

Abstract
Duchenne muscular dystrophy (DMD) is a dystrophin-deficient lethal muscle disease. To date, the catastrophic muscle wasting phenotype has only been seen in dystrophin-deficient humans and dogs. While Duchenne-like symptoms have been observed in more than a dozen dog breeds, the mutation is often not known and research colonies are rarely established. Here we report an independent canine DMD model originally derived from the Pembroke Welsh corgi breed. The affected dogs presented clinical signs of muscular dystrophy. Immunostaining revealed the absence of dystrophin and up-regulation of utrophin. Histopathologic examination showed variable fiber size, central nucleation, calcification, fibrosis, neutrophil and macrophage infiltration and cardiac focal vacuolar degeneration. Carrier dogs also displayed mild myopathy. The mutation was identified as a long interspersed repetitive element-1 (LINE-1) insertion in intron 13 which introduced a new exon containing an in-frame stop codon. Similar mutations have been seen in human patients. A colony was generated by crossing carrier females with normal males. Affected puppies had a normal birth weight but they experienced a striking growth delay in the first 5 days. In summary, the new corgi DMD model offers an excellent opportunity to study DMD pathogenesis and to develop novel therapies.
Introduction
Duchenne muscular dystrophy (DMD) results from mutations in the dystrophin gene (1). DMD is the most common childhood lethal muscle disease with a prevalence of 1.3 to 2.63 per 10,000 male births (2–5). In normal striated muscle, dystrophin localizes at the cytosolic side of the sarcolemma. It protects the sarcolemma from contraction-associated shearing stress. The absence of dystrophin compromises myofiber integrity. Damaged muscle cells undergo degeneration and necrosis, and are eventually replaced by adipose and fibrous tissues. Patients gradually lose their mobility and inevitably die during early adulthood.
It has been more than two decades since the dystrophin gene mutation was recognized as the molecular cause of DMD. Our understanding of DMD pathogenesis remains incomplete and the disease remains incurable. Among many reasons that may have halted the progress is the shortage of appropriate animal models. While dystrophin deficiency leads to severe muscle atrophy and early death in humans, the characteristic clinical manifestation is not observed in dystrophin-null mice and cats (6–8). In contrast, dystrophin-deficient dogs show signs that are generally consistent with those of human patients (9).
Duchenne-like muscular dystrophy has been reported in at least 15 different dog breeds including beagle (10, 11), Belgian groenendaeler shepherd (12), Brittany spaniel (13), Cavalier King Charles spaniel (14), German short-haired pointer (15), golden retriever (11, 16, 17), grand basset griffon vendeen (18), Irish terrier (19), Japanese spitz (20), labrador retriever (21), miniature schnauzer (22), old English sheepdog (23), rat terrier (24), samoyed (25) and weimaraner (26). However, a majority of studies have been limited to descriptive clinical presentations. In most cases, the disease causing mutations are not identified and research colonies have not been established. Nearly all our current knowledge of canine DMD derives from the golden retriever muscular dystrophy (GRMD) dog, a model caused by a single point mutation in the dystrophin gene (11, 16, 17).
A wide variety of dystrophin gene mutations have been found in human patients (27–29). These include deletions, insertions, duplications, and point mutations. While most in-frame mutations result in mild Becker muscular dystrophy (BMD) and most out-of-frame mutations lead to DMD, a significant number of patients do not fit into the reading-frame rule. Recent studies further suggest the existence of genetic modifiers that may substantially alter the clinical phenotype (30). Collectively, the pathogenic process of DMD is much more complex than we may have appreciated previously. A thorough understanding of DMD pathogenesis requires comprehensive analysis in multiple models. Considering the genetic and clinical similarities between affected dogs and human patients, establishing additional canine models will be extremely helpful.
Here we report a novel DMD dog model originally derived from a spontaneous mutation in the Pembroke Welsh corgi breed (abbreviated as “corgi” in this manuscript). The affected dogs showed clinical and histological changes resembling human DMD. Immunofluorescence staining revealed a lack of sarcolemmal dystrophin staining. We also identified the mutation as a long interspersed repetitive element-1 (LINE-1) insertion in intron 13. Similar mutations have been observed in human patients (31–38). Most importantly, we have successfully established a research colony. The corgi model described here complements the existing GRMD model and will greatly benefit translational studies on DMD.
