

Abstract
The p53 tumor suppressor exerts a variety of cell-autonomous effects that are aimed to thwart tumor development. In addition, however, there is growing evidence for cell non-autonomous tumor suppressor effects of p53. In the present study, we investigated the impact of stromal p53 on tumor growth. Specifically, we found that ablation of p53 in fibroblasts enabled them to promote more efficiently the growth of tumors initiated by PC3 prostate cancer-derived cells. This stimulatory effect was dependent on the increased expression of the chemokine SDF-1 in the p53-deficient fibroblasts. Notably, fibroblasts harboring mutant p53 protein were more effective than p53-null fibroblasts in promoting tumor growth. The presence of either p53-null or p53-mutant fibroblasts led also to a markedly elevated rate of metastatic spread of the PC3 tumors. These findings implicate p53 in a cell non-autonomous tumor suppressor role within stromal fibroblasts, through suppressing the production of tumor-stimulatory factors by these cells. Moreover, expression of mutant p53 by tumor stroma fibroblasts might exert a gain of function effect, further accelerating tumor development.
Keywords: p53, Fibroblast, SDF-1
Introduction
Stromal input is critical for the development of many organs [1]. Likewise, the progressing tumor can be viewed as a growing organ with contribution of the immune system, vasculature, fibroblasts and extracellular matrix [2]. Indeed, tumorigenesis is regulated by reciprocal interactions between cancer cells and the dynamic microenvironment (stroma) in which they reside.
Cancer associated fibroblasts (CAFs) are a major component of the tumor microenvironment [3]. While fibroblasts derived from the stroma of prostate carcinomas appear normal, large tumors nevertheless evolve following their co-inoculation into mice together with transformed, non-tumorigenic epithelial cells, whereas fibroblasts from a healthy prostate fail to support tumor growth [4]. A direct pro-metastatic effect of CAFs, through mechanism(s) involving the chemokine CCL5, was recently reported for mesenchymal stem cells co-injected into mice together with breast cancer cells [5]. CAFs are an established source of classical growth factors, known to possess tumor-promoting roles [3] including migration, proliferation and invasiveness [6-11].
p53 is a potent tumor suppressor. The TP53 gene is mutated in about half of all human cancers. Most of p53 research has focused on its cell-autonomous functions. However, it is conceivable that p53 may also impact tumor-stroma cross-talk [12]. Indeed, Kiaris et al [13] found that inoculation of cancer cells into p53-null mice reduced the latency for tumor development relative to wild-type mice, and that the fibroblastic compartment is sufficient to modulate both tumor latency and the morphology of the resulting tumors in a p53-dependent manner [13]. Thus, p53 activity in the host stroma may exert an inhibitory influence on cancer progression, and attenuation of this activity may favor tumor progression.
Further indication that stromal p53 may negatively affect tumor growth is provided by studies reporting a loss of p53 function in tumor-associated stroma. In a mouse model of prostate cancer, Hill et al. [1] showed that tumor progression imposed a strong selective pressure for loss of p53 in CAFs. Furthermore, several studies described TP53 gene mutations or LOH (loss of heterozygosity) in CAFs of various human cancers [14-20]. These findings, however, are still subject to ongoing debate [21-24]. Moreover, activation of p53 was shown to be attenuated in CAFs, even though the TP53 gene sequence was unaltered [25]. Finally, we reported that tumor cells acquire the ability to inhibit p53 induction in adjacent fibroblasts [26]. Interestingly, CAFs were more susceptible to this inhibitory mechanism than their normal counterparts, suggesting that the ability of tumor cells to emit signals that quench p53 activation in adjacent fibroblasts and the ability of the tumor-associated fibroblasts to respond to such inhibitory signals co-evolve during tumor progression [26]. Altogether, stromal p53 is implicated as a relevant inhibitor of epithelial tumor progression.
As a first step towards elucidating the mechanistic basis for the tumor-inhibitory effects of stromal fibroblasts, we found that p53 can repress the expression of the chemokine SDF-1/CXCL12 in cultured human and mouse fibroblasts [27]. Since SDF-1 is known to exert a variety of pro-cancer effects, particularly at late stages of the disease, its downregulation within the tumor microenvironment might contribute to tumor inhibition by stromal p53. In the present study, we explored the in-vivo relevance of the loss of p53 in stromal fibroblasts, and the role of SDF-1 in such context. Furthermore, we investigated the biological impact of the presence of mutant p53 in the fibroblasts surrounding the tumor.
Experimental procedures
Cell culture
Primary mouse embryonic fibroblasts (MEFs) were prepared from wild type (WT), p53 knockout and p53 mutant (515A) [28, 29] C57/BL day 13.5 embryos according to standard protocols [30]. All MEFs were grown in DMEM (Gibco, Carlsbad, CA) supplemented with beta mercaptoethanol (60μM, Sigma, Israel). PC3 human prostate adenocarcinoma-derived cells co-expressing luciferase and GFP (kindly provided by Dr. Amnon Peled, Hebrew University, Jerusalem) were grown in RPMI with sodium pyruvate. The identity of these cells was authenticated at start of project, by the National Center of Forensic Medicine, Israel, by matching the genetic profile against the ATCC profile. All media were supplemented with 10% FCS, L-glutamine solution (Beit HaEmek), non-essential amino acids and antibiotics.
Tumor models
Animal experiments were approved by the Weizmann Institutional Animal Care and Use Committee. Procedures were performed under anesthesia [100 mg/kg ketamine i.p. (Fort Dodge Animal Health, Fort Dodge, IA); 20 mg/kg xylazin i.p. (XYL-M2, V.M.D., Arendonk, Antwerp, Belgium). PC3 cells were inoculated either alone or together with MEFs, either subcutaneously in the back of male SCID mice (C.B-17/Icr-scid-bg; Harlan laboratories, Israel) or orthotopically: a small transverse incision was performed under anesthesia in the abdomen next to the linea alba, the prostate was exposed and cells were injected into the left side of the prostate in a volume of 20μl.
In-vivo imaging
Tumor development was monitored using the IVIS spectrum bioluminescence imaging system (Caliper Life Sciences, Hopkinton, MA). Mice were imaged once a week, following IP injection of 1.5mg D-luciferin (Caliper Life Sciences.). Measurements were performed when signal reached maximal plateau, ROI was defined for each tumor and average radiance (p/sec/cm2/sr) values were extracted for quantification. To detect spontaneous metastasis, light emission from the primary tumor was shielded with black tape to prevent detector saturation. Each mouse was scanned separately in supine position, using high exposure parameters.
See Supplementary Materials and Methods for additional information.
Results
Loss of p53 in stromal fibroblasts enhances tumor growth
The effect of stromal p53 on tumor development was studied in a xenograft model composed of the human prostate epithelial cancer cell line PC3 coinjected with fibroblasts of different p53 status. We employed PC3 cells co-expressing firefly luciferase and GFP, enabling us to monitor tumor development in-vivo over time by bioluminescence imaging. Importantly, the bioluminescence signals are contributed solely by the cancer cells, and therefore their intensity reflects directly the number of cancer cells within the tumor.
PC3 cells were inoculated into male SCID mice, either alone or together with wild type (WT) or p53 knockout (p53-KO) MEFs. Bioluminescence imaging was performed weekly. The first set of experiments employed subcutaneous injection into the back of the mouse. As shown in Fig 1 and 2A-C, addition of WT MEFs to PC3 cells appeared to exert a small positive effect on their in-vivo growth, although the difference between tumors induced by such combination and tumors induced by injection of PC3 cells alone was not statistically significant. Most notably, p53-KO MEFs significantly increased the bioluminescence of PC3 tumors, relative to WT MEFs. Statistically significant difference between the two groups of tumors was evident already after 4 weeks, and was maintained until the end of the experiment. Hence, absence of p53 in the MEFs markedly promoted tumor growth. Histopathological examination revealed that tumors generated by PC3 cells alone exhibited a modest stromal response (Fig 1D). Tumors generated by co-inoculation of PC3 cells with MEFs displayed a higher density of stroma; however, there was no detectable difference between WT and p53-KO fibroblasts in that regard.
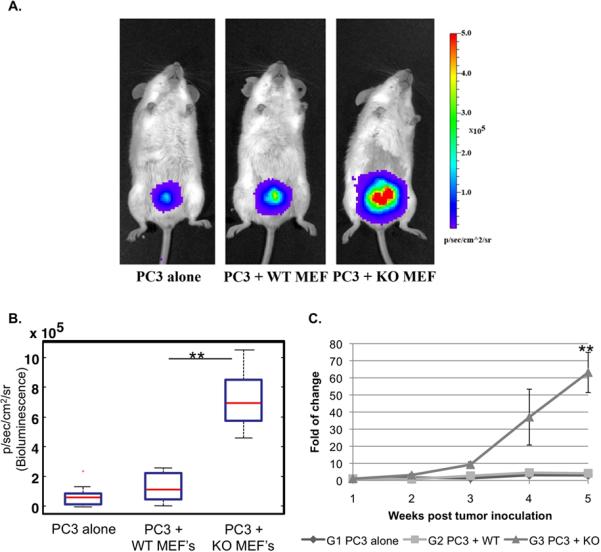
Fig 1. Effect of stromal p53 on PC3 tumor growth.

PC3 cells (2×106) expressing luciferase were inoculated subcutaneously, either alone or together with WT MEFs or p53-KO MEFs (1×106), in the back of male SCID mice (n=8 per group; total inoculation volume 30 μl). Every 1-2 weeks tumor load was measured by bioluminescence imaging of luciferase activity. A. Color-coded luciferase bioluminescence images acquired 6 weeks after inoculation. B. Distribution of luciferase bioluminescence values for individual mice 6 weeks post tumor inoculation is presented. P-value; * = 0.0097 ‡ = 0.16 C. Kinetics of tumor growth. For each time point the fold of change in luciferase bioluminescence intensity of each mouse was calculated relative to the initial luciferase signal of the same mouse, which was measured one week post inoculation. The average tumor load of all mice in the same group was calculated for each time point and plotted. P-values: 1 = 0.318, 2 = 0.025, 3 = 0.01; p-values relate to the last time point (6 weeks). D. H&E staining of histological sections from a representative tumor of each group, acquired 7 weeks post inoculation.
Fig 2. Effect of stromal p53 on orthotopic PC3 tumor growth.
PC3 cells (1×106) expressing luciferase were inoculated either alone or together with WT MEFs or p53-KO MEFs (0.5×106), in the prostate left lobe of male SCID mice (n=10 per group; total inoculation volume 20μl). Tumor growth was measured weekly by bioluminescence imaging. A. Color-coded representative bioluminescence images, 5 weeks after inoculation. B. Distribution of luciferase bioluminescence values in each group, 5 weeks after inoculation. ** p= 9×10-5 C. Kinetics of tumor growth. Analysis performed as in Fig1C.** p= 2×10-4 ;p-values relate to the last time point (5 weeks).
Tumor vascularization is a critical component of tumor development; furthermore, p53 can exert anti-angiogenic activities, e.g. by inducing the expression of TSP-1 [31] and downregulating VEGF [32]. However, CD34 staining of tumor specimens revealed comparable vascularization in all three experimental groups (Supplementary Fig 1)
To further substantiate the impact of fibroblast p53 status on tumor growth, we repeated the experiment in an orthotopic model employing direct inoculation of the tumor and stroma cells into the prostate. As shown in Fig 2, p53-null MEFs strongly enhanced tumor growth also in this model, whereas WT MEFs failed to do so.
Thus, in both subcutaneous and orthotopic prostate tumors, loss of p53 in fibroblasts adjacent to the cancer cells can accelerate tumor growth.
Excess exogenous fibroblasts abolish the differential impact of their p53 status on tumor growth
We next wished to explore whether the relative numerical ratio of cancer cells to fibroblasts affects the impact of fibroblast p53 on tumor growth. An experiment similar to that illustrated in Fig. 1 was performed, except that different PC3-to-fibroblasts ratios (1:2 versus 2:1) were compared. The first bioluminescence imaging was carried out 3 weeks post-inoculation. Again, p53-KO MEFs significantly increased cancer cell proliferation at both PC3:MEF ratios tested, (Supplementary Fig 2 A,B). However, when tumor monitoring was continued for up to 12 weeks post-injection, the differential effect of the p53-KO MEFs was gradually lost when the fibroblasts were in excess over the PC3 cells (Supplementary Fig. 2 C,D). Under such conditions, both WT and p53-KO MEFs strongly stimulated tumor growth. In contrast, when the number of fibroblasts was limiting (ratio of cancer cells to MEFs = 2:1), the differential stimulatory effect of the p53-KO MEFs was maintained for the entire duration of the experiment. These observations are consistent with the notion that MEFs emit a signal, presumably a secreted factor(s), which enhances tumor growth, and whose expression is higher in p53 KO MEFs than in WT MEFs. A sufficient excess of WT MEFs presumably compensates for the relatively low level of factor(s) per individual fibroblast, allowing the factor(s) to reach a critical local concentration sufficient for optimal tumor promotion.
p53-mediated repression of SDF-1 expression in stromal fibroblasts attenuates tumor growth
p53 represses the expression and secretion of the chemokine SDF-1/CXCL12 in cultured fibroblasts, resulting in the attenuation of adjacent cancer cell migration and invasion invitro [27]. Furthermore, in myofibroblasts isolated from surgically resected breast cancer specimens, SDF-1 expression is significantly upregulated relative to myofibroblasts from normal breast tissue; moreover, these cancer-associated fibroblasts can enhance SDF-1-dependent growth of human breast cancer cells in-vitro and in a mouse model [9, 33]. PC3 cells express functional CXCR4 (the SDF-1 receptor) [34-36]; hence, it was plausible that the SDF-1/CXCR4 axis play a role also in our in-vivo model. We therefore investigated whether quenching of SDF-1 expression by p53 in the stromal cells might underlie, at least in part, the differential effects of WT and p53-null fibroblasts on tumor growth. SDF-1 expression was stably knocked down in a pool of WT and p53-KO MEFs with the aid of a recombinant retrovirus expressing SDF-1-specific shRNA (sh-SDF-1). When assayed invitro, WT and p53-KO MEFs expressing sh-SDF-1 displayed a 8-fold and 13-fold reduction, respectively, in the amounts of SDF-1 mRNA, as compared to MEFs expressing control shRNA (sh-lacZ)(Fig. 3A).
Fig 3. p53-mediated repression of SDF-1 expression in stromal fibroblasts attenuates tumor growth in-vivo.

A. In-vitro evaluation of the efficacy of SDF-1 knockdown. WT and p53-KO MEFs were infected with retroviruses expressing either SDF-1 shRNA (sh-SDF1) or control shRNA (sh-lacZ). Following 48 hr of selection with puromycin, the pool of cells were harvested and RNA expression analysis was carried out. Real-time qPCR was preformed on cDNA prepared from total RNA using primers specific for SDF-1 and beta actin. Relative levels of SDF-1 mRNA are shown after normalization to the β-actin control. C,B. PC3 cells (2×106) expressing luciferase were inoculated subcutaneously either alone or together with the indicated types of MEFs as in Fig. 1 (n=8 per group). Prior to inoculation, expression of SDF-1 in the MEFs was knocked down by stable infection with a recombinant retrovirus expressing SDF-1 shRNA (sh-SDF1). Tumor progression was monitored as in Fig. 1. The data shown is from the same experiment as in Fig 1.
B. Kinetics of tumor growth. Analysis was performed as in Fig. 1C. p-values: 1= 0.336, 2= 0.025.
C. Luciferase bioluminescence of each group of mice, determined 6 weeks post inoculation. On each box, the central mark is the median, the edges of the box are the 25th and 75th percentiles, whiskers extend to the most extreme data points not considered outliers.
Next, these fibroblasts were compared with regard to their effect on PC3 tumor growth invivo. As expected, control p53-KO MEFs expressing sh-lacZ significantly augmented tumor growth relative to control WT MEFs expressing sh-lacZ (Fig 3B and C). However, SDF-1 knockdown strongly attenuated the positive impact of the p53-KO MEFs on tumor growth; furthermore, the difference between WT and p53-KO MEFs became statistically insignificant. This suggests that SDF-1, produced abundantly by p53-KO MEFs but less so by WT MEFs, is at least partly responsible for the stimulatory effect of p53-KO MEFs on PC3 tumor growth. Thus, p53-mediated repression of SDF-1 expression in stromal fibroblasts represents a molecular mechanism for cell non-autonomous tumor suppression by stromal p53.
Mutant p53 in fibroblasts exerts a gain of function effect on tumor growth
As discussed above, it has been reported that cancer-associated fibroblasts may sometimes harbor p53 mutations. When expressed within cancer cells, such mutant p53 (mut-p53) proteins may exert cell-autonomous oncogenic gain of function (GOF), resulting in enhanced proliferation, resistance to apoptosis, invasiveness and metastatic potential [37-41]. We therefore evaluated whether mut-p53 protein may also exert cell non-autonomous GOF effects when expressed in stromal cells. Specifically, we asked whether the presence of mut-p53 in fibroblasts adjacent to tumor cells would further enhance tumor development beyond the effects elicited by p53-null fibroblasts. To that end MEFs were prepared from the 515A mut-p53 “knock-in” mouse [29]; these mice express constitutively, within all their cells, p53R172H, the mouse equivalent of the human cancer-associated hot-spot mutant p53R175H. WT, p53-KO and p53-mutant (515A) MEFs were then employed for PC3 tumor progression analysis as in Fig. 1 As seen in Fig 4A and B, p53-mutant MEFs accelerated tumor growth significantly more than p53-KO MEFs, suggesting that stromal mut-p53 has a GOF effect on the growth of PC3 epithelial tumors.
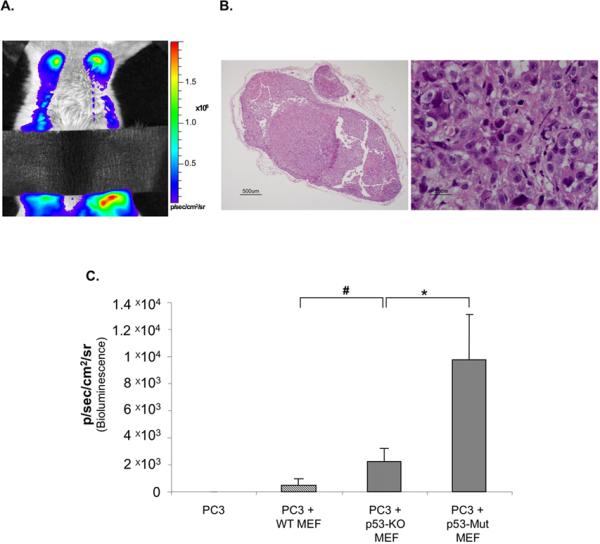
Fig 4. Mutant p53 in stromal fibroblasts exerts a gain of function effect on tumor growth.

PC3 cells (2×106) expressing luciferase were inoculated subcutaneously, either alone or together with p53-KO or p53-mutant MEFs (1×106) as in Fig. 1. (n=12 per group).
A. Luciferase bioluminescence of each group of mice, determined 7 weeks post inoculation. Boxes are as in Fig. 3C. * p= 0.02
B. The average luciferase bioluminescence of all mice in the same group was calculated for each time point and plotted.
Interestingly, p53-mutant MEFs exhibited a further upregulation of SDF-1 expression, beyond the levels found in p53-KO MEFs (Fig. 5A). Furthermore, reconstitution of p53-KO MEFs with cancer-associated human p53 mutants revealed that p53R175H brought about a modest, yet significant, elevation of SDF-1 mRNA levels (Fig. 5B). A similar trend was observed at the level of the secreted protein (Supplementary Fig 3A). This elevation appeared to be dependent on the nature of the p53 mutant, as expression of a different hotspot human p53 mutant, p53R273H, did not cause a similar increase in SDF-1 mRNA. Moreover, p53-mutant MEFs also overexpressed another chemokine, CXCL1 (Fig. 5C and Supplementary Fig 3B), known to be associated with tumorigenesis [42-44]. Hence, mutant p53 exerts a GOF effect on SDF-1 and CXCL1 expression in fibroblasts, which may contribute to their ability to augment tumor growth. Thus, if p53 mutations occur within the stroma, the ensuing non cell-autonomous GOF may provide a selective advantage to adjacent cancer cells.
Fig 5. Mutant p53 augments the expression of SDF-1 and CXCL1 mRNA in fibroblasts.

A. Primary MEFs were isolated from 13.5 days old embryos of wt, p53-KO or mutant p53 (515A) mice and harvested for RNA and protein analysis at passages 3-4. Real-time qPCR was preformed on cDNA prepared from total RNA using primers specific for SDF-1, CXCL1 and HPRT. Relative levels of SDF-1 mRNA are shown after normalization for HPRT mRNA. Lower panel shows a Western blot analysis of WTp53 and mutant p53 levels.
B. p53-KO MEFs were infected with recombinant retroviruses encoding two different cancer-associated human p53 mutants (p53R175H and p53R273H, respectively) or with control retrovirus. Following puromycin selection the cells were harvested for RNA and protein analysis. Real-time qRT-PCR was preformed and relative SDF-1 mRNA levels determined as in (A). Lower panel shows a Western blot analysis of exogenous mutant p53 levels.
C. The same RNA samples as in (A) were analyzed for relative CXCL1 mRNA levels.
Loss of p53 in the primary tumor stroma promotes metastasis
Next, we evaluated the impact of stromal p53 on metastasis. Employing the same experimental model as in Fig. 1, PC3 cells were inoculated either alone or together with WT, p53-KO or p53-mutant MEFs, and metastatic spread was monitored by masking the high bioluminescence arising from the primary tumor and imaging of each mouse separately (Fig. 6A). The first metastatic events became detectable 7 weeks post injection, and were localized to the inguinal lymph nodes only, as confirmed subsequently by H&E staining (Fig 6B). At later time points, the disease could be found to spread along the lymph node axis, in some cases also reaching the lungs (Fig 6A). Importantly, when assayed 7 weeks post infection, mice with tumors generated by inoculation of PC3 cells alone did not exhibit any detectable metastases (Fig. 6C and Supplementary Table 1). A low metastasis incidence was observed in mice with tumors generated by co-inoculation of PC3 cells together with WT MEFs. In contrast, when the primary tumors were induced in the presence of p53-null fibroblasts, a clearly higher metastatic load was evident. Interestingly, the mutant p53 MEFs exhibited a substantial further increase in metastasis incidence. Hence, loss of WT p53 in the stroma of the primary tumor can increase metastatic spread, and this is further exacerbated by acquisition of stromal p53 mutations.
Fig 6. Loss of WTp53 and expression of mutant p53 in the primary tumor stroma promote metastasis.
PC3 cells (2×106) expressing luciferase were inoculated subcutaneously into mice, either alone or together with WT, p53-KO or p53-mutant MEFs (1×106) as in Fig. 1 (n=12 per group).
A. Color-coded luciferase bioluminescence image acquired with the IVIS system 7 weeks post inoculation, showing bioluminescence from the lymph nodes and lungs. Black tape was used to block the high bioluminescence originating from the primary tumor.
B. Representative H&E staining of the excised metastasized inguinal lymph node.
C. Average bioluminescence of lymph node metastases calculated for each group of mice 7 weeks post inoculation. p-values: # =0.056; *=0.025.
Discussion
Tumor associated fibroblasts have been implicated in various steps of tumor progression. Notably, within the tumor microenvironment, such fibroblasts often acquire altered properties driven by epigenetic and sometimes perhaps also genetic events. In particular, recent studies have raised the possibility that tumor-associated fibroblasts may undergo changes that affect the functionality of their endogenous p53 protein. In the present work, we sought to determine whether and how the status of p53 in tumor-associated fibroblasts impacts tumor progression. Our findings support the conjecture that, in addition to the classical cell-autonomous role of p53 as a tumor suppressor within incipient tumor cells, it can also exert cell non-autonomous effects on tumor growth and metastasis when expressed by tumor-associated fibroblasts. These results raise the intriguing possibility that reactivation of p53 in tumor fibroblasts with defective p53 function may inhibit further tumor growth; such possibility may be approached experimentally through the use of MEFs derived from mice with switchable p53 [45].
One may question the biological relevance of studying the consequences of genetic alterations in stromal p53. Thus, while several reports have concluded that p53 mutations and consistent patterns of loss-of-heterozygosity are common events in stromal cells obtained from microdissected tumors as well as in cultured CAFs [14, 16, 18, 24], others have seriously challenged those conclusions [21-23]. Nonetheless, there remains a possibility that only a small number of CAFs actually acquire genetic alterations, making their detection difficult, yet this minority population may still exert a significant effect on the growing tumor. Alternatively it is conceivable that, unlike the characteristic clonality of advanced cancer cells, fibroblasts with mutant p53 may arise at a high frequency but not be extensively clonally expanded. This might result in a heterogeneous population of cells carrying a plethora of different p53 mutations, rendering such mutations hard to detect by standard sequencing approaches. Even more likely is the possibility that in many cases the primary sequence of the p53 gene is not altered at all in the CAFs, but its expression is quenched to some degree through epigenetic mechanisms. In that regard, it is noteworthy that reduced basal levels of p53 and compromised activation of p53 by genotoxic stress were found in a set of cultured breast CAFs [46], as well as in stromal cells expanded in vitro from several mouse tumor models [25], while no structural alterations in the p53 gene were found. Furthermore, recent work from our laboratory demonstrated that tumor cells acquire the ability to suppress p53 activation in adjacent fibroblasts [26]. Hence, studying the effects of fibroblasts with abnormal p53 status may indeed be biologically relevant. Under standard cell culture conditions, p53-null MEFs usually proliferate faster than WT MEFs. Furthermore, unlike their WT counterparts, p53-null MEFs don't undergo cellular senescence in culture. One could therefore argue that, if the same phenotypic differences are maintained also in-vivo, then the greater effect of p53-KO MEFs on tumor growth may be due to their ability to increase rapidly in number, while the WT MEFs presumably fail to do so. Yet, such conclusion was not supported by histological comparison of tumors induced with p53-KO MEFs to those induced by WT MEFs, performed by either H&E staining (which monitors both endogenous host stroma and the exogenous MEFs) or by specific detection of GFP-positive MEFs (Supplementary Fig 4). Furthermore, if p53-KO MEFs are indeed more abundant in the tumor relative to WT MEFs, this may reflect a biologically relevant scenario, where in the course of tumor progression there is selection for stromal cells with defective p53 function. Such selection may enable the cancer cells to elicit a more effective stromal reaction and create a microenvironment that is more supportive of their proliferation and survival.
Regardless of the above considerations, our data strongly suggest that there is also a profound qualitative difference between WT and p53-KO MEFs with regard to their competence to promote tumor growth. This appears to be underpinned, at least in part, by elevated production of secreted tumor-promoting factors by the p53-deficient fibroblasts. In the PC3 model, SDF-1 is a major contributor to this differential effect. Thus depletion of SDF-1 did not affect the proliferation rate of either WT or p53-KO MEFs (data not shown), but it strongly compromised the ability of p53-KO MEFs to promote tumor growth while hardly affecting the impact of WT MEFs on this process.
Several recent studies have raised the possibility that stromal p53 inactivation or mutation may increase tumor metastasis [17, 47, 48]. Kang et al. reported that elevated expression of prosaposin by prostate cancer cells led to induction of p53 within the tumor stroma, which resulted in increased Tsp-1 production by the stromal fibroblasts both locally and in the lungs, with consequent attenuation of metastatic spread [48]. Patocs et al [17] reported that p53 mutation rates in stromal cells of sporadic breast cancers are positively correlated with lymph node metastasis. Similarly, Hasebe et al [47] reported that enhanced p53 immunohistochemical staining in breast cancer stromal cells correlates with nodal metastasis and worse prognosis. Although the TP53 gene was not sequenced in that study, enhanced p53 staining is often indicative of the presence of mutations and excessive accumulation of mutant p53 protein. These correlative studies suggest that loss of stromal p53 function may augment metastatic spread.
Our data provide a direct experimental link between stromal p53 dysfunction and metastasis. Specifically, we found that loss or mutation of p53 in co-injected fibroblasts accelerated the formation of lymph node metastases. Since fibroblasts with p53 gene aberrations accelerated also primary tumor growth, their effect on metastasis may largely be a consequence of increased tumor size. However, p53-defective stroma may also have additional, more direct effects on the metastasis process. For instance, by modulating the expression of secreted factors, stromal p53 at the site of the primary tumor may restrict the invasive capacity of the tumor cells and their ability to disseminate away into the lymphatic system. The downregulation of SDF-1 by WT p53, previously shown to restrict the migration and invasiveness of adjacent cancer cells in vitro [27], is a likely candidate to play a role also in this process.
Our findings also reveal a GOF effect of mutant p53 on tumor growth when acting within the tumor-associated stroma. As noted above, there is still an uncertainty with regard to the actual existence and relative abundance of p53 mutations within the stromal compartment of tumors. However, if such mutations do indeed exist, our study provides a possible mechanism whereby they may benefit the growing tumor. Previous studies have demonstrated that tumors arising within mutant p53 knock-in mice tend to be more aggressive and metastatic [29, 49]. Of note, all stromal cells in such mice also carry the same p53 mutation. It will be of interest to determine whether, and to what extent, the presence of mutant p53 in the stroma of those mice contributes to the observed increase in tumor aggressiveness.
In conclusion, the findings described in this study suggest that p53 may act within stromal fibroblasts to restrict the availability of secreted factors that are required to support cancer cell proliferation, survival and invasiveness. Loss of p53 function in such fibroblasts may therefore contribute to tumor growth as well as to metastasis. This contribution can be augmented, via a GOF mechanism, when p53 becomes mutated in such fibroblasts. Some cancer cells, exemplified here by PC3, may be more sensitive to the restrictive effect of stromal p53, and may therefore benefit more significantly from local alterations in stromal p53 function. Subsequently, probably as part of the tumor progression process, cancer cells may acquire an ability to suppress p53 function in adjacent stromal cells [27]. Alternatively, they may develop an increased capacity to elicit a vigorous stromal response, ensuring by mass action an abundant supply of the necessary factors even if stromal p53 is not inactivated. Either way, these findings raise the possibility that pharmacological boosting of WT p53 activity in stromal cells may help restrict tumor growth and metastasis.
Supplementary Material
Acknowledgements
We thank Amnon Peled for the gift of cell lines and helpful discussions, Reuven Agami for the gift of pRetroSuper, Bradley Sarak, Sylvia Wilder, Vyacheslav Kalchenko, Tamara Berkutzki, Raya Eilam and Alon Harmelin for their help and opinions. Nava Nevo for the assistance with the orthotopic tumor model. Supported in part by grant R37 CA40099 from the National Cancer Institute, EC FP7 funding (INFLACARE, agreement 223151), The Robert Bosch Stiftung, the Canceropole Lyon Auvergne Rhône-Alpes-Weizmann Institute Program, and the Yad Abraham Center for Cancer Diagnosis and Therapy (to MO) and by grant R01 CA75334 from the US National Cancer Institute, the European Commission 7th Framework Integrated Project ENCITE, and the European Research Council Advanced grant 232640-IMAGO (to MN). The EC is not liable for any use that may be made of the information contained herein. MO is the incumbent of the Andre Lwoff Professorial Chair in Molecular Biology. M.N. is incumbent of the Helen and Morris Mauerberger Chair in Biological Sciences.
References
- 1.Hill R, et al. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell. 2005;123(6):1001–11. doi: 10.1016/j.cell.2005.09.030. [DOI] [PubMed] [Google Scholar]
- 2.Tlsty TD, Hein PW. Know thy neighbor: stromal cells can contribute oncogenic signals. Curr Opin Genet Dev. 2001;11(1):54–9. doi: 10.1016/s0959-437x(00)00156-8. [DOI] [PubMed] [Google Scholar]
- 3.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 4.Olumi AF, et al. Carcinoma-associated fibroblasts direct tumoprogression of initiated human prostatic epithelium. Cancer Res. 1999;59(19):5002–11. doi: 10.1186/bcr138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karnoub AE, et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449(7162):557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 6.Anderberg C, et al. Paracrine signaling by platelet-derived growth factor-CC promotes tumor growth by recruitment of cancer-associated fibroblasts. Cancer Res. 2009;69(1):369–78. doi: 10.1158/0008-5472.CAN-08-2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Augsten M, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multi-modal stimulator of prostate tumor growth. Proc Natl Acad Sci U S A. 2009;106(9):3414–9. doi: 10.1073/pnas.0813144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maeda T, Desouky J, Friedl A. Syndecan-1 expression by stromal fibroblasts promotes breast carcinoma growth in vivo and stimulates tumor angiogenesis. Oncogene. 2006;25(9):1408–12. doi: 10.1038/sj.onc.1209168. [DOI] [PubMed] [Google Scholar]
- 9.Orimo A, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 10.Taniwaki K, et al. Stroma-derived matrix metalloproteinase (MMP)-2 promotes membrane type 1-MMP-dependent tumor growth in mice. Cancer Res. 2007;67(9):4311–9. doi: 10.1158/0008-5472.CAN-06-4761. [DOI] [PubMed] [Google Scholar]
- 11.Zhu CQ, et al. Integrin alpha 11 regulates IGF2 expression in fibroblasts to enhance tumorigenicity of human non-small-cell lung cancer cells. Proc Natl Acad Sci U S A. 2007;104(28):11754–9. doi: 10.1073/pnas.0703040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bar J, Moskovits N, Oren M. Involvement of stromal p53 in tumor-stroma interactions. Semin Cell Dev Biol. 2010;21(1):47–54. doi: 10.1016/j.semcdb.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiaris H, et al. Evidence for nonautonomous effect of p53 tumor suppressor in carcinogenesis. Cancer Res. 2005;65(5):1627–30. doi: 10.1158/0008-5472.CAN-04-3791. [DOI] [PubMed] [Google Scholar]
- 14.Wernert N, et al. Presence of genetic alterations in microdissected stroma of human colon and breast cancers. Anticancer Res. 2001;21(4A):2259–64. [PubMed] [Google Scholar]
- 15.Fukino K, et al. Combined total genome loss of heterozygosity scan of breast cancer stroma and epithelium reveals multiplicity of stromal targets. Cancer Res. 2004;64(20):7231–6. doi: 10.1158/0008-5472.CAN-04-2866. [DOI] [PubMed] [Google Scholar]
- 16.Fukino K, et al. Genomic instability within tumor stroma and clinicopathological characteristics of sporadic primary invasive breast carcinoma. Jama. 2007;297(19):2103–11. doi: 10.1001/jama.297.19.2103. [DOI] [PubMed] [Google Scholar]
- 17.Patocs A, et al. Breast-cancer stromal cells with TP53 mutations and nodal metastases. N Engl J Med. 2007;357(25):2543–51. doi: 10.1056/NEJMoa071825. [DOI] [PubMed] [Google Scholar]
- 18.Kurose K, et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet. 2002;32(3):355–7. doi: 10.1038/ng1013. [DOI] [PubMed] [Google Scholar]
- 19.Narendran A, et al. Mutant p53 in bone marrow stromal cells increases VEGF expression and supports leukemia cell growth. Exp Hematol. 2003;31(8):693–701. doi: 10.1016/s0301-472x(03)00159-0. [DOI] [PubMed] [Google Scholar]
- 20.Paterson RF, et al. Molecular genetic alterations in the laser-capture-microdissected stroma adjacent to bladder carcinoma. Cancer. 2003;98(9):1830–6. doi: 10.1002/cncr.11747. [DOI] [PubMed] [Google Scholar]
- 21.Campbell I, Polyak K, Haviv I. Clonal mutations in the cancer-associated fibroblasts: the case against genetic coevolution. Cancer Res. 2009;69(17):6765–8. doi: 10.1158/0008-5472.CAN-08-4253. discussion 6769. [DOI] [PubMed] [Google Scholar]
- 22.Campbell IG, et al. Breast-cancer stromal cells with TP53 mutations. N Engl J Med. 2008;358(15):1634–5. doi: 10.1056/NEJMc086024. author reply 1636. [DOI] [PubMed] [Google Scholar]
- 23.Qiu W, et al. No evidence of clonal somatic genetic alterations in cancer-associated fibroblasts from human breast and ovarian carcinomas. Nat Genet. 2008;40(5):650–5. doi: 10.1038/ng.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eng C, et al. Genomic alterations in tumor stroma. Cancer Res. 2009;69(17):6759–64. doi: 10.1158/0008-5472.CAN-09-0985. [DOI] [PubMed] [Google Scholar]
- 25.Dudley AC, et al. Attenuated p53 activation in tumour-associated stromal cells accompanies decreased sensitivity to etoposide and vincristine. Br J Cancer. 2008;99(1):118–25. doi: 10.1038/sj.bjc.6604465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bar J, et al. Cancer cells suppress p53 in adjacent fibroblasts. Oncogene. 2009;28(6):933–6. doi: 10.1038/onc.2008.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moskovits N, et al. p53 Attenuates cancer cell migration and invasion through repression of SDF-1/CXCL12 expression in stromal fibroblasts. Cancer Res. 2006;66(22):10671–6. doi: 10.1158/0008-5472.CAN-06-2323. [DOI] [PubMed] [Google Scholar]
- 28.Jacks T, et al. Tumor spectrum analysis in p53-mutant mice. Curr Biol. 1994;4(1):1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 29.Lang GA, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell. 2004;119(6):861–72. doi: 10.1016/j.cell.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Zindy F, et al. Expression of the p16INK4a tumor suppressor versus other INK4 family members during mouse development and aging. Oncogene. 1997;15(2):203–11. doi: 10.1038/sj.onc.1201178. [DOI] [PubMed] [Google Scholar]
- 31.Dameron KM, et al. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265(5178):1582–4. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 32.Pal S, Datta K, Mukhopadhyay D. Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res. 2001;61(18):6952–7. [PubMed] [Google Scholar]
- 33.Allinen M, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32. doi: 10.1016/j.ccr.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 34.Singh S, et al. CXCL12-CXCR4 interactions modulate prostate cancer cell migration, metalloproteinase expression and invasion. Lab Invest. 2004;84(12):1666–76. doi: 10.1038/labinvest.3700181. [DOI] [PubMed] [Google Scholar]
- 35.Taichman RS, et al. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62(6):1832–7. [PubMed] [Google Scholar]
- 36.Zhang S, et al. Chemokine CXCL12 and its receptor CXCR4 expression are associated with perineural invasion of prostate cancer. J Exp Clin Cancer Res. 2008;27:62. doi: 10.1186/1756-9966-27-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dittmer D, et al. Gain of function mutations in p53. Nat Genet. 1993;4(1):42–6. doi: 10.1038/ng0593-42. [DOI] [PubMed] [Google Scholar]
- 38.Adorno M, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 2009;137(1):87–98. doi: 10.1016/j.cell.2009.01.039. [DOI] [PubMed] [Google Scholar]
- 39.Muller PA, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell. 2009;139(7):1327–41. doi: 10.1016/j.cell.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 40.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9(10):701–13. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 41.Oren M, Rotter V. Mutant p53 Gain-of-Function in Cancer. Cold Spring Harb Protoc. 2010;2(2):a001107. doi: 10.1101/cshperspect.a001107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang D, et al. CXCL1 induced by prostaglandin E2 promotes angiogenesis in colorectal cancer. J Exp Med. 2006;203(4):941–51. doi: 10.1084/jem.20052124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitsuyama K, et al. Increased circulating concentrations of growth-related oncogene (GRO)-alpha in patients with inflammatory bowel disease. Dig Dis Sci. 2006;51(1):173–7. doi: 10.1007/s10620-006-3104-4. [DOI] [PubMed] [Google Scholar]
- 44.Yang G, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(44):16472–7. doi: 10.1073/pnas.0605752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Martins CP, Brown-Swigart L, Evan GI. Modeling the therapeutic efficacy of p53 restoration in tumors. Cell. 2006;127(7):1323–34. doi: 10.1016/j.cell.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Hawsawi NM, et al. Breast carcinoma-associated fibroblasts and their counterparts display neoplastic-specific changes. Cancer Res. 2008;68(8):2717–25. doi: 10.1158/0008-5472.CAN-08-0192. [DOI] [PubMed] [Google Scholar]
- 47.Hasebe T, et al. p53 expression in tumor-stromal fibroblasts is closely associated with the nodal metastasis and outcome of patients with invasive ductal carcinoma who received neoadjuvant therapy. Hum Pathol. 2009 doi: 10.1016/j.humpath.2009.07.021. [DOI] [PubMed] [Google Scholar]
- 48.Kang SY, et al. Prosaposin inhibits tumor metastasis via paracrine and endocrine stimulation of stromal p53 and Tsp-1. Proc Natl Acad Sci U S A. 2009;106(29):12115–20. doi: 10.1073/pnas.0903120106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Olive KP, et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell. 2004;119(6):847–60. doi: 10.1016/j.cell.2004.11.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.