

Abstract
The effect of folate-targeted liposomal doxorubicin (FTL-Dox) has been well characterized in folate receptor (FR) over-expressing tumors in vitro, particularly in KB human carcinoma cells. However, there are few studies evaluating the in vivo efficacy of FTL-Dox in KB murine xenograft models. In this study, we investigated the antitumor activity of FTL-Dox injected intravenously in mice bearing KB tumors. Folate ligands comprising of folate-polyethyleneglycol-distearoylphosphatidylethanolamine (FA-PEG-DSPE) were synthesized with different MW PEG. To design an optimum FTL-Dox formulation for therapeutic studies, we prepared various FTLs and characterized their in vitro targeting and in vivo tissue biodistribution. Mice were administered a single intravenous injection of free Dox, non-targeted PEGylated liposomal Dox (PL-Dox), or FTL-Dox. FTLs and PLs accumulated similarly in tumor tissue, despite FTLs’ faster clearance from circulation. Mice treated with FTL-Dox 20 mg/kg had a slightly greater tumor growth inhibition and almost a 50% increase in life span than mice receiving PL-Dox 20 mg/kg (P = 0.0121; log-rank test). We conclude that FTLs administered systemically have the potential to enhance the delivery of anticancer drugs in vivo; however, their removal by FR expressing normal tissues may have to be blocked if the benefits of tumor targeting are to be realized.
Keywords: drug delivery, folate receptor, KB tumor, ligand-targeting
Introduction
Several strategies for tumor-specific delivery of chemotherapy are currently under evaluation because of the possibility to reduce dose limiting toxicity and bypass multi-drug resistance, both of which hamper the therapeutic efficacy of potent anticancer drugs. An often proposed target for directed cancer therapy is the folate receptor (FR). FR is a 38 kDa glycosyl-phosphatidylinositol membrane anchored glycoprotein that is over-expressed on various human cancers; however, FR is present at low levels in most normal epithelial tissues (Lu and Low, 2002). Folic acid (FA) binds to the folate receptor (FR) with high affinity (Kd = 0.1 nM) and gets internalized via receptor-mediated endocytosis (Antony, 1992). Folate is an attractive ligand because of its low immunogenicity, ease of modification, and low cost (Lu and Low, 2002).
Folate targeted liposomes (FTL) have successfully delivered chemotherapeutic agents as well as genes, antisense oligonucleotides, and radionuclides into FR over-expressing tumor cells (Hilgenbrink and Low, 2005). FTLs encapsulating doxorubicin (FTL-Dox) have increased cellular uptake and cytotoxicity as compared to non-targeted PEGylated liposomal doxorubicin (PL-Dox) and unencapsulated doxorubicin (Dox) in vitro (Lee and Low, 1995). These effects result from more efficient delivery of Dox after internalization into FR over-expressing tumor cells (Goren et al., 2000).
Although FTL drugs are widely studied in KB human carcinoma cells and other FR+ cancer cell lines in vitro, there are very few reports on FTL drug therapy in vivo, especially in solid tumor mouse models such as the KB tumor mouse model. The KB tumor cell line is often misrepresented as being isolated from an oral carcinoma (O’Neill, 2009) but is actually derived from contaminating HeLa cells in the primary tumor isolate. The KB tumor has become a model tumor for investigating factors that impact FR targeted delivery in an animal. Recently, Pan and coworkers have found that KB tumor-bearing mice treated with FTL-Dox via multiple i.p. injections had greater tumor growth inhibition (p < 0.01) and a 31% increase in lifespan compared to mice treated with PL-Dox (Pan et al., 2003). However, there are no studies to date on the in vivo therapeutic activity of FTL drugs after intravenous administration in KB tumor-bearing mice.
We formulated various FTLs and validated their binding and targeting to KB cells in vitro and in vivo. We utilized the best FTL formulation to investigate the therapeutic efficacy of FTL-Dox in a KB tumor mouse xenograft model after a single intravenous injection and observed a statistically significant tumor response to the FTL at the highest dose tested.
Methods
Materials
Folic acid, cholesterol (Chol), and fluorescein isothiocyanate (FITC) were purchased from Sigma-Aldrich (St. Louis, MO). Distearoylphosphatidylcholine (DSPC), distearoyl-phosphatidylethanolamine (DSPE) and methoxy-polyethylene glycol (MW2000)-DSPE (mPEG2000-DSPE) were purchased from Genzyme (Cambridge, MA). Amino-PEG2000-DSPE was purchased from Avanti Polar Lipids (Alabaster, AL). PEG3350-bis-amine was purchased from Shearwater Polymers (Huntsville, AL). Doxorubicin (Dox) was acquired from LGM Pharmaceuticals (Boca Raton, FL). KB cells (ATTC number CCL-17™) and RPMI 1640 media were purchased from University of California, San Francisco Cell Culture Facility. The KB cell line was established from a HeLa cell contamination of a tumor cell line (O’Neill, 2009). It is used as a tumor model because it forms a tumor in nude mice with reproducible characteristics and over expresses the folate receptor. The folate free RPMI 1640 media was a product of InVitrogen (Carlsbad, CA). Folate-free chow was purchased from Harlan Teklad (Madison, WI).
Cell Culture
KB cells were maintained in folate-free RPMI 1640 media supplemented with 10% fetal calf serum that provides the only source of folic acid. The cells were cultured as a monolayer in 5% CO2 at 37 °C.