Materials and Method
Animals
The initial affected dog was evaluated at the College of Veterinary Medicine at Oklahoma State University (39). Muscle was sent to Auburn University for immunohistochemisty to confirm that dystrophin was deficient (Supplementary Figure 1). A small colony was established at Oklahoma State and subsequently sent to the University of Missouri. In establishing the colony at Oklahoma State, the original dogs were outbred to nonchondysplastic breeds. Some dogs were then provided to Auburn University to establish a colony. While at Auburn University, the colony has also been outbred to beagles. Thus, the current colony includes only mixed-breed dogs. To our knowledge, the Auburn colony is now the only source of affected dogs. All experimental dogs used in this study were produced by in-house breeding at Auburn University. All animal experiments were approved by the Animal Care and Use Committees of the three universities and were in accordance with NIH guidelines.
Histopathology studies
Tissues from four normal dogs, three affected dogs and two carriers were used in histopathology studies (Supplementary Table 1). Haematoxylin and eosin (HE) staining was used to reveal general histopathological lesions. The percentage of centrally nucleated myofibers were quantified in HE stained muscle cross-sections as we described before (40). The minimum myofiber diameter was determined from the digitized images with the Image J software (version 1.36b, NIH). Three random fields were analyzed for each muscle section. Three different sections were studied for each muscle sample. Fibrosis and muscle calcification were evaluated with Masson trichrome staining and Alizarin red staining, respectively, according to our published protocols (41). Macrophage infiltration was evaluated by non-specific esterase (α-naphthyl butyrate esterase) staining according to a published protocol (42, 43). Macrophages are dark brown cells scatted between myofibers.
Immunohistochemical staining for inflammatory cells
Immune cells were detected with the Vectastain ABC kit (Vector Laboratories, Burlingame, CA) according to a previously published protocol (44). Canine specific antibodies were obtained from AbD Serotec (Raleigh, NC). The antibody dilutions are 1:500, 1:1000, 1:200, 1:8000 and 1:2000 for B cell, CD4+ T cell, CD8+ T cell, neutrophil and macrophage, respectively (44).
Immunofluorescence staining for dystrophin, utrophin and the dystrophin-associated glycoprotein complex (DGC)
Dystrophin was examined with ten epitope-specific antibodies including a rabbit polyclonal antibody against spectrin-like repeats 4–6 (1:400; Santa Cruz, CA), a mouse monoclonal antibody against spectrin-like repeats 6–8 (Dys-1, Novocastra, Newcastle, UK), a mouse monoclonal antibody against spectrin-like repeat 11 (Mandys8, 1:200; Sigma, St Louis, MO), a mouse monoclonal antibody against spectrin-like repeat 10/11 (Mandys1=DD414, 1:10; a gift from Dr. Glenn Morris, The Robert Jones and Agnes Hunt Orthopaedic Hospital, UK) (45), a mouse monoclonal antibody against spectrin-like repeat 14 (Mandys110=DD417, 1:10; a gift from Dr. Glenn Morris) (45), a mouse monoclonal antibody against spectrin-like repeat 15 (Mandys101=DD#419, 1:10; a gift from Dr. Glenn Morris) (45), a mouse monoclonal antibody against spectrin-like repeat 17 (Manex44A, 1:300; a gift from Dr. Glenn Morris) (45), a mouse monoclonal antibody against spectrin-like repeat 14–18 (Mandys105, 1:10; a gift from Dr. Glenn Morris) (45), a mouse monoclonal antibody against hinge 3 (Manex50, 1:2000; a gift from Dr. Glenn Morris) (45), and a mouse monoclonal antibody against the C-terminal domain (Dys-2, 1:30; Novocastra). Utrophin was examined with a mouse monoclonal antibody against the utrophin N-terminal domain (VP-U579, 1:20; Vector Laboratories, Burlingame, CA). nNOS was detected with a polyclonal antibody (1:2,000; Santa Cruz Biotechnology, Santa Cruz, CA). β-dystroglycan was revealed with a mouse monoclonal antibody (NCL-b-DG, 1:50; Novocastra). β-sarcoglycan was revealed with a mouse monoclonal antibody (NCL-b-SARC, 1:50; Novocastra). Syntrophin was revealed with a pan-syntrophin mouse monoclonal antibody that recognizes the syntrophin PDZ domain (ab11425, 1:200; Abcam, Cambridge, MA). Dystrobrevin was revealed with a mouse monoclonal antibody (#610766, 1:200; BD Biosciences, San Diego, CA). Immunostaining was performed essentially as we described before (42, 46, 47).
