes to its malignant transformation (reviewed in Refs. 1,2,3,4,5,6). Examples of altered PRL-PRLr action in human breast cancers include elevated levels of PRL (7,8,9), accumulation of PRLr due to its impaired proteolysis (6,10,11,12), or mutations that yield a constitutively active PRLr (13,14).
All effects that PRL elicits upon cells require the function of PRLr. The long form of PRLr mediates all known branches of PRL signaling; various shorter splicing isoforms that differ in the primary structure of the intracellular domain either enable partial signaling or appear to be inactive and may function as negative regulators (reviewed in Refs. 1 and 6). The polypeptide chain that constitutes the intracellular tail of PRLr serves as a platform for assembling signaling mediators. Some of these mediators are bound to the receptor constitutively (e.g. JAK2); for the recruitment of others, additional ligand-inducible posttranslational modification (such as Tyr phosphorylation for signal transducer and 5 activator of transcription proteins or lysine polyubiquitination for Adaptin Protein 50) might be required (6).
Sensitivity of cells to PRL is controlled by the cell surface expression of PRLr, which, in turn, is under negative regulation by phosphorylation-dependent ubiquitination and subsequent proteolytic turnover (reviewed in Ref. 6). A central event in this regulation is facilitated by the β-Trcp E3 ubiquitin ligase that is recruited in a manner that requires phosphorylation of PRLr on Ser349 within its degron (15,16,17). Ubiquitination of PRLr accelerates the rate of its endocytosis and lysosomal degradation (17). Whereas basal phosphorylation of Ser349 is mediated by glycogen synthase kinase 3β (11), the nature of the ligand-inducible Ser349 kinase remains to be determined. Nonetheless, phosphorylation of Ser349 inevitably accelerates subsequent ubiquitination and proteolysis of PRLr; and inhibition of these processes increases the transformation characteristics of mammary epithelial cells (12). Therefore, identification of novel partners of PRLr is expected to gain important new insights into the modes of PRL-initiated signaling and its outcomes for cell and tissue physiology.
Our preliminary proteomics screen identified pyruvate kinase M2 (PKM2) as a novel interacting protein of PRLr. PKM2 is an enzyme that plays an important role in cell metabolism, catalyzing the conversion of phosphoenolpyruvate to pyruvate. Unlike its alternate splice isoform pyruvate kinase M1 (PKM1), which is found in normal tissues, PKM2 has been shown to be uniquely expressed in highly proliferating embryonic cells and in cancer cells (18). Furthermore, dissimilar to PKM1, PKM2 activity could be inhibited by binding to the phosphotyrosines in a manner that is dependent on the integrity of a key residue within PKM2, lysine 433 (19). Furthermore, tyrosine phosphorylation of PKM2 on Y105 by fibroblast growth factor receptor 1 as well as other oncogenic tyrosine kinases including JAK2 also inhibited PKM2 activity (20). Expression of PKM2 (but not PKM1) and integrity of K433 were also shown to be important for cell proliferation and tumorigenicity (21).
Lactate accumulation was observed associated with PKM2 inhibition after oncogene activation (21). Although the mechanisms of intracellular increase in lactate levels are still unresolved, current paradigm views the inhibition of PKM2 as a key event in preventing the progression of metabolites through the Krebs cycle and in promoting the utilization of metabolites for the production of lipids, amino acids, and nucleic acids (22). Here we demonstrate that PRL elicits an inhibition of PKM2 in a manner that depends on JAK2 activity and Tyr phosphorylation and that this inhibition plays an important role in PRL-stimulated cell proliferation.
Results
Given the central role of PRLr in PRL signaling, we sought to identify proteins that interact with the long form of this receptor, which confers a multitude of signaling events (1,6). Proteomics-based analysis of proteins that coimmunoprecipitated with Flag-tagged PRLr expressed in human kidney embryo 293T cells (Fig. 1A) was focused on prominent bands that exhibit an apparent molecular mass of 55–65 kDa and should contain the approximately 62-kDa nonglycosylated PRLr precursor. This analysis yielded a number of peptides representing a diverse set of proteins (see Supplemental Table 1 published on The Endocrine Society’s Journals Online web site at http://mend.endojournals.org). Presence of peptides derived from PRLr itself and from the phosphoserine/threonine-binding protein 14-3-3 that has been previously described as an interactor of PRLr (23) validated our screen.
Figure 1.

A, Whole-cell lysates from human embryonic kidney 293T cells transfected to express Flag-tagged PRLr or the corresponding control vector (pCDNA3) and treated with PRL were immunoprecipitated (IP) with anti-Flag M2 agarose followed by stringent washes to minimize nonspecific interactions. The proteins that copurified with PRLr were resolved by SDS-PAGE and visualized by Colloidal Coomassie staining. Indicated proteins (Band 1) were excised, digested with trypsin, and analyzed by LC-MS/MS. The results were searched against the NIH database using SEQUEST software. B, Material from the experiment shown in panel A was analyzed by immunoblotting (IB) using anti-Flag and anti-PKM2 antibodies. C, Lysates from the MCF10a-Δp53 cells stably expressing Flag-PRLr [wild type (WT) or SA mutant], previously characterized in Ref. 12 were immunoprecipitated using Flag antibody and analyzed by IB using anti-PRLr antibody or anti-PKM2 antibody. Levels of PKM2 in the whole-cell extracts (WCE) are also shown. D, 293T cells transfected with Flag-tagged PRLr and HA-tagged PKM2 as indicated were lysed and IP-IB assays using anti-Flag and anti-HA antibody were carried out as depicted.
Among other putative interactors of PRLr, peptides representing pyruvate kinase 3, isoform 1 variant, also termed pyruvate kinase M2 (PKM2), was most frequently detected. The PKM2 splice isoform of pyruvate kinase is known to be expressed in highly proliferating embryonic cells and cancer cells and has been suggested to play an important role in cell proliferation and tumorigenesis (18,21,22,24,25). Immunoblotting analysis of proteins coprecipitated with anti-Flag antibody from 293T cells expressing Flag-PRLr indeed confirmed the presence of associated endogenous PKM2 (Fig. 1B). Similar results were obtained when these reactions were performed on the lysates from derivatives of near-normal human mammary epithelial MCF10a cells that stably expressed Flag-PRLr (Fig. 1C; cells described in detail in Refs. 12, 16, and 17). A greater amount of PKM2 was coprecipitated from the lysates of cells that expressed the ubiquitination-deficient PRLrS349A mutant (SA) compared with that from cells that harbor wild-type PRLr (Fig. 1C); this difference is likely reflective of elevated levels of total PRLr in the cells that express poorly degradable PRLr mutant (11,15). Interaction between PRLr and PKM2 was further demonstrated in 293T cells expressing Flag-PRLr and HA-tagged PKM2 by direct and reciprocal coimmunoprecipitation followed by the immunoblotting analysis (as seen in Fig. 1D).
We further sought to determine whether endogenous PKM2 is capable of interacting with endogenous PRLr. Coimmunoprecipitation of endogenous PRLr and PKM2 using commercially available antibodies was observed in the lysates from 293T cells treated or not with PRL (Fig. 2A). Due to low efficacy of these reactions, we developed a novel polyclonal antibody against PRLr (N30) and repeated the analyses in cells that express higher levels of PRLr. To characterize the novel anti-PRLr antibody, we used human breast cancer cells MDA-MB231 that express low levels of endogenous PRLr unless exposed to adenoviral delivery of increasing levels of human PRLr (26). Direct immunoblotting analysis of lysates from these cells demonstrated that N30 antibody readily detected mature human PRLr. Conversely, PRLr immunoprecipitated with N30 antibody was detected by commercially available monoclonal anti-PRLr antibody (Fig. 2B).
Figure 2.

A, Immunoprecipitation (IP) of endogenous PRLr from lysates from 293T cells treated with or without human PRL (purchased from the National Hormone and Peptide program and used at 100 ng/ml for 30 min) was carried out using anti-PRLr antibody (H-300, Santa Cruz) or naïve rabbit serum (NRS). Levels of PKM2 in whole-cell extracts (WCE) are also shown. B, MDA-MB-231 cells were infected with adenoviruses for delivery of human PRLr at increasing multiplicity of infection (MOI). WCEs were prepared and aliquots were resolved by SDS-PAGE and subjected to direct immunoblotting (IB) using anti-PRLr (N30, upper panel). Additional aliquots of the extracts were immunoprecipitated with N30 antibody and analyzed by IB using monoclonal anti-PRLr antibody from Invitrogen. NS, Nonspecific band. C, Lysates from the T47D cells (treated with or without 100 ng/ml of PRL for 15 min as indicated) were immunoprecipitated using either NRS or anti-PRLr N30 polyclonal antibody. Levels of PRLr and PKM2 in these reactions were detected by IB as indicated. Levels of PKM2, PRLr, and total and phosphorylated in the WCEs are also shown. IgG, Immunoglobulin heavy chain.
We then used N30 antibody for both immunoprecipitation and immunoblotting analyses of lysates from T47D cells treated with or without PRL. These experiments further demonstrated that endogenous PKM2 is coprecipitated with endogenous PRLr (Fig. 2C). Although under these conditions PRL treatment robustly activated signal transducer and activator of transcription 5, PRL did not noticeably alter the association of PKM2 to PRLr. These results, together with the data from proteomics screen where cells were not treated with PRL, suggest that PKM2 is indeed constitutively associated with PRLr.
In normal tissues that preferentially express PKM1, stimulation of activity of this kinase by PRL has been previously demonstrated (27,28,29,30). However, the effect of PRL on PKM2, the predominant pyruvate kinase splice isoform, in malignant cells has not been assessed. In human embryo kidney 293T cells, which express PKM2 but not PKM1 (21). and display low levels of endogenous PRLr (Ref. 15 and Fig. 2A), exposure to PRL led to a statistically significant inhibition of pyruvate kinase activity (Fig. 3A). The extent of this inhibition was comparable to the inhibition reported in cancer cells treated with the IGF (19).
Figure 3.

A, Pyruvate kinase activity was determined in lysates (50 μg) from 293T cells treated with PRL (100 ng/ml for indicated time points) by a coupled enzymatic-based spectrophotometric assay as described in Materials and Methods. Average data from four independent experiments (each in triplicate) are presented as percent of activity measured in cells that did not receive PRL (±sd). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). B, Transient expression of indicated Flag-tagged PRLr species in 293T cells was verified by immunoblotting (IB) using indicated antibodies. C, Activity of pyruvate kinase was determined (as in panel A) in lysates from 293T cells transfected with vector control (pCDNA3) or vectors for expression of PRLr [wild type (WT) or S349A mutant] and treated (100 ng/ml PRL for 20 min, white bars) or not (gray bars) with PRL. Here and in similar subsequent figures, the average data from three independent experiments (each in triplicate) are presented as percent of activity of nontreated control cells (±sd). Absolute values of PKM activity in untreated cells transfected with different PRLr constructs were comparable (data not shown). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). D, Pyruvate kinase activity was measured in the lysates from MCF10a-derived cells expressing wild-type or S349A mutant of PRLr (described in details in Ref. 12) as in panel B. E, Lactate levels in the lysates from cells used in panel C were determined using a fluorescence-based lactate measurement assay. Asterisks signify that the difference in lactate levels between the treated and untreated samples is significant as determined by Student’s t test (P < 0.05).
Given that PRL induces tyrosine phosphorylation of PRLr (reviewed in Ref. 31), that tyrosine kinase signaling is associated with inhibition of PKM2 (19,20), and that PKM2 interacts with PRLr (Figs. 1 and 2), we proposed that the expression levels of PRLr and the degree of PRLr-mediated phosphotyrosine signaling may influence the extent of PKM2 inhibition by PRL. To examine this possibility, we altered the extent of PRLr expression by transfecting 293T cells with either wild-type PRLr or with its proteolytically stable PRLrS349A mutant (Fig. 3B), which may be capable of interacting with a greater number of PKM2 molecules (Fig. 1C). Transient expression of this more stable and more abundant mutant caused a noticeably augmented PRL-induced inhibition of PKM2 activity (Fig. 3C).
We sought to corroborate these data in more physiologically relevant mammary epithelial cells and used MCF10a cell line derivatives that stably expressed PRLr (wild-type or stable PRLrS349A mutant, Fig. 1C). In these cells, a robust inhibition of pyruvate kinase activity by PRL was observed; the relative level of this inhibition corresponded to the relative levels of PRLr (wild type vs. S349) expressed in these cells (Fig. 3D). Consistent with previous observations (21), the magnitude of pyruvate kinase inhibition was inversely related to accumulation of lactate levels in the cells (Fig. 3E).
We further tested whether inhibition of PKM2 is affected by PRLr levels using derivatives of human breast cancer T47D cells that express high levels of PRLr and were stably transfected with control short hairpin RNA (shRNA) (shCON) or shRNA against PRLr (shPRLr; Fig. 4A, previously characterized in details in Ref. 12). Whereas PRL treatment significantly inhibited pyruvate kinase activity and increased lactate levels in control cells, knockdown of PRLr prevented these effects (Fig. 4, B and C). Collectively, these results indicate that inhibition of PKM2 and accumulation of lactate in response to PRL may be influenced by the abundance of PRLr.
Figure 4.
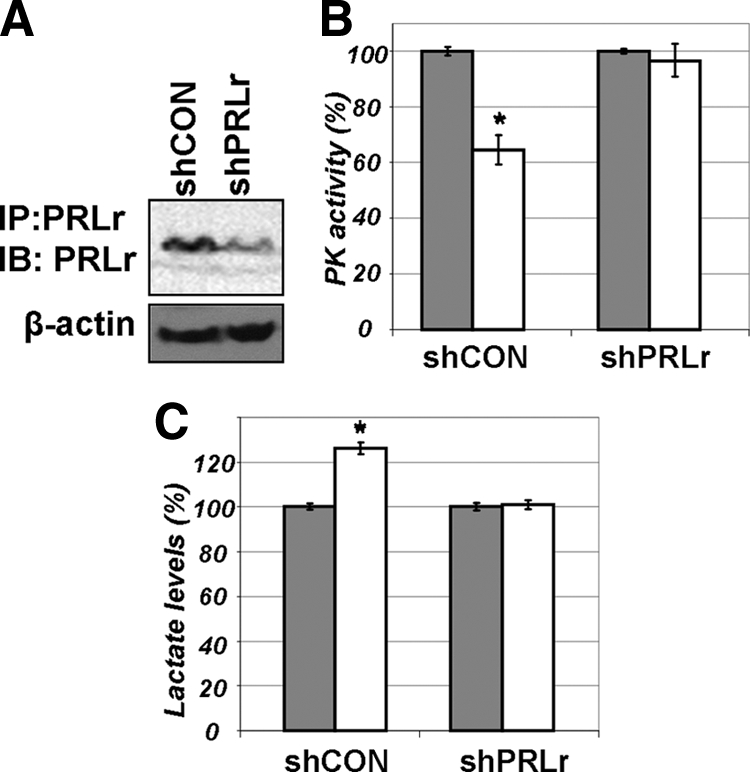
A, Levels of endogenous PRLr in T47D-derived cells that harbor shCON or shRNA against PRLr (shPRLr) were determined by immunoprecipitation (IP)-immunoblotting (IB) as described in details in Ref. 12. B, Pyruvate kinase activity was measured in the lysates from T47D-derived cells that harbor shCON or shRNA against PRLr (shPRLr). Cells (that were described in details in Ref. 12) were left untreated (gray bars) or treated with PRL (100 ng/ml for 20 min, white bars). Absolute values of PKM activity in untreated cells transfected with different shRNA constructs were comparable (data not shown). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). C, Lactate levels in the lysates from cells used in panel B were determined as outlined in Fig. 3E.
We then investigated how PRL inhibits PKM2 activity. Given that binding of PKM2 to phosphotyrosines was shown to elicit such inhibition (19) and that PRLr that interacts with PKM2 undergoes tyrosine phosphorylation (32,33), we next tested the role of this phosphorylation in PKM2 inhibition. To this end, we used two converging approaches: expression of PRLr mutants that either cannot undergo Tyr phosphorylation or that display a constitutive Tyr phosphorylation. Expression of a PRLr mutant that lacks Tyr residues in its intracellular domain compromised PRL-induced inhibition of PKM2 (Fig. 5B) and increase in intracellular lactate levels in 293T cells (Fig. 5C) despite the fact that this mutant was expressed at a level comparable to that of wild-type PRLr (Fig. 5A). Furthermore, expression of a gain-of-function PRLrI170L mutant [which is found in some breast cancers and benign tumors and exhibits constitutive Tyr phosphorylation that could be further stimulated by ligand (13,14)] resulted in a robust inhibition of PKM2 and accumulation of lactate even in the absence of the ligand; treatment with PRL further augmented these effects (Fig. 5, D and E). These results suggest that Tyr phosphorylation of PRLr is involved in inhibition of PKM2 by PRL. This inhibition could be either direct or mediated by additional recruitment of proteins containing phosphorylated Tyr residues that would interact with PKM2 and decrease its activity.
Figure 5.

A, Transient expression of indicated hemaggltinin (HA)-tagged and V5-tagged PRLr species in 293T cells was verified by immunoblotting (IB) using indicated antibodies. B, Activity of pyruvate kinase (PK) was determined (as in Fig. 3A) in lysates from 293T cells transfected with vector control (pCDNA3) or vectors for expression of PRLr [wild type or YF mutant (all intracellular tyrosines mutated to phenylalanine, described in Ref. 16)] and treated (100 ng/ml PRL for 20 min, white bars) or not (gray bars) with PRL. Absolute values of PKM activity in untreated cells transfected with either PRLrWT or PRLrYF were comparable (data not shown). Average data from four independent experiments (each in triplicate) are presented as percent of activity measured in cells that did not receive PRL (± sd). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). C, Lactate levels in the lysates from cells used in panel B were determined as outlined in Fig. 3E. D, PK activity was determined in lysates from 293T cells transfected with vector control (pCDNA3) or vectors for expression of PRLr (wild-type or I170L mutant). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). E, Lactate levels in the lysates from cells used in panel D were determined as outlined in Fig. 3E.
What are the kinases that may contribute to PRL-stimulated inhibition of PKM2? JAK2 and Src are two major tyrosine kinases activated by PRL (reviewed in Ref. 1). Pretreatment of PRLr expressing 293T cells with JAK inhibitor AG490 (but not with Src inhibitor PP1) attenuated inhibition of PKM2 by PRL (Fig. 6A). Furthermore, knockdown of JAK2 (but not of another Janus kinase, Tyk2) prevented PRL-mediated decrease in PKM2 activity and increase in the levels of intracellular lactate (Fig. 6, B and C). Efficacy of these constructs in down-regulating the respective levels of JAK2 and Tyk2 has been previously characterized (34).
Figure 6.

A, Pyruvate kinase (PK) activity was determined in lysates from 293T cells pretreated with ethanol (Vehicle), JAK inhibitor AG490 (AG490, Calbiochem, 50 μm), or Src inhibitor PP1 (PP1, Calbiochem, 10 μm). Cells were then were left untreated (gray bars) or treated with PRL (100 ng/ml for 20 min, white bars). Absolute values of PKM activity in the absence of PRL in vehicle, AG490, and PPI-treated cells were comparable (data not shown). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). B, PK activity was determined in lysates from 293T cells transfected with shCON, shRNA targeting JAK2 (shJAK2), or shRNA targeting Tyk2 (shTyk2) and either left untreated (gray bars) or treated with PRL (100 ng/ml for 20 min; white bars). Efficacy of these shRNAs (80–90% knockdown) have been previously reported (34). Absolute values of PKM activity in untreated cells transfected with different shRNA constructs were comparable (data not shown). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). C, Lactate levels in the lysates from cells used in panel B were determined. D, PK activity was determined in lysates from 293T cells transfected with vector control (pcDNA3) or vectors for expression of JAK2 (wild type or TEL-JAK2 fusion or V617F mutant). Cells were then were left untreated (gray bars) or treated with PRL (100 ng/ml for 20 min, white bars). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test).
In addition, expression of constitutively active JAK2 proteins such as TEL-JAK2 fusion protein and JAK2V617F mutant found in hematological malignancies (35) led to a robust suppression of PKM2 activity even in cells that did not receive PRL (Fig. 6D). These results suggest that PRL-induced JAK2 activity may be required and sufficient for inhibition of PKM2.
We next sought to investigate the role of PRL-mediated inhibition of PKM2 in cell proliferation. In rat lymphoma Nb2-11C cells that are exquisitely sensitive to PRL-stimulated growth (36), expression of murine PKM2 further promoted cell proliferation in the presence of PRL but not in the absence of this hormone (Fig. 7A). Remarkably, when PKM2K433E mutant [which is insensitive to inhibition by phospho-Tyr (19,21)] was introduced into the cells, it did not further increase PRL-stimulated proliferation despite being expressed at levels comparable with the wild-type protein (Fig. 7B). This indicates that inhibition of PKM2 may contribute to PRL-induced cell proliferation.
Figure 7.

A, The rate of proliferation of rat lymphoma Nb2–11C cells electroporated with a vector control (blue squares), wild-type murine Flag-PKM2 (PKM2WT, green diamonds), or murine Flag-PKM2K433E (PKM2KE, red circles). Cells (1 ×106) were plated in triplicate for each sample and time point and cultured in the absence (dashed lines, light colors) or presence of PRL (200 ng/ml, solid lines and intense colors). Trypan blue-negative live cells were counted at 24 and 48 h after plating. Average data from three independent experiments (each in triplicate) are presented as percent of number of initially seeded cells (± sd). Asterisks signify that the difference in growth rates between the treated and untreated samples is significant as determined by Student’s t test (P < 0.05). B, Material from experiment shown in panel A was analyzed by immunoblotting (IB) using anti-Flag and anti-β-actin antibodies. C, Pyruvate kinase (PK) activity was determined (as in Fig. 2A) in lysates from PRL-deficient MCF7 cells transfected with a control shRNA or shRNA targeting PKM2 [alone or in addition to an expression vector expressing a nontargetable murine PKM2 protein (wild type or K433E mutant)] and treated (200 ng/ml PRL for 30 min, white bars) or not (gray bars) with PRL. Bar data reflect percent changes whereas average absolute PKM activity numbers (in arbitrary units) are depicted above the bars in either blue (untreated cells) or red (PRL-treated cells). Asterisks denote statistical significance of obtained differences (P < 0.05 by Student’s t test). PRL-deficient MCF7 cells (37) and control and PKM2-specific shRNA lentiviral vectors [∼70–80% knockdown efficacy (21)] have been previously described. D, Lactate levels in the lysates from cells used in panel C were determined. Bar data reflect percent changes whereas average absolute lactate concentrations (in arbitrary units) are depicted above the bars in either blue (untreated cells) or red (PRL-treated cells). E, The rate of proliferation of MCF7-derived cells treated (solid lines, intense colors) or not (dashed lines, light colors) with PRL as described in panel C was determined as outlined in panel A. These cells were transduced with either control shRNA (blue squares) or shRNA against human PKM2 (all other symbols). Among the latter, cells received empty vector (black/gray triangles) or murine wild-type PKM2 (green diamonds) or murine PKM2KE mutant (red/orange circles). Average data from three independent experiments (each in triplicate) are presented as percent of number of initially seeded cells (±sd). Asterisks denote statistically significant differences (P < 0.05 by Student’s t test). F, The levels of expression of Flag-tagged PKM2 species in MCF7 cells described in panel E was analyzed by IB.
To further investigate this possibility we used a subclone derived from the human breast cancer MCF7 cells; this subclone was deficient in producing its own PRL and, therefore, very sensitive to the effects of exogenously added hormone (described in detail in Ref. 37). Knockdown of PKM2 in these cells abrogated PRL-induced suppression of pyruvate kinase activity (Fig. 7C), accumulation of lactate (Fig. 7D), and PRL-stimulated proliferation (Fig. 7E). We then used a functional knock-in approach (described in detail in Refs. 19 and 21) and expressed murine Flag-tagged PKM2 insensitive to these specific shRNAs against human enzyme (Fig. 7F). Whereas expression of wild-type murine PKM2 rescued PRL-stimulated reciprocal changes in pyruvate kinase activity and lactate levels as well as in cell proliferation, expression of the phosphotyrosine-insensitive PKM2K433E mutant failed to do so (Fig. 7, C–E). These data suggest that inhibition of PKM2 by PRL is important for PRL-stimulated cell proliferation.
Discussion
A metabolic switch that occurs within a tumor cell as a fundamental factor in tumorigenesis has been proposed by Warburg et al. (38) in the first half of the last century. Warburg (39) observed that although tumor cells take up more glucose than normal cells, they do not utilize this glucose for energy production via oxidative phosphorylation. Instead, byproducts of glucose catabolism (that proceeds via different relatively energetically inefficient pathways) play an important role in the anabolic processes and enable the synthesis of building blocks (i.e. lipids, amino acids, nucleic acids) important for cell proliferation. Recent reemergence of Warburg’s ideas is in part favored by new insights in the role of PKM2 expression in metabolic alterations in rapidly proliferating cells. Specifically, demonstration that replacement of PKM2 by PKM1 in tumor cells suppressed the glycolytic activity of these cells and inhibited their proliferation places expression of PKM2 at the center of mechanisms underlying the Warburg effect (19,20,21).
PKM2 has been thought of as a key enzyme promoting aerobic glycolysis in tumors, the Warburg paradox: although tumor cells take up more glucose than normal cells, they do not utilize this glucose for energy production via oxidative phosphorylation. Instead, tumor cells channel the byproducts of glucose metabolism for the synthesis of building blocks (i.e. lipids, amino acids, nucleic acids) important for a growing tumor (reviewed in Ref. 40). The increased lactate levels associated with PKM2 inhibition in cancer cells may reflect a compensatory increase in glutamine metabolism (22).
Whereas tyrosine phosphorylation of PKM2 by the oncogenes Src (41) as well as the inhibition of PKM2 was implicated in the pro-proliferative effects of the E7 oncoprotein of human papilloma virus (42), recent work by Cantley and co-workers (19,21) represented a new step for PKM2 characterization as a key regulator of the tumor metabolome. Although counterintuitive, both expression of PKM2 and its inhibition by pro-oncogenic factors appear to be required for efficient proliferation of tumor cells.
Given the ability of PKM2 to undergo inhibition mediated by interacting phosphotyrosine groups is essential for preventing progression of metabolic reactions through the citric acid cycle as well for cell proliferation and tumorigenesis, characterization of extracellular stimuli capable of PKM2 inhibition is of significant interest. One related mode of this regulation has been recently uncovered by Hitosugi et al. (20), who showed that direct tyrosine phosphorylation on PKM2 itself (mediated by the kinase activity of the fibroblast growth factor receptor-1) plays an important role in inhibition of PKM2. In addition, it is plausible that interaction of PKM2 with proteins that undergo Tyr phosphorylation can mediate such inhibition. Whereas numerous growth factors implicated in cancer are known for their ability to promote intracellular tyrosine phosphorylation, their effects on PKM2 activity and the role of PKM2 inhibition in effects that these factors elicit upon cells remain largely unknown.
From a proteomic screen, we have identified PKM2 as a putative interactor of PRLr. Collectively our data demonstrate that PKM2 constitutively binds to PRLr and undergoes a phosphotyrosine-mediated inhibition in response to PRL. This inhibition is dependent on PRLr levels and its tyrosine phosphorylation and requires activity of JAK2. Although we cannot exclude a possibility that JAK2 might be able to directly phosphorylate PKM2, it is evident that phosphorylation of intracellular domain of PRLr is involved in inhibition of PKM2 by PRL. This inhibition could either result from direct interaction of phosphorylated Tyr residues within PRLr with PKM2 or be mediated by additional recruitment of proteins containing phosphorylated Tyr residues that would interact with PKM2 and decrease its activity.
Importantly, we demonstrate that Tyr phosphorylation-mediated inhibition of PKM2 plays a key role in PRL-stimulated cell proliferation of lymphoma and breast cancer cells. These results strongly suggest the importance of metabolic alterations in mitogenic effects of PRL. Despite significant appreciation for PRL as a metabolic regulator (43), the importance of this role of PRL in mammary cell transformation and breast tumorigenesis largely remains to be elucidated.
In addition, such a mode of regulation might be common for other inducers of signal transduction that stimulate cell growth and division in a manner that requires induction of tyrosine phosphorylation. Further studies are needed to investigate whether receptors for other extracellular mitogens of polypeptide origin also interact with PKM2 and to determine the role of such interaction in proliferative and tumorigenic effects.
Materials and Methods
Cell lines
MCF10A derivative cell line, in which p53 expression is knocked down [MCF10AΔp53 (44)] was a generous gift of Alan Eastman (Dartmouth College, Hanover, NH). Generation of the MCF10AΔp53 cells stably expressing wild-type or S349A mutant PRLr was previously described (17). Human embryo kidney 293T cells and human breast cancer MDA-MB231 and T47D cells were kindly provided by Dr. Z. Ronai (Burnham Institute, San Diego, CA). Generation of T47D stably transduced with control shRNA (Sigma Chemical Co., St. Louis, MO; catalog no. SHC002, which is a lentiviral pLKO.1-puro vector containing an irrelevant shRNA insert that does not target human and mouse genes) or shRNA against PRLr (Open Biosystems, Huntsville, AL; catalog no. RHS3979-98492771) have been described elsewhere (12). Characterization of vectors for knockdown of JAK2 and Tyk2 were described elsewhere (26,34). Human breast cancer MCF7-derived cells that are deficient in production of endogenous PRL (described in detail in Ref. 37) were a generous gift of Linda Schuler (University of Wisconsin, Madison, WI). Rat lymphoma Nb2-11C cells that are exquisitely sensitive to PRL-stimulated growth (45) were kindly provided by Charles V. Clevenger (Northwestern University, Evanston, IL). Cells were cultured as previously described (11,12,46). Proliferation of cells was determined by counting live Trypan blue-negative cells as previously described (46). Average data from three independent experiments (each in triplicate) were calculated. The statistical differences were analyzed using Student’s two-tailed t test.
DNA constructs and gene delivery
Constructs based on pCDNA3 for expression of Flag-tagged, HA-tagged, or V5-tagged PRLr (wild-type or S349A mutant as well as the YF mutant in which all intracellular tyrosine residues have been mutated to Phe) kindly provided by Charles V. Clevenger (Northwestern University) were previously described (16,17). Vector for expression of constitutively active mutant of PRLr (PRLrI170L) was generated on the backbone of the HA-PRLr by site-directed mutagenesis followed by verification of its integrity by dideoxy sequencing.
Vector for expression of human HA-tagged PKM2 (47,48) was a kind gift from Hodaka Fujii (Osaka University, Osaka, Japan). Vectors for expression of Flag-tagged murine PKM2 (wild-type or K433E mutant) and for knockdown of human PKM2 were previously described in detail elsewhere (19,21). These shRNA constructs [delivery of which would routinely decrease PKM2 protein levels by 70–80% (Refs. 19 and 21 and data not shown)] were generously provided by Lewis Cantley (Harvard University, Cambridge, MA). ShRNA constructs for knocking down of Tyk2 and JAK2 were previously described (26,34); in our hands, delivery of these constructs routinely achieved 80–90% decrease in the levels of respective proteins monitored by immunoblotting (Refs. 26 and 34 and data not shown). Constructs for expression of JAK2 (wild-type or V617F mutant or TEL-JAK2 fusion protein) were a generous gift of Wei Tong (University of Pennsylvania, Philadelphia, PA).
Construction and titering of adenoviruses for expression of human PRLr were described previously (26). MDA-MB-231 cells were plated 24 h before infection and had grown to 90% confluence at the time of infection. Virus mixtures with desired MOI were made in serum-free medium according to the cell number and added to cells. Cells were washed with cold PBS 24 h after infection and subjected to future assays.
Nbc2–11C cells were electroporated using Gene Pulser II (Bio-Rad Laboratories, Hercules, CA). 293T cells were transfected using Lipofectamine Plus (Invitrogen, Carlsbad, CA) following the manufacturer’s recommendations or a calcium phosphate precipitate-based procedure as described elsewhere (49). MCF7-derived cells were transduced with puromycin resistance marker-containing lentiviral vectors for knocking down PKM2 and expressing murine PKM2. These cells were selected in puromycin (1 μg/ml) for 2 wk and maintained in this concentration of puromycin and used for experiments within the following 4 wk.
Chemicals and antibodies
Human recombinant prolactin (PRL) was kindly provided for a fee by Dr. A.F. Parlow (National Hormone and Peptide Program, Torrance, CA). JAK inhibitor AG490 (AG490, 50 μm), or Src inhibitor PP1 (PP1, 10 μm) were purchased from Calbiochem (La Jolla, CA). Other reagents were purchased from Sigma (St. Louis, MO).
Antibodies against Flag tag (M2; Sigma, HA (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), β-actin, and PKM2 were purchased from Affinity BioReagents, Inc. (Golden, CO) and Serotec (Oxford, UK), respectively. Secondary antibodies conjugated with infrared fluorescent probes were purchased from LI-COR Biosciences (Lincoln, NE). Detection was carried out using the LI-COR’s Odyssey Infrared Imaging System. Immunoprecipitation and immunoblotting procedures were carried out as previously described (49).
For analyses of PRLr, commercially available antibodies (Invitrogen and Santa Cruz) were used. In addition, a novel polyclonal anti-PRLr (N30) antibody against a keyhole limpet hemocyanin-conjugated peptide QLPPGKPEIFKCRSPN representing the amino terminus of the processed human PRLr was raised in rabbits. Immunoprecipitation using this antibody was carried out on 1 mg of whole-cell lysates.
Immunopurification of PRLr and associated proteins
Fifty 100-mm plates containing 293T cells were transfected with Flag-PRLr plasmid or pCDNA3 (10 μg/plate) via the calcium phosphate method. Cells were treated with PRL (100 ng/ml for 30 min) and harvested, and the lysates were prepared using Tris-HCl buffer (50 mm, pH 7.4) containing 150 mm NaCl, 50 mm NaF, 1% Nonidet P-40, and a protease inhibitors cocktail (all reagents from Sigma). The cell lysate was centrifuged at 4 C, 14,000 rpm for 15 min, and the protein concentration was measured. Protein (90 mg) was precleared by incubating with 1 ml of Protein A+G agarose (Invitrogen) at 4 C for 2 h. Cell lysate was centrifuged at 10,000 rpm for 5 min at 4 C, and the supernatant was collected and incubated overnight with 0.5 ml of anti-Flag M2 affinity gel (Sigma, catalog no. A2220). The beads were thoroughly washed and eluted using Flag peptide as previously described (50). The eluate was resolved on SDS-PAGE and visualized by Colloidal Coomassie staining. Selected protein bands were excised, digested with trypsin, and analyzed by liquid chromatography-tandem mass chromatograpy by Midwest Bio Services, LLC (Overland Park, KS). The results were searched against the NIH database using SEQUEST software.
Analysis of pyruvate kinase activity and lactate content
Cells were starved overnight in media without serum and then treated or not with PRL (100 ng/ml) as indicated. Cells were harvested and the lysates were prepared as outlined above. The activity of pyruvate kinase activity was measured by a coupled enzymatic assay using lactate dehydrogenase as described in detail in Ref. 51. Briefly, cell lysate (50 μg) was incubated with lactate dehydrogenase (25 units) in HEPES buffer (10 mm, pH 7.5) containing KCl (50 mm), MgCl2 (10 mm), ADP (2 mm), phosphoenolpyruvate (10 mm), and reduced nicotinamide adenine dinucleotide (0.5 m). The change in absorbance at 340 nm due to the conversion of reduced nicotinamide adenine dinucleotide to nicotinamide adenine dinucleotide was measured using Varian CARY 1E UV-Vis Spectrophotometer. Average data from four independent experiments (each in triplicate) were used for calculating the mean and presented as percent of activity measured in cells that did not receive PRL (±sd). Lactate levels in the cell lysates were determined using a fluorescence-based lactate measurement kit (BioVision) according to the manufacturer’s recommendations.
Supplementary Material
Acknowledgments
We thank Drs. Schuler (University of Wisconsin-Madison), Eastman (Dartmouth College), Clevenger (Northwestern University), Cantley (Harvard University), Tong (Children’s Hospital of Philadelphia), and Fujii (New York University) for reagents; Drs. K.G. Suresh Kumar, S. Srivinasan, and M. Guha (University of Pennsylvania) for technical help and advice, and the members of the Diehl, Greenberg, and Kushner laboratories (University of Pennsylvania) for helpful discussion.
Footnotes
This work was supported by National Institutes of Health Grants CA115281 (to S.Y.F.), CA118740 (to H.R.), and T32CA09140 (to B.V.).
Disclosure summary: Authors report no conflict of interest that is relevant to the topic of this manuscript outside of grant support (see above).
First Published Online October 20, 2010
Abbreviations: HA, Hemagglutinin; JAK2, Janus kinase 2; PKM1 and 2, pyruvate kinase M1 and M2; PRL, prolactin; PRLr, PRL receptor; shCON, control shRNA; shRNA, short hairpin RNA.
References
- Clevenger CV, Furth PA, Hankinson SE, Schuler LA 2003 The role of prolactin in mammary carcinoma. Endocr Rev 24:1–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger CV, Zheng J, Jablonski EM, Galbaugh TL, Fang F 2008 From bench to bedside: future potential for the translation of prolactin inhibitors as breast cancer therapeutics. J Mammary Gland Biol Neoplasia 13:147–156 [DOI] [PubMed] [Google Scholar]
- Clevenger CV, Gadd SL, Zheng J 2009 New mechanisms for PRLr action in breast cancer. Trends Endocrinol Metab 20:223–229 [DOI] [PubMed] [Google Scholar]
- Carver KC, Arendt LM, Schuler LA 2009 Complex prolactin crosstalk in breast cancer: new therapeutic implications. Mol Cell Endocrinol 307:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KU, Rui H 2008 Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J Mammary Gland Biol Neoplasia 13:93–103 [DOI] [PubMed] [Google Scholar]
- Swaminathan G, Varghese B, Fuchs SY 2008 Regulation of prolactin receptor levels and activity in breast cancer. J Mammary Gland Biol Neoplasia 13:81–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankinson SE, Willett WC, Michaud DS, Manson JE, Colditz GA, Longcope C, Rosner B, Speizer FE 1999 Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 91:629–634 [DOI] [PubMed] [Google Scholar]
- Tworoger SS, Eliassen AH, Rosner B, Sluss P, Hankinson SE 2004 Plasma prolactin concentrations and risk of postmenopausal breast cancer. Cancer Res 64:6814–6819 [DOI] [PubMed] [Google Scholar]
- Tworoger SS, Hankinson SE 2008 Prolactin and breast cancer etiology: an epidemiologic perspective. J Mammary Gland Biol Neoplasia 13:41–53 [DOI] [PubMed] [Google Scholar]
- Li Y, Clevenger CV, Minkovsky N, Kumar KG, Raghunath PN, Tomaszewski JE, Spiegelman VS, Fuchs SY 2006 Stabilization of prolactin receptor in breast cancer cells. Oncogene 25:1896–1902 [DOI] [PubMed] [Google Scholar]
- Plotnikov A, Li Y, Tran TH, Tang W, Palazzo JP, Rui H, Fuchs SY 2008 Oncogene-mediated inhibition of glycogen synthase kinase 3 β impairs degradation of prolactin receptor. Cancer Res 68:1354–1361 [DOI] [PubMed] [Google Scholar]
- Plotnikov A, Varghese B, Tran TH, Liu C, Rui H, Fuchs SY 2009 Impaired turnover of prolactin receptor contributes to transformation of human breast cells. Cancer Res 69:3165–3172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbay E, Degerli N, Gulluoglu BM, Kaya H, Sen M, Bardakci F 2004 Could prolactin receptor gene polymorphism play a role in pathogenesis of breast carcinoma? Curr Med Res Opin 20:533–540 [DOI] [PubMed] [Google Scholar]
- Bogorad RL, Courtillot C, Mestayer C, Bernichtein S, Harutyunyan L, Jomain JB, Bachelot A, Kuttenn F, Kelly PA, Goffin V, Touraine P 2008 Identification of a gain-of-function mutation of the prolactin receptor in women with benign breast tumors. Proc Natl Acad Sci USA 105:14533–14538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kumar KG, Tang W, Spiegelman VS, Fuchs SY 2004 Negative regulation of prolactin receptor stability and signaling mediated by SCF(β-TrCP) E3 ubiquitin ligase. Mol Cell Biol 24:4038–4048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan G, Varghese B, Thangavel C, Carbone CJ, Plotnikov A, Kumar KG, Jablonski EM, Clevenger CV, Goffin V, Deng L, Frank SJ, Fuchs SY 2008 Prolactin stimulates ubiquitination, initial internalization, and degradation of its receptor via catalytic activation of Janus kinase 2. J Endocrinol 196:R1–R7 [DOI] [PubMed] [Google Scholar]
- Varghese B, Barriere H, Carbone CJ, Banerjee A, Swaminathan G, Plotnikov A, Xu P, Peng J, Goffin V, Lukacs GL, Fuchs SY 2008 Polyubiquitination of prolactin receptor stimulates its internalization, postinternalization sorting, and degradation via the lysosomal pathway. Mol Cell Biol 28:5275–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurek S 2007 Pyruvate kinase type M2: a key regulator within the tumour metabolome and a tool for metabolic profiling of tumours. Ernst Schering Found Symp Proc 99–124 [DOI] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC 2008 Pyruvate kinase M2 is a phosphotyrosine-binding protein. Nature 452:181–186 [DOI] [PubMed] [Google Scholar]
- Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J 2009 Tyrosine phosphorylation inhibits PKM2 to promote the Warburg effect and tumor growth. Sci Signal 2:ra73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC 2008 The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452:230–233 [DOI] [PubMed] [Google Scholar]
- Dang CV 2009 PKM2 tyrosine phosphorylation and glutamine metabolism signal a different view of the Warburg effect. Sci Signal 2:pe75 [DOI] [PubMed] [Google Scholar]
- Olayioye MA, Guthridge MA, Stomski FC, Lopez AF, Visvader JE, Lindeman GJ 2003 Threonine 391 phosphorylation of the human prolactin receptor mediates a novel interaction with 14-3-3 proteins. J Biol Chem 278:32929–32935 [DOI] [PubMed] [Google Scholar]
- Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E 2002 Pyruvate kinase type M2: a crossroad in the tumor metabolome. Br J Nutr 87(Suppl 1):S23–S29 [PubMed] [Google Scholar]
- Mazurek S, Boschek CB, Hugo F, Eigenbrodt E 2005 Pyruvate kinase type M2 and its role in tumor growth and spreading. Semin Cancer Biol 15:300–308 [DOI] [PubMed] [Google Scholar]
- Neilson LM, Zhu J, Xie J, Malabarba MG, Sakamoto K, Wagner KU, Kirken RA, Rui H 2007 Coactivation of janus tyrosine kinase (Jak)1 positively modulates prolactin-Jak2 signaling in breast cancer: recruitment of ERK and signal transducer and activator of transcription (Stat)3 and enhancement of Akt and Stat5a/b pathways. Mol Endocrinol 21:2218–2232 [DOI] [PubMed] [Google Scholar]
- Arunakaran J, Balasubramanian K, Srinivasan N, Aruldhas MM, Govindarajulu P 1993 Effects of prolactin and androgens on enzymes of carbohydrate metabolism in seminal vesicles of castrated mature bonnet monkeys, Macaca radiata. Biochem Mol Biol Int 31:211–218 [PubMed] [Google Scholar]
- Arunakaran J, Balasubramanian K, Srinivasan N, Aruldhas MM, Govindarajulu P 1992 Effects of prolactin and androgens on enzymes of carbohydrate metabolism in prostate of castrated bonnet monkeys Macaca radiata (Geoffroy). Indian J Exp Biol 30:8–11 [PubMed] [Google Scholar]
- Kumaran B, Gunasekar PG, Aruldhas MM, Govindarajulu P 1988 Role of prolactin on neural and glial cellular enzymes involved in carbohydrate metabolism. I. Studies on immature male bonnet monkeys. Brain Res 450:325–333 [DOI] [PubMed] [Google Scholar]
- Costello LC, Franklin RB 2002 Testosterone and prolactin regulation of metabolic genes and citrate metabolism of prostate epithelial cells. Horm Metab Res 34:417–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger CV, Kline JB 2001 Prolactin receptor signal transduction. Lupus 10:706–718 [DOI] [PubMed] [Google Scholar]
- Rui H, Djeu JY, Evans GA, Kelly PA, Farrar WL 1992 Prolactin receptor triggering. Evidence for rapid tyrosine kinase activation. J Biol Chem 267:24076–24081 [PubMed] [Google Scholar]
- Rui H, Kirken RA, Farrar WL 1994 Activation of receptor-associated tyrosine kinase JAK2 by prolactin. J Biol Chem 269:5364–5368 [PubMed] [Google Scholar]
- Kumar KG, Varghese B, Banerjee A, Baker DP, Constantinescu SN, Pellegrini S, Fuchs SY 2008 Basal ubiquitin-independent internalization of interferon α receptor is prevented by Tyk2-mediated masking of a linear endocytic motif. J Biol Chem 283:18566–18572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentino L, Pierre J 2006 JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 71:713–721 [DOI] [PubMed] [Google Scholar]
- Gertler A, Walker A, Friesen HG 1985 Enhancement of human growth hormone-stimulated mitogenesis of Nb2 node lymphoma cells by 12-O-tetradecanoyl-phorbol-13-acetate. Endocrinology 116:1636–1644 [DOI] [PubMed] [Google Scholar]
- Schroeder MD, Symowicz J, Schuler LA 2002 PRL modulates cell cycle regulators in mammary tumor epithelial cells. Mol Endocrinol 16:45–57 [DOI] [PubMed] [Google Scholar]
- Warburg O, Wind F, Negelein E 1927 The metabolism of tumors in the body. J Gen Physiol 8:519–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O 1956 On respiratory impairment in cancer cells. Science 124:269–270 [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB 2009 Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324:1029–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presek P, Reinacher M, Eigenbrodt E 1988 Pyruvate kinase type M2 is phosphorylated at tyrosine residues in cells transformed by Rous sarcoma virus. FEBS Lett 242:194–198 [DOI] [PubMed] [Google Scholar]
- Zwerschke W, Mazurek S, Massimi P, Banks L, Eigenbrodt E, Jansen-Dürr P 1999 Modulation of type M2 pyruvate kinase activity by the human papillomavirus type 16 E7 oncoprotein. Proc Natl Acad Sci USA 96:1291–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Jonathan N, Hugo ER, Brandebourg TD, LaPensee CR 2006 Focus on prolactin as a metabolic hormone. Trends Endocrinol Metab 17:110–116 [DOI] [PubMed] [Google Scholar]
- Levesque AA, Kohn EA, Bresnick E, Eastman A 2005 Distinct roles for p53 transactivation and repression in preventing UCN-01-mediated abrogation of DNA damage-induced arrest at S and G2 cell cycle checkpoints. Oncogene 24:3786–3796 [DOI] [PubMed] [Google Scholar]
- Gout PW, Beer CT, Noble RL 1980 Prolactin-stimulated growth of cell cultures established from malignant Nb rat lymphomas. Cancer Res 40:2433–2436 [PubMed] [Google Scholar]
- Tang W, Li Y, Yu D, Thomas-Tikhonenko A, Spiegelman VS, Fuchs SY 2005 Targeting β-transducin repeat-containing protein E3 ubiquitin ligase augments the effects of antitumor drugs on breast cancer cells. Cancer Res 65:1904–1908 [DOI] [PubMed] [Google Scholar]
- Tani K, Yoshida MC, Satoh H, Mitamura K, Noguchi T, Tanaka T, Fujii H, Miwa S 1988 Human M2-type pyruvate kinase: cDNA cloning, chromosomal assignment and expression in hepatoma. Gene 73:509–516 [DOI] [PubMed] [Google Scholar]
- Tani K, Fujii H, Nagata S, Miwa S 1988 Human liver type pyruvate kinase: complete amino acid sequence and the expression in mammalian cells. Proc Natl Acad Sci USA 85:1792–1795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman VS, Slaga TJ, Pagano M, Minamoto T, Ronai Z, Fuchs SY 2000 Wnt/β-catenin signaling induces the expression and activity of βTrCP ubiquitin ligase receptor. Mol Cell 5:877–882 [DOI] [PubMed] [Google Scholar]
- Kumar KG, Barriere H, Carbone CJ, Liu J, Swaminathan G, Xu P, Li Y, Baker DP, Peng J, Lukacs GL, Fuchs SY 2007 Site-specific ubiquitination exposes a linear motif to promote interferon-α receptor endocytosis. J Cell Biol 179:935–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmeyer HU 1963 Methods of enzymatic analysis. Weinheim, Germany: Verlag Chemie; New York: Academic Press [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.