Synthesis and Characterization of FA-PEG-DSPE Ligands
FA-PEG2000-DSPE was synthesized as described previously (Gabizon et al., 1999). As shown in Scheme 1, FA-PEG3350-DSPE was synthesized according to a modified method from Stephenson and coworkers (Stephenson et al., 2004). First DSPE was converted to N-succinyl-DSPE by succinic anhydride. Then amino-PEG3350 was conjugated to N-succinyl-DSPE through the activated ester. Finally, FA-PEG3350-DSPE was obtained by coupling FA to the terminal amino group of DSPE-PEG3350-NH2. Detailed synthetic procedures are described below.
Scheme 1.

Synthesis of FA-PEG3350-DSPE. Reagents and conditions: a) succinic anhydride (2 equiv.), triethylamine (4 equiv.), CHCl3, r.t., 24 h; b) (i) N-hydroxylsuccinimide (1.2 equiv.), DMAP (1 equiv.), DCC (1.2 equiv.), r.t., 4 h; (ii) PEG3350-bis-amine (2.2 equiv.), CHCl3, r.t., overnight; c) Folic acid (1 equiv.), DSPE-PEG3350-NH2 (3 equiv.), DCC (3 equiv.), DMSO-Py (10/1, v/v), r.t., 16 h.
N-Succinyl-DSPE
To a solution of DSPE (2 g) and triethylamine (1.5 mL, 4 equiv.) in dry ethanol-free chloroform (75 mL) was added succinic anhydride (535 mg, 2 equiv.) at room temperature (r.t.) with stirring. The reaction was complete after stirring at r.t. for 24 h according to TLC analysis. The reaction mixture was diluted with 35 mL methanol and extracted with 1 M HCl (22 mL) in a separation funnel. The organic layer was dried with anhydrous sodium sulfate, filtered, concentrated, and precipitated with acetone at −20 °C. The precipitate was collected and placed under high vacuum overnight. White solid was obtained. Rf = 0.47 in chloroform-methanol-ammonium hydroxide (65:25:4 v/v). The structure of the product was confirmed with 1H NMR and MALDI mass spectrometry.
DSPE-PEG3350-NH2
To a solution of N-succinyl-DSPE (400 mg) and N-hydroxysuccinimide (65.1 mg, 1.2 equiv.) in dry chloroform (10 mL) were added dimethylaminopyridine (57.6 mg, 1 equiv.), triethylamine (263 μL), and dicyclohexylcarbodiimide (DCC) (116.8 mg, 1.2 equiv.) at r.t. under argon. After 4 h reaction in the dark at r.t., a solution of PEG3350-bis-amine (3.5 g, 2.2 equiv.) in dry chloroform (40 mL) was added to the reaction mixture. The reaction was kept at r.t. overnight. The precipitate was filtered off and the filtrate was concentrated by rotary evaporation and purified by flash column chromatography. Elution method: solvent A: chloroform, solvent B: methanol; segment 1: 0% –10% B, 240 mL; segment 2: 10%–15% B, 360 mL; segment 3: 15%–15% B, 240 mL. Rf = 0.6 in chloroform-methanol (5:1 v/v). The structure of the product was confirmed with 1H NMR and MALDI mass spectrometry.
FA-PEG3350-DSPE
To a solution of folic acid (188.5 mg) and DSPE-PEG3350-NH2 (600 mg, 3 equiv.) in dimethyl sulfoxide (20 mL) and pyridine (2 mL) was added DCC (88 mg, 3 equiv.) at r.t. under argon in the dark. After the completion of the reaction (16 h according to TLC), the product was precipitated by diethyl ether at 4 °C. The crude product was dissolved in 2 mL dimethyl sulfoxide and 1 mL pyridine, loaded on flash column, and purified with the following solvent elution scheme. Solvent A: chloroform, solvent B: MeOH-NH4OH 25/4; segment 1: 0%–10% B, 96 mL; segment 2: 10%–20% B, 144 mL; segment 3: 20%–20% B, 144 mL. Rf = 0.85 in chloroform-methanol-ammonium hydroxide (65:25:4 v/v). The structure of the product was confirmed with 1H NMR and MALDI mass spectrometry.
Synthesis of FITC-DSPE
FITC-DSPE was synthesized by the direct coupling of FITC to DSPE (Scheme 2). To a solution of DSPE (50 mg) in dry chloroform (5 mL) and triethylamine (37.3 μL) was added FITC (52.1 mg, 2 equiv.) in dimethyl sulfoxide. The mixture was allowed to react overnight at r.t. in the dark. The mixture was concentrated by rotary evaporation, and purified by flash column chromatography. Elution method: solvent A: chloroform, B: methanol; segment 1: 0%–20% B, 24 mL; segment 2: 20%–20% B, 48 mL; segment 3: 20%–30% B, 72 mL. Fractions of pure product were pooled, evaporated, and dried under high vacuum overnight in the dark. Rf = 0.38 in chloroform-methanol (4:1 v/v). The structure of the product was confirmed with 1H NMR and MALDI mass spectrometry.
Scheme 2.

Synthesis FITC-DSPE. Reagents and conditions:a) DSPE (1 equiv.), FITC (2 equiv.), triethylamine (4 equiv.), CHCl3, r.t., overnight.
Effect of Ligand Concentration and PEG on FTL Cell Association
To investigate how ligand density effects FTL cell association, liposomes composed of DSPC/Chol (55:40) were formulated with varying mole ratios (0.01, 0.03, 0.1, 0.5 ) of FA-PEG2000-DSPE and 0.2 mole ratio of FITC-DSPE. To examine the effect of PEG on FTL cell association, the following liposome formulations were prepared by mixing the molar ratio of the components in chloroform: F2000L(0.03) - DSPC/Chol/FA-PEG2000-DSPE (55:40:0.03), F2000PL (0.03) - DSPC/Chol/mPEG2000-DSPE/FA-PEG2000-DSPE (55:40:4.5:0.03), F3350L(0.03) -DSPC/Chol/FA-PEG3350-DSPE (55:40:0.03), and F3350PL(0.03) - DSPC/Chol/mPEG2000-DSPE/FA-PEG3350-DSPE (55:40:4.5:0.03). These formulations were also fluorescently labeled with 0.2 mole ratio of FITC-DSPE to total lipid amount. All lipid mixtures (10 μmol lipid) were dissolved in chloroform and dried into a thin film by rotary evaporation then placed under high vacuum overnight. The films were subsequently hydrated with Hepes buffer (10 mM Hepes, 140 mM NaCl pH 7.4) at 60 °C and vortexed. The liposomes were then sonicated at 60 °C and extruded through 200 nm and 100 nm polycarbonate membranes (Avestin, Ottawa, CA). The liposome diameter and particle size distribution were sized by dynamic light scattering (Zetasizer 3000, Malvern Instruments, Westborough, MA). The average liposome diameter was 100–120 nm for all formulations. A schematic diagram of the FTL formulations are shown in Figure 1, and the liposome compositions are summarized in Table 1.
Figure 1.

Schematic diagram of the liposome formulations.
Table 1.
Summary of Liposome Components in Mole Ratios Used in these Studies
| Lipid | Formulation | ||||
|---|---|---|---|---|---|
| PL | F 2000 L | F 2000 PL | F 3350 L | F 3350 PL | |
| DSPC | 55 | 55 | 55 | 55 | 55 |
| Chol | 40 | 40 | 40 | 40 | 40 |
| mPEG2000-DSPE | 5 | - | 4.5–5 | - | 4.5–5 |
| FA-PEG2000-DSPE | - | 0.01–0.5 | 0.03–0.3 | - | - |
| FA-PEG3350-DSPE | - | - | - | 0.03–0.3 | 0.03–0.3 |
KB cells grown as a monolayer were washed with phosphate buffered saline ((PBS; 2.16 g/L Na2HPO4 7H20, 0.2 g/L KH2PO4, 0.2 g/L KCl, 8.0 g/L NaCl) then incubated with 10 mM EDTA for 5 min at 37 °C to resuspend the cells. Then 106 cells were transferred to FACS tubes and centrifuged at 800 rpm for 5 min at r.t. The supernatant was aspirated and the pellets were washed with PBS. The cells were incubated with the different folate targeted liposome formulations (10 nmol lipid) in serum free folate-free RPMI 1640 medium at 37 °C for 3 hr with gentle mixing. The cells were washed twice with PBS. Then the cells were resuspended in 500 μL PBS and analyzed with a FACS Calibur flow cytometer (Becton Dickson) with a 488nm Argon laser. 10,000 events were recorded.
For some of the FTL formulations, the cell association was also measured after 24 hr incubation. KB cells were plated in 6 well plates at a density of 5×105 cells per well. After 48 hr, the media was aspirated, and the cells were incubated with 1 mL of each formulation (10 nmol lipid) in serum free, Folate Free RPMI 1640 medium at 37 °C. After 24 hr, the supernatant was aspirated, and the wells were washed with PBS. The cells (~106) were resuspended with 1 mL trypsin/EDTA. Then the cells were transferred to FACS tubes and centrifuged at 800 rpm for 5 min at r.t. The supernatant was aspirated, and the pellets were washed twice with PBS. Then the cells were resuspended in 500 μL PBS and analyzed by flow cytometry as stated above.
Animals
Balb/c nu/nu mice (8–10 weeks) were obtained from Simonsen Laboratories, Inc. (Gilroy, CA). Animal maintenance and experiments adhered to the “Principles of Laboratory Animal Care” (NIH publication #85-23, revised in 1985) under a protocol approved by the Committee on Animal Research at the University of California, San Francisco.
Circulation Profile and Biodistrubution of FTLs in KB Tumor-bearing Mice
Non-targeted PEGylated liposomes (PLs) were formulated as listed in Table 1. FTLs incorporating FA-PEG-DSPE at three different mole ratios were formulated. The liposome compositions were DSPC/Chol/mPEG2000-DSPE/FA-PEG-DSPE (55:40:5:n) where n = 0.03, 0.1, or 0.3. The lipids (10 μmol total) were dissolved in chloroform and mixed with 200 μL of 5×107 CPM/ml of iodo-PHB-DPPE (Abra et al., 1982). The lipid mixtures were dried into a thin film with N2 gas and then placed under high vacuum overnight. The films were subsequently hydrated with Hepes buffer (10 mM Hepes, 140 mM NaCl pH 7.4) at 60 °C and vortexed. The liposomes were sonicated at 60 °C and then extruded through 200 nm and 100 nm polycarbonate membranes. The liposome diameter and particle size distribution were measured by dynamic light scattering. The mean liposome diameter was 90–110 nm for all formulations. Balb/c nu/nu mice were placed on a folate-free diet one week before tumor inoculation and were maintained on the special feed throughout the study. KB (106) cells suspended in 50 μL of folate-free RPMI 1640 medium without serum were inoculated subcutaneously in the right hind flank of each mouse. On Day 15 after tumor inoculation, each formulation (~1 μmol lipid in 200 μL) was injected into the tail vein of the mice (n = 3 mice/group). At 3 and 24 hrs post-injection, blood from submandibular puncture was collected. At 48 hr post-injection, the liver, spleen, tumor, muscle tissue, and blood (from cardiac puncture) were collected and weighed. The amount of radioactivity present was quantified by gamma scintillation counting (Wallac Wizard 1480 automatic gamma counter, PerkinElmer, Waltham, MA).
Antitumor Activity and Survival Studies
FTLs composed of DSPC/Chol/mPEG2000-DSPE/FA-PEG-DSPE (55:40:5:0.03) and NTLs were formulated as listed in Table 1. The lipid mixtures were dissolved in chloroform, dried into a thin film by rotary evaporation, and placed under high vacuum overnight. The lipid films were rehydrated with 1 mL of 250 mM ammonium sulfate at 60 °C, vortexed, and then subjected to three freeze thaw cycles. The liposomes were sonicated at 60 °C and extruded through 200 nm and 100 nm polycarbonate membranes. The formulations were added to a dialysis cassette (10, 000 MWCO) and dialyzed against 5% dextrose at 4 °C. Then Dox was remote loaded into the liposomes at a 0.1/1 drug to lipid molar ratio for 1 hr at 65 °C. To separate the liposome encapsulated Dox from unencapsulated Dox, a column packed with Dowex 50 Wx4 resin was used. The encapsulated Dox concentration was determined after dissolving a liposome sample into acidified isopropyl alcohol (90% isopropyl alcohol, 75 mM HCl) and comparing the fluorescence (ex 490 nm, em 585 nm) to a standard curve prepared in the same solvent. The remote loading efficiency was ~ 90% for all formulations. The liposome diameter and particle size distribution were measured by dynamic light scattering. The mean liposome diameter was ~120 nm for all formulations.
Balb/c nu/nu mice were placed on a folate-free diet two weeks before tumor inoculation and maintained on the special feed throughout the study. KB cells (106), resuspended in 50 μL folate free RPMI 1640 medium without serum, were inoculated subcutaneously in the right hind flank of each mouse. On Day 8 after tumor implantation, mice were randomly distributed into treatment groups of 7–8 mice. On Day 10 after tumor implantation, the tumors were 30–60 mm3. Each formulation (200 μL) was administered by a single tail vein injection. Tumor volume was determined by measuring the tumor in three dimensions with calipers and calculated using the formula: tumor volume = length × width × height. Mice were sacrificed due to tumor burden (volume ≥2000 mm3) or decrease in body weight (> 15% loss). The percent tumor growth delay (%TGD) was calculated from the equation %TGD = (T−C)/C × 100, where T is the average time for the tumor volume of a treatment group to reach a designated volume of 500 mm3 and C is the mean time for the control group to reach the designated volume of 500 mm3. Mouse survival was analyzed by using MedCalc 8.2.1.0 for Windows (MedCalc Software, Mariakerke, Belgium).
Results
Effect of Ligand Density and PEG on Folate Targeting to KB Cells in vitro
To determine the optimum ligand concentration for folate targeting to KB cells, we evaluated the cellular association of FITC labeled FTLs with varying mole percentages of FA-PEG2000-DSPE by flow cytometry. As depicted in Figure 2, FTL association with KB cells was dependent on ligand concentration. F2000L with 0.03 mole ratio of FA-PEG2000-DSPE -F2000L(0.03) - had the greatest mean fluorescence (~14 fold above blank) and the most association to KB cells compared to the other formulations (p<0.05). In fact, increasing the ligand density to 0.1 mole ratio decreased the cellular association to 11 fold above blank. In addition, F2000L(0.5) had a similar cellular association as F2000L (0.01). Therefore, these results demonstrated that as little as 0.03 mole ratio (~0.03 mol%) of the ligand was optimal for binding.
Figure 2.

Cell association of FTLs of varying FA-PEG(2000)-DSPE mole ratios with KB cells. Cells were incubated with liposomes labeled with FITC-DSPE and containing Folate-PEG(2000)-DSPE at either 0.01, 0.03, 0.1, or 0.5 mole ratio for 3 hours at 37 °C and analyzed by flow cytometry. A. Flow cytometry spectrum. B. Mean fluorescence of formulations. Data shown as average ± standard deviation of at least two measurements. * Statistical significance (p < 0.05) between cells treated with F2000L(0.03) and other F2000L groups as measured by ANOVA followed by Student-Newman-Keuls post-hoc test of the geometric mean of the fluorescence.
We next examined the cell association of FTLs with and without mPEG2000-DSPE by flow cytometry. The data in Figure 3(A and C) demonstrate that additional PEG significantly decreased the cellular association of FTLs. F2000PL had about 5 fold less association than F2000L (p < 0.05), and F3350PL had approximately 3 fold less cell association than F3350L (p < 0.05). The cellular association of F2000PL and F3350PL were similar. Co-incubation of F2000L and F3350L with 2mM folic acid further reduced the cellular association, thus demonstrating the specific binding of the FTLs to the KB cells. The cellular association of PLs was similar to the cell association of FTLs + 2 mM FA (data not shown). After 24 hr incubation of cells with the formulations (Figure 3B and D), the cellular association of F2000PL was approximately 9 fold less than F2000L (p < 0.05) whereas the cellular association of F3350PL was only about 2 fold less than F3350L (p < 0.05). Again, the KB cellular association of F2000PL and F3350PL were similar.
Figure 3.

Cell association of FTLs with and without mPEG2000-DSPE. Cells were incubated with liposomes labeled with FITC-DSPE and containing Folate-PEG(2000)-DSPE or Folate-PEG(3350)-DSPE at 0.03 mole ratio (~0.03 mole %) and analyzed by flow cytometry. A, C. 3 hour incubation at 37 °C. B, D. 24 hr incubation at 37 °C. Data shown as average ± standard deviation of at least two measurements. * p < 0.05 as measured by ANOVA followed by Student-Newman-Keuls post-hoc test of the geometric mean of the fluorescence.
Blood Concentration Profile and Biodistribution of FTLs
The circulation profile and biodistribution properties of the PEGylated FTL formulations with various ligand densities were studied in KB tumor mouse model. Figure 4 illustrates the circulation profile of the various FTL formulations. There were significantly more PLs present in the circulation at the 3 hr and 24 hr time points compared to all the FTL formulations (p < 0.05). There was no significant difference among the various FTL formulations at 3, 24 or 48 hr although there was a trend that FTLs with lower mol% of the targeting ligand circulated longer.
Figure 4.

Blood circulation profile of FTLs with varying FA-PEG-DSPE mole percentages in KB tumored Balb/c nu/nu mice. Blood was collected by submandibular puncture at 3 hr and 24 hr and by heart puncture at 48 hr after i.v. injection of 1 μmol lipid/mouse (n=3 mice). The values are presented as mean ± standard deviation. *Statistical significance between PL compared to all other formulations at 3 hr and 24 hr time points (p < 0.05) as measured by ANOVA followed by Student-Newman-Keuls post-hoc test.
We also investigated the biodistribution of the various PEGylated FTL formulations in the blood, tumor, liver, spleen and muscle tissue 48 hr post-injection (Figure 5). Although there were some differences in the blood circulation profiles of the various formulations as described above, there were no significant differences in the tumor accumulation. F3350PL(0.03%) had significantly more liver uptake compared to the PL and other FTL formulations (p < 0.05). Thus, the FTL with the lower ligand density and the longer PEG spacer had enhanced uptake by the liver. Despite the longer systemic circulation of PLs, all the formulations had similar levels of tumor accumulation.
Figure 5.
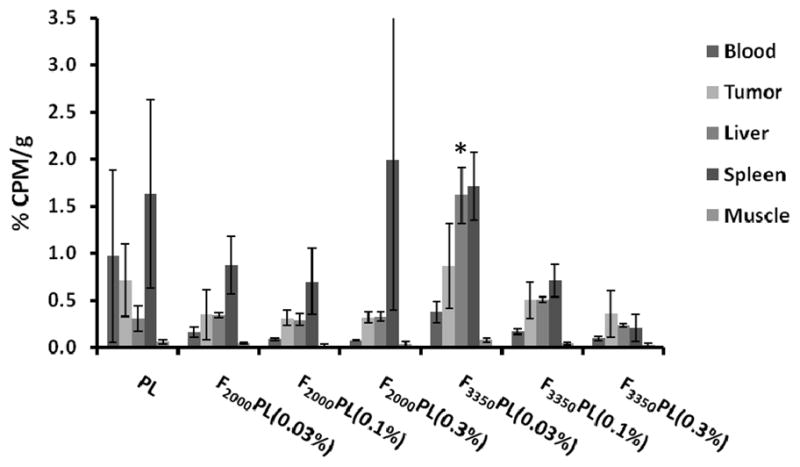
Biodistribution of radiolabeled FTLs and PLs in KB tumored Balb/c nu/nu mice sacrificed 48 hr after i.v. injection of 1 μmol lipid/mouse (n=3 mice). The values are mean ± standard deviation. *Statistical significance between F3350PL(0.03%) compared to all other formulations (p < 0.05) as measured by ANOVA followed by Student-Newman-Keuls post-hoc test.
Effect of FTL-Dox on Tumor Growth and Survival Rate
The antitumor activity of Dox encapsulated in PLs (PL-Dox) and in F3350PL(0.03 mol%), F3350PL(0.03%)-Dox, after a single intravenous tail injection in KB tumor- bearing mice was evaluated. As depicted in Figure 6, all the liposomal formulations were more effective than PBS and Dox 10 mg/kg as expected. F3350PL(0.03%)-Dox 10 mg/kg and PL-Dox 10 mg/kg had similar antitumor activity and had the same TGD of 43% (Table 2). F3350PL(0.03%)-Dox 20 mg/kg had the greatest therapeutic efficacy with 86% TGD. NTL-Dox 20 mg/kg had the second best therapeutic effect with 64% TGD. However, the difference between the tumor volumes of F3350PL(0.03%)-Dox 20 mg/kg and PL-Dox 20 mg/kg groups were not statistically significant.
Figure 6.

Antitumor activity of PL-Dox and F3350PL(0.03%)-Dox. KB tumored Balb/c nu/nu mice (n=7 or 8) were treated with a single i.v. injection on Day 10 (as indicated by arrow). Error bars represent SEM.
Table 2.
Quantification of Antitumor and Survival Data
| Formulation | TGD* (%) | MST‡ (Days) | ILS# (%) |
|---|---|---|---|
| PBS | - | 47 ± 19 | - |
| Dox 10 mg/kg | 7 | 58 ± 21 | 21 |
| PL-Dox 10 mg/kg | 43 | 67 ± 19 | 42 |
| F3350PL(0.03%)-Dox 10 mg/kg | 43 | 55 ± 7 | 16 |
| PL-Dox 20 mg/kg | 64 | 55 ± 22 | 16 |
| F3350PL(0.03%)-Dox 20 mg/kg | 86 | 78 ± 12 | 65 |
TGD - tumor growth delay
MST - mean survival time
ILS - increase in life span
The effect of the NTL-Dox and FTL-Dox formulations at the maximum tolerated dose on the survival rate of the KB tumored mice is shown in Figure 7. Mice treated with F3350PL(0.03%)-Dox 20 mg/kg had the longest mean survival time and the highest percent increase in life span (Table 2). The F3350PL(0.03%)-Dox 20 mg/kg group lived significantly longer than the PL-Dox 20 mg/kg group (P = 0.0121; log-rank test). These findings in combination with the antitumor data indicate that F3350PL(0.03%)-Dox 20 mg/kg is therapeutically more effective than PL-Dox 20 mg/kg in treating KB tumors even after only a single intravenous tail vein injection. Table 3 summarizes the statistical comparisons of the survival data.
Figure 7.

Effect of PL-Dox and F3350PL(0.03%)-Dox on the survival of KB tumored Balb/c nu/nu mice. Mice (n=7 or 8) were treated with a single i.v. injection on Day 10. The F3350PL(0.03%)-Dox 20 mg/kg group lived significantly longer than PL-Dox 20 mg/kg group (p = 0.0121; log-rank test).
Table 3.
Multiple Statistical Comparison of Survival Data*
| PBS | Dox 10 mg/kg | PL- Dox 10 mg/kg | F3350PL(0.03%)- Dox 10 mg/kg | PL- Dox 20 mg/kg | F3350PL(0.03%)- Dox 20 mg/kg | |
|---|---|---|---|---|---|---|
| PBS | - | NS | 0.0437 | NS | NS | 0.049 |
| Dox 10 mg/kg | NS | - | NS | NS | NS | NS |
| PL-Dox 10 mg/kg | 0.0437 | NS | - | NS | NS | NS |
| F3350PL(0.03%)-Dox 10 mg/kg | NS | NS | NS | - | NS | NS |
| PL- Dox 20 mg/kg | NS | NS | NS | NS | - | 0.0121 |
| F3350PL(0.03%)- Dox 20 mg/kg | 0.049 | NS | NS | NS | 0.0121 | - |
p values determined by long-rank test. p > 0.05 considered not significant (NS).
Discussion
The purpose of this study was to evaluate folate-targeted liposome drug delivery and assess whether this targeting strategy has the potential to enhance cancer therapy. We focused our efforts on the KB tumor cell line since it is one of the highest FR over-expressing cancer cell lines, and it is a model cell line for FR targeted delivery.
We first synthesized folate lipid conjugates then prepared and characterized various FTLs formulations. We found that including only 0.03 mole ratio (~0.03 mol%) of the folate ligand in the liposome formulations resulted in the best liposome cell association to KB cells in vitro (Figure 2). There is a possibility that liposomes containing a greater folate surface density were internalized into a low pH compartment more readily than liposomes with a lower folate surface density and the fluorescein lipid was more highly quenched. We think this unlikely since this result is consistent with previous findings (Reddy et al., 2002). Reddy and coworkers hypothesized that only a small density of ligands are optimal for binding because folates can bind to each other at high surface densities and inhibit binding to FRs (Reddy et al., 2002). Additionally, an optimal folate ligand density can depend on a number of factors: the accessibility of the folate moiety, PEG length, PEG-folate chemical linkage, and type of folate-lipid conjugate (Gabizon et al., 2003, Reddy et al., 2002, Shmeeda et al., 2006, Zhao et al., 2007). PEG is widely used in liposome formulations because it is known to prolong the retention of the liposomes in the systemic circulation and to enhance tumor accumulation (Allen and Cullis, 2004). We investigated whether the presence of PEG in the liposome formulation decreases FTL cell association since there are divergent views on the influence of PEG on folate-mediated targeting (Gabizon et al., 2004, Gabizon et al., 1999, Lee and Low, 1994). We found that the presence of PEG in the liposome decreases FTL cell association. We also determined that PEGylated liposomes with FA attached via a PEG2000 spacer or a PEG3350 spacer had equivalent levels of binding to KB cells (Figure 3).
An increase in folate ligand density decreased the circulation lifetime of FTLs. In spite of this more rapid elimination of FTLs (Figures 4 and 5), the PLs and FTLs equivalently distributed to the tumor tissue. The fact that FTLs were cleared faster from blood but accumulated similarly in the tumor tissue as PLs suggests that the FTLs were targeting to KB cells in vivo. It is interesting that F3350PL at the optimal binding density of 0.03 mol % is targeted to the liver significantly more than the other formulations yet has a comparable level of tumor accumulation after 48 hrs. Similar findings have been previously reported (Lu et al., 2007, Gabizon et al., 2003). Putting the mice on a folate-free diet may have led to up-regulation of FR in the liver, spleen, and kidney in addition to the tumor tissue (Gabizon et al., 2004). This may lead to increased clearance of FTL drugs and further compromise FTL accumulation in tumor tissue.
There are few studies reporting on the antitumor efficacy of FTL therapeutics in vivo. Pan and coworkers have found that F3350PL-Dox 5 mg/kg administered via three i.p. injections improved the survival of mice bearing murine lymphocytic L1210-JF FR+ cell ascites more than mice receiving PL-Dox 5mg/kg (P = 0.0259; log-rank test) (Pan et al., 2002). KB tumor-bearing mice treated with F3350PL-Dox 10 mg/kg via six i.p. injections had greater tumor growth inhibition and a slight increase in lifespan compared to mice treated with PL-Dox (Pan et al., 2003). However, in mice bearing murine lung carcinoma M109 FR+ tumors, F5000PL-Dox 8 mg/kg administered after a single intravenous injection had a lower tumor-killing effect than PL-Dox 8 mg/kg (Yamada et al., 2008). Therefore, the literature is unclear whether there is a therapeutic advantage of folate-targeted delivery.
Although KB cells have been widely utilized for investigating folate targeting to FR+ cells, there are no studies reporting on the efficacy of FTL therapeutics after intravenous administration in KB tumor-bearing mice. This motivated us to further investigate the therapeutic efficacy of FTL-Dox in a KB tumor mouse model (Figures 6 and 7). We found that KB tumor-bearing mice treated with a single i.v. injection of F3350PL(0.03%)-Dox 20 mg/kg had a slightly greater tumor growth inhibition and longer life span than mice treated with PL-Dox at the same dose (P = 0.0121; log-rank test). This result is surprising given that the FTLs were cleared faster than PLs. However, F3350PL(0.03%)-Dox 10 mg/kg was slightly less effective than PL-Dox 10 mg/kg. The enhanced therapeutic efficacy of F3350PL (0.03%)-Dox compared to PL-Dox at the higher dose could be explained by saturation of RES uptake mechanisms since at the higher dose, there is an increase in the lipid concentration. Therefore, more of the FTLs are able to reach the tumor tissue.
The pharmacokinetic, biodistribution, and antitumor results imply that minimizing folate targeting to the liver and other tissues of the RES may help to improve the antitumor efficacy of FTL drugs. Gabizon and coworkers investigated the effect of co-dosing FTLs with free FA on FTL biodistribution. They determined that co-dosing with free FA significantly reduced FTL liver uptake as well as clearance from the blood, but had neglible effect on FTL accumulation in tumors (Gabizon et al., 2003). They explained that free folic acid inhibits FTLs from being taken up by Kuppfer cells in the liver via receptor mediated endocytosis. However, co-dosing with free FA did not affect the tumor accumulation of FTLs because the distribution of the FTLs in tumor tissue is primarily due to passive extravasation via the enhanced permeation and retention (EPR) effect (Maeda, 2001) rather than ligand-mediated targeting. Therefore, increasing the circulation time of FTLs might enhance tumor localization of folate-targeted liposomes. One tactic to accomplish this may be to decrease the folate ligand density below the optimal concentration so as to minimize binding to non-target, FR expressing tissues, and thus enhance the circulation time and tumor accumulation. When all of the folate liposome targeting reports are viewed together, it appears that a multivalent folate-ligand targeted particle does not provide as great of a therapeutic benefit as does a folate-drug conjugate in which there is a single folate is attached to a single agent (Leamon et al., 2008). A variety of folate drug conjugates are proceeding into clinical trials. Thus there may be a fundamental problem with accessibility and entry of folate-targeted liposomes into tumor cells; certainly success in vitro has not yet been reflected in the in vivo assessment of FTL liposome drugs. Alternatively, masking the folate ligand with a longer PEG that can be removed from the liposome surface may enable the FTLs to avoid clearance in the liver (Kale and Torchilin, 2007). The decreased binding avidity of the FTLs may also lead to their increased penetration into the solid tumor (Allen, 2002).
Conclusions
These studies indicate that under certain conditions folate targeting can improve delivery of liposome drug carriers to tumors with concomitant improvement in antitumor activity compared to the non-targeted liposomal drug. We observed that in vitro optimization of folate targeting does not necessarily translate to an enhancement in in vivo targeting to FR+ tumors. Further modification of FTLs to reduce liver and spleen uptake may increase FTL circulation time and therefore enhance the tumor accumulation and antitumor efficacy of FTL therapeutics.
Acknowledgments
This work was supported by NIH RO1 GM061851. Kareen Riviere was supported by a PhRMA Foundation Pre-Doctoral Fellowship and a UNCF-Merck Dissertation Fellowship.
Footnotes
Declaration of Interest: The authors report no conflicts of interest.
References
- ABRA RM, SCHREIER H, SZOKA FC. The use of a new radioactive-iodine labeled lipid marker to follow in vivo disposition of liposomes: comparison with an encapsulated aqueous space marker. Res Commun Chem Pathol Pharmacol. 1982;37:199–213. [PubMed] [Google Scholar]
- ALLEN TM. Ligand-targeted therapeutics in anticancer therapy. Nat Rev Cancer. 2002;2:750–63. doi: 10.1038/nrc903. [DOI] [PubMed] [Google Scholar]
- ALLEN TM, CULLIS PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–22. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- ANTONY AC. The biological chemistry of folate receptors. Blood. 1992;79:2807–20. [PubMed] [Google Scholar]
- GABIZON A, HOROWITZ AT, GOREN D, TZEMACH D, MANDELBAUM-SHAVIT F, QAZEN MM, ZALIPSKY S. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10:289–98. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- GABIZON A, HOROWITZ AT, GOREN D, TZEMACH D, SHMEEDA H, ZALIPSKY S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9:6551–9. [PubMed] [Google Scholar]
- GABIZON A, SHMEEDA H, HOROWITZ AT, ZALIPSKY S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56:1177–92. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- GOREN D, HOROWITZ AT, TZEMACH D, TARSHISH M, ZALIPSKY S, GABIZON A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res. 2000;6:1949–57. [PubMed] [Google Scholar]
- HILGENBRINK AR, LOW PS. Folate receptor-mediated drug targeting: from therapeutics to diagnostics. J Pharm Sci. 2005;94:2135–46. doi: 10.1002/jps.20457. [DOI] [PubMed] [Google Scholar]
- KALE AA, TORCHILIN VP. Enhanced transfection of tumor cells in vivo using “Smart” pH-sensitive TAT-modified pegylated liposomes. J Drug Target. 2007;15:538–45. doi: 10.1080/10611860701498203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEAMON CP, REDDY JA, VETZEL M, DORTON R, WESTRICK E, PARKER N, WANG Y, VLAHOV I. Folate targeting enables durable and specific antitumor responses from a therapeutically null tubulysin B analogue. Cancer Res. 2008;68:9839–44. doi: 10.1158/0008-5472.CAN-08-2341. [DOI] [PubMed] [Google Scholar]
- LEE RJ, LOW PS. Delivery of liposomes into cultured KB cells via folate receptor-mediated endocytosis. J Biol Chem. 1994;269:3198–204. [PubMed] [Google Scholar]
- LEE RJ, LOW PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233:134–44. doi: 10.1016/0005-2736(94)00235-h. [DOI] [PubMed] [Google Scholar]
- LU Y, LOW PS. Folate-mediated delivery of macromolecular anticancer therapeutic agents. Adv Drug Deliv Rev. 2002;54:675–93. doi: 10.1016/s0169-409x(02)00042-x. [DOI] [PubMed] [Google Scholar]
- LU Y, WU J, GONIT M, YANG X, LEE A, XIANG G, LI H, LIU S, MARCUCCI G, RATNAM M, LEE RJ. Role of formulation composition in folate receptor-targeted liposomal doxorubicin delivery to acute myelogenous leukemia cells. Mol Pharm. 2007;4:707–12. doi: 10.1021/mp070058l. [DOI] [PubMed] [Google Scholar]
- MAEDA H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- O’NEILL ID. Continued misrepresentation of KB cells as being of oral cancer phenotype requires action. Oral Oncology. 2009;45:e117–e118. doi: 10.1016/j.oraloncology.2009.02.005. [DOI] [PubMed] [Google Scholar]
- PAN XQ, WANG H, LEE RJ. Antitumor activity of folate receptor-targeted liposomal doxorubicin in a KB oral carcinoma murine xenograft model. Pharm Res. 2003;20:417–22. doi: 10.1023/a:1022656105022. [DOI] [PubMed] [Google Scholar]
- PAN XQ, ZHENG X, SHI G, WANG H, RATNAM M, LEE RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood. 2002;100:594–602. doi: 10.1182/blood.v100.2.594. [DOI] [PubMed] [Google Scholar]
- REDDY JA, ABBURI C, HOFLAND H, HOWARD SJ, VLAHOV I, WILS P, LEAMON CP. Folate-targeted, cationic liposome-mediated gene transfer into disseminated peritoneal tumors. Gene Ther. 2002;9:1542–50. doi: 10.1038/sj.gt.3301833. [DOI] [PubMed] [Google Scholar]
- SHMEEDA H, MAK L, TZEMACH D, ASTRAHAN P, TARSHISH M, GABIZON A. Intracellular uptake and intracavitary targeting of folate-conjugated liposomes in a mouse lymphoma model with up-regulated folate receptors. Mol Cancer Ther. 2006;5:818–24. doi: 10.1158/1535-7163.MCT-05-0543. [DOI] [PubMed] [Google Scholar]
- STEPHENSON SM, LOW PS, LEE RJ. Folate receptor-mediated targeting of liposomal drugs to cancer cells. Methods Enzymol. 2004;387:33–50. doi: 10.1016/S0076-6879(04)87003-4. [DOI] [PubMed] [Google Scholar]
- YAMADA A, TANIGUCHI Y, KAWANO K, HONDA T, HATTORI Y, MAITANI Y. Design of folate-linked liposomal doxorubicin to its antitumor effect in mice. Clin Cancer Res. 2008;14:8161–8. doi: 10.1158/1078-0432.CCR-08-0159. [DOI] [PubMed] [Google Scholar]
- ZHAO XB, MUTHUSAMY N, BYRD JC, LEE RJ. Cholesterol as a bilayer anchor for PEGylation and targeting ligand in folate-receptor-targeted liposomes. J Pharm Sci. 2007;96:2424–35. doi: 10.1002/jps.20885. [DOI] [PubMed] [Google Scholar]