Western blot
A ~3 mm3 piece of muscle was pulverized in liquid nitrogen in a homogenization buffer containing 62.5 mM Tris-HCl pH 6.8, 5 mM ethylenediaminetetraacetic acid (EDTA), 10% sodium dodecyl sulfate (SDS) and 1% protease inhibitor (Roche, IN). After boiling for 2 min, the homogenate was spun at 14,000 rpm for 2 min (Eppendorf centrifuge, model 5417C). The supernatant was used for western blots. Protein concentration was determined using a DC protein assay kit (Bio-Rad, Hercules, CA). 50 μg muscle lysate was electrophoresed on a 6% SDS-polyacrylamide gel. Protein was transferred to a nitrocellulose membrane. Dystrophin was detected with a monoclonal antibody against the N-terminus of the dystrophin protein (MANEX1A, 4C7, 1:500; a gift from Dr. Glenn Morris) (45). As a loading control, membrane was also probed with an anti-α-actin antibody (1:3,000; Sigma, St Louis, MO).
PCR genotyping
Samples of umbilical cord (5 mm long) from a newborn puppy were lysed at 95°C in a buffer containing 25 mM NaOH and 0.2 mM EDTA. After neutralization with 40 mM Tris-HCl, the sample was centrifuged at 14,000 rpm for 2 min. The supernatant was collected as genomic DNA and used in PCR. Three PCR primers were used together in the PCR reaction. These include (1) a common forward primer corresponding to the dystrophin intron 13 sequence prior to the LINE element insertion (DL1255, 5’ TGTCATTTCCTCAGGTTACTGCTT), (2) a reverse primer specific to the inserted LINE element (DL1257, 5’ TGAAGCAAATGGTGGGTATTTG), and (3) a reverse primer corresponding to the dystrophin intron 13 sequence after the LINE element insertion (DL1256, 5’ GCAATGGCCTATCAGTACAGTCC) (Figure 5A). The PCR conditions are (1) 95°C for 2 min, (2) 30 cycles of 98°C for 10 sec, 60°C for 20 sec and (3) 72°C for 2 min. The normal allele yields a 286 bp band and the affected allele yields a 166 bp band.
Figure 5. Mutation in the corgi model.

a, Schematic outline of the normal canine dystrophin gene, mRNA and protein between exons 13 and 14. b, Schematic outline of the mutation in affected corgi. Gray letters, from normal canine dystrophin; Black letters, from LINE element insertion; Small letters, nucleotides in intron; Bolded small letters, conserved splicing signal in intron; Capital letters, nucleotides in exon; Bolded capital letters, stop codon. The cartoons are not drawn to scale.
Serum creatine kinase (CK) assay
Fresh serum was collected from the jugular vein. The CK level was determined with a CK liqui-UV test kit from Stanbio Laboratory (Boerne, TX).
Statistical analysis
Data are presented as mean ± standard error of mean. Statistical analysis was performed with the SPSS software (SPSS, Chicago, IL). Statistical significance for multiple group comparison was determined by one-way ANOVA followed by Bonferroni post hoc analysis. Statistical significance for two-group comparison was determined by t test. Statistical significance for growth curve and breeding data was analyzed with the SAS software (SAS Institute, Cary, NC) using the general linear model and one-side test, respectively. Difference was considered significant when P < 0.05.
Results
Affected corgi dogs show clinical features of muscular dystrophy
Signs of muscular dystrophy were observed in affected dogs. Compared to age-matched normal corgis, affected dogs showed feeding difficulties, delayed growth, generalized muscle wasting, short-stride shuffling and stiff gait, drooling, easy fatigability and exercise intolerance (Supplementary video 1). Muscle atrophy was clearly visible in the head, body and limb muscles (Figure 1). The tibio-tarsal jointswere “dropped” to a plantigrade position (Figure 1). Some affected dogs were barely able to support themselves on their paws and had difficulty walking.
Figure 1. Clinical manifestation of corgi muscular dystrophy.

Representative photographs of 10-m-old corgi in standing position. Top, Rachel, a normal corgi. Bottom, Jerry, a dystrophic corgi. Asterisk indicates proximal limb muscle; Cross indicated distal limb muscle; Arrow indicates the tibial-tarsal joint. The affected dog Jerry shows limb atrophy and his tibial-tarsal joints are dropped to the ground.
Dystrophin is absent in affected corgi dogs
To determine the cause of muscular dystrophy in the corgi breed, we performed comprehensive immunofluorescence staining using antibodies that recognize dystrophin, utrophin, and different components of the DGC. Ten dystrophin antibodies were used in the study.
They recognize spectrin-like repeats 4–6, 6–8, 10/11, 11, 14, 15, 17, 14–18, hinge 3 and the C-terminal domain (Figure 2 and Supplementary Figure 1, data not shown for spectrin-like repeat 14 antibody). All of them reacted with canine dystrophin in normal dog muscle (Figure 2a). In affected dogs, dystrophin staining was lost at the sarcolemma (Figure 2b). Occasionally, revertant fibers were observed (Supplementary Figure 1). While utrophin was not detected in normal muscle, it was clearly visible at the sarcolemma in affected dogs (Figure 2). Compared to that of normal dog muscle, the β-dystroglycan, β-sarcoglycan, dystrobrevin and syntrophin levels were substantially reduced but not eliminated in affected dog muscle. nNOS was not detected at the sarcolemma in affected dogs (Figure 2b). In summary, the immunostaining profile suggests a diagnosis of Duchenne-like muscular dystrophy.
Figure 2. Characterization of dystrophin, utrophin and dystrophin-associated protein expression in normal and affected dog skeletal muscle by immunofluorescence staining.

Immunofluorescence staining was performed on serial muscle sections with antibodies specific for dystrophin N-terminal domain, dystrophin spectrin-like repeats R4–6, R6–8, R11 and R17, dystrophin hinge 3 (H3) and dystrophin C-terminal domain, utrophin, β-dystroglycan, β-sarcoglycan, dystrobrevin and syntrophin. a, Representative immunofluorescence images from skeletal muscle of a normal corgi dog. b, Representative immunofluorescence images from skeletal muscle of a dystrophic corgi dog. Scale bar applies to all the images in the same panel.
Affected dogs display prominent muscle histopathologic lesions
On HE staining, normal corgi skeletal muscle showed minimal variation in fiber size, peripheral nuclei and absence of mononuclear cell infiltration. Dystrophic corgi skeletal muscle contained abnormally large and small fibers, prominent inflammatory cell infiltration and a high frequency of centrally nucleated myofibers (Figure 3a). On Masson trichrome staining, we observed extensive endomysial and perimysial fibrosis in affected dog muscle. While calcium overloading was not detected in normal muscle, highly calcified myofibers were frequently seen in dystrophic muscle (Figure 3a, arrowhead). Furthermore, Alizarin red staining revealed an elevated interstitial calcium level in affected dog muscle (Figure 3a, asterisk). Using non-specific esterase staining, we also detected conspicuous macrophage infiltration (Figure 3a, arrow).
Figure 3. Characterization of muscle histopathology in carrier and dystrophic corgi dogs.


a, Representative images of HE, Masson trichrome (for fibrosis), Alizarin red (for calcium) and non-specific esterase (for macrophages) staining of serial muscle sections from normal limb muscle, dystrophic limb muscle and the dystrophic diaphragm. High magnification images of the boxed areas are shown for Masson trichrome and non-specific esterase staining. Arrowhead, individual myofiber with strong calcium staining; Asterisk, enhanced interstitial calcium staining in dystrophic muscle; Arrow, macrophages. b, Representative images of HE, Alizarin red, Masson trichrome and non-specific esterase staining from limb muscle of a carrier dog. High magnification images of the boxed areas are shown for Masson trichrome and non-specific esterase staining. Arrowhead, centrally localized nuclei; White arrow, focal inflammation; Black arrow, macrophages. c, Quantification of central nucleation from the limb muscle of normal, carrier and affected dogs. Asterisk, significantly different from the other groups. Black bar, normal (N = 1,259 fibers); gray bar, carrier (N = 875 fibers); white bar, affected (N = 1,553 fibers). d, Histograms of minimum myofiber diameter from normal (black bars, N = 394 fibers) and affected (white bars, N = 310 fibers) dog limb muscle. e, Representative HE, Masson trichrome and Alizarin red stained images from the heart of normal and affected dogs. High power HE stained images correspond to the boxed areas in each row. Arrow, vacuolar degeneration seen only in dystrophic heart. Please note, the Masson trichrome positive area in the normal heart corresponds to vessel location.
Similar histology studies were performed on muscle samples obtained from carrier dogs. Despite the lack of fiber size variation, fibrosis and calcification, carrier muscles contained perceptible levels of centrally nucleated myofibers (Figure 3b, arrowhead) and focal macrophage infiltration (Figure 3b, arrow).
On quantification, ~ 0.2 % of normal muscle contained centrally located nuclei. This number increased to ~ 4 % and 23 % in carrier and affected muscles, respectively (Figure 3c). Myofiber size quantification revealed a much broader distribution in affected dogs (Figure 3d). In normal muscle, 60 % myofibers had a minimum diameter between 32 to 40 μm. In affected muscle, only 40 % were in this range (Figure 3d).
Cardiomyopathy is a salient feature in DMD. We examined heart histopathology of a 16-m-old dystrophic corgi dog (Figure 3e). We observed patches of cardiomyocytes with large empty space in the cytoplasm resembling vacuolar degeneration (Figure 3e, arrow). In the same region, there was also fibrosis and calcification (Figure 3e).
Macrophages and neutrophils are the primary inflammatory cells in dystrophic corgi muscle
Canine specific immune cell antibodies were used to characterize inflammation (Figure 4a). Similar to normal muscle controls, B cell, CD4+ and CD8+ T cells were rarely detected in affected dog muscles (Figure 4b, data not shown for the diaphragm and the heart). Neutrophils and macrophages were the dominant immune cells in dystrophic muscles. In limb muscles and the diaphragm of affected dogs, scattered neutrophils and large aggregates of macrophages were observed (Figure 4c). In carrier dog limb muscles, neutrophils and macrophages were also detected occasionally (Figure 4d).
Figure 4. Characterization of immune cell infiltration in the limb muscle, diaphragm and the heart.


a, Representative photomicrographs of immunohistostaining in the dog spleen. For CD8+ T cell and the secondary antibody alone, high power images of the boxed areas are shown. Positive cells stain in dark purple color. b, Representative photomicrographs of immunohistostaining for B cells, CD4+ T cells and CD8+ T cells in the limb muscle of dystrophic corgi dogs. Yellow arrowhead, a rare B cell. c, photomicrographs of immunohistostaining for neutrophils and macrophages in the limb muscle and diaphragm of dystrophic corgi dogs. Top panel, low magnification images; Bottom panel, high magnification images. Purple arrowhead, neutrophil in dystrophic limb muscle; Green arrowhead, neutrophil in dystrophic diaphragm muscle. Neutrophils are less abundant than macrophages. d, Representative photomicrographs of immunohistostaining for neutrophils and macrophages in the limb muscle of normal and carrier dogs. Arrow, centrally located nuclei; Blue arrowhead, a neutrophil in the carrier limb muscle; Red arrowhead, a macrophage in the carrier limb muscle. e, photomicrographs of immunohistostaining for neutrophils and macrophages in the heart of normal and dystrophic corgi dogs. Top panel, low magnification images; Bottom panel, high magnification images. Left two columns, normal heart; Right two columns, dystrophic heart. Pink arrowhead, sporadic neutrophil in the normal heart; Orange arrowhead, sporadic macrophage in the normal heart; Scale bar applies to all the images in the same row.
Neutrophil and macrophage infiltration showed a slightly different pattern in the heart. In the normal heart, residential neutrophils and macrophages were seen (Figure 4e) (neutrophils and macrophages were essentially not detected in normal skeletal muscle, data not shown). In dystrophic dog heart , both neutrophils and macrophages appeared as large aggregates spread out through out the entire heart section (Figure 4e).
A LINE-1 element insertion aborts dystrophin expression in dystrophic corgi dogs
To identify the mutation in the affected corgi dogs, we designed primers covering the entire canine dystrophin cDNA and sequenced the cDNA extracted from affected dogs. We noted a 167 bp insertion between exons 13 and 14 (Figure 5). The insert contained an in-frame stop codon (TAG). A BLAST search identified the insert as part of a canine LINE-1 element (GenBank AB012217, 99% similarity).
To determine the location of the LINE element insertion in the canine dystrophin gene, we initially performed PCR using primers specific to the inserted sequence and the adjacent exons. Unfortunately, we were not able to get definitive PCR products suggesting that the insertion was not adjacent to an exon. Additional PCR primers were designed according to the canine genome data to entirely cover canine dystrophin intron 13 (approximately 25,000 bp). Endogenous splicing acceptor and donor signals were intact in intron 13. A LINE-1 element insertion was identified beginning 1,048 bp into intron 13 and spanning approximately 480 bp. The insertion began immediately after “cag”, a cryptic splicing acceptor signal in canine intron 13 (Figure 5b, Supplementary Figure 2). A conserved splicing donor signal “gtaagt” appeared after the first 167 bp of the insertion. The LINE-1 element insertion ended after approximately 312 additional base pairs. The distal end of the insertion contained a poly A tract which has been difficult to sequence precisely. Hence, the total length of the insertion remained approximate.
Establishment of a dystrophic corgi colony
In an effort to utilize the corgi model for preclinical studies, we established a colony and characterized dogs in the colony (Figure 6, Tables 1 and 2). We first developed a PCR genotyping protocol (Figure 6a-c). A pair of primers specific to dystrophin intron 13 and an additional reverse primer specific to the insert were used in the PCR reaction. The wild type allele is expected to yield a 286 bp band and the mutant allele to yield two bands of 166 bp and ~ 766 bp. With optimized PCR conditions, only the 166 bp band, derived from the insertion, was detected as the diagnostic band for the affected allele (Figure 6c).
Figure 6. Establishment of a corgi muscular dystrophy colony.

a, Schematic representation of the genotyping PCR primer location and expected PCR product size from the normal allele. Arrows, the location of the PCR primers used for genotyping. DL1255 is the common forward primer. DL1256 is a reverse primer corresponding to a region distal of the inserted LINE-1 element in dystrophin intron 13. The normal allele results in a 286 bp PCR product. b, Schematic representation of the genotyping PCR primer location and expected PCR product size from the affected allele. A canine repetitive element LINE-1 (gray color) is inserted in intron 13. The insertion introduces a pair of splicing signals (SA, splicing acceptor; SD, splicing donor). A new exon is created and it contains an in-frame stop codon (asterisk). Arrows, the location of the PCR primers used for genotyping. DL1257 is the mutation specific reverse primer. The mutated allele is expected to yield a 167 bp and a ~766 bp PCR product. c, PCR genotyping results from a litter. Optimized PCR condition allows for the amplification of the 167 bp band only from the affected allele. Two are normal puppies, one is an affected puppy and one is a carrier. The last lane represents the negative water control in which no DNA template was added in the reaction. d, Representative whole muscle lysate western blot from limb muscles of normal, carrier and affected puppies. α-Actin is the loading control. e, Representative dystrophin immunofluorescence staining images from neonatal puppies. Top panels, skeletal muscle. Under the same exposure condition, staining is much weaker in carrier puppy skeletal muscle. Right corner inset in the carrier puppy panel is a longer exposure photomicrograph of the boxed area. Bottom panels, heart. Asterisk, a dystrophin negative region in the carrier puppy heart. f, Serum CK quantification from newborn puppy and 1-month-old dog. Sample sizes for normal newborn male, affected newborn male, normal newborn female, carrier newborn female, normal 1-m-old male and affected 1-m-old male are 8, 3, 12, 3, 4 and 3, respectively. Asterisk, significantly different from other groups in the same panel. g, Growth curve of normal and affected male puppies. Trend lines and sample size are indicated. h, Growth curve of normal and carrier female puppies. Trend lines and sample size are shown.
Table 1.
Breeding record
| Parent Information | Male Normal | Femalea Normal | Male Normal | Femaleb Carrier |
|---|---|---|---|---|
| Mating | 2 | 6 | ||
| Litter size | 4, 7 | 2c, 3, 5, 6d, 6, 9 | ||
| Average litter size | 5.5 | 5.2 | ||
| Puppy number | ||||
| Total | 11 | 31 | ||
| Male | 5 (45.5%) | 14 (45.2%) | ||
| Female | 6 (54.5%) | 16 (51.6%) | ||
| Unknown gender | 0 | 1 (3.2%)e | ||
| Puppy genotype | ||||
| Normal | 11 (100%) | 18 (58.1%) | ||
| Affectedf | 0 (0%) | 5 (16.1%) | ||
| Carrier | 0 (0%) | 7 (22.6%) | ||
| Unknown | 0 (0%) | 1 (3.2%)e | ||
| Neonatal deathg | ||||
| Normal | 0 | 3h | ||
| Affected | NA | 2 (stillborn) | ||
| Carrier | NA | 1 (stillborn) | ||
| Unknown genotype | NA | 1 (stillborn)e | ||
Two females (Adrian and Madonna) were used in breeding.
Four female (Crash, Rachel, Monica, and Bitty) were used in breeding. Three litters were from carrier Monica.
This litter was born on 7/6/2007. Both puppies were female (one is normal, the other is a carrier) and both were stillborn.
This litter was born on 6/25/2007. Only two survived from this litter (1 normal male and 1 carrier). Two affected males were stillborn. One normal male died at day 3 and one normal female died at day 8.
This was a stillbirth and puppy’s gender was not recorded and tissue is not available for genotyping.
All affected are male.
Neonatal death also include five cases of stillbirth. Four deaths were from the first litter of carrier Rachel (see c) and two from the first litter of carrier Crash (see d). These deaths occurred in 2007. These two carriers were not used for breeding since then. One stillbirth occurred in later 2009 and it was the third litter from carrier Monica. This was also the largest litter (total 9 puppies).
One normal male is stillborn, one normal male died at day 3 and one normal female died at day 8.
Table 2.
Body weight and neonatal growth rate
| Male | Female | ||||||
|---|---|---|---|---|---|---|---|
| Normal (N=14) | Affected (N=3) | Normal/ Affected | Normal (N=15) | Carrier (N=5) | Normal/ Carrier | ||
| Body weight (gram) | Day 1 | 283±12 | 257±16 | 1.10 | 270±11 | 246±29 | 1.10 |
| Day 5 | 419±25 | 302±31 | 1.39 | 405±24 | 302±46 | 1.34 | |
| Day 10 | 618±42 | 409±39 | 1.51 | 624±34 | 464±49 | 1.34 | |
| Day 20 | 997±60 | 751±89 | 1.33 | 1044±67 | 814±80 | 1.28 | |
| Day 45 | 2664±150 | 2056±109 | 1.30 | 2459±121 | 2517±93 | 0.98 | |
| Growth rate (gram/day) | Day 1 to 5 | 34.44 | 10.17 | 3.39 | 33.64 | 14.18 | 2.37 |
| Day 6 to 10 | 40.79 | 23.20 | 1.76 | 44.88 | 33.68 | 1.33 | |
| Day 11 to 20 | 38.26 | 33.76 | 1.13 | 42.50 | 34.12 | 1.25 | |
| Day 20 to 45 | 67.84 | 55.09 | 1.23 | 58.81 | 70.93 | 0.83 | |
| Day 1 to 45 | 49.10 | 38.96 | 1.26 | 47.83 | 49.27 | 0.97 | |
Next, we compared the breeding profile (Table 1). Two rounds of normal mating yielded ~5.5 puppies per litter. Five puppies were male and six were female. None died during the first 45 days (the end of observation). Breeding carrier females with a normal male yielded an average litter size of ~ 5.2 puppies per litter with approximately equal numbers of males and females. 58% of offspring from carrier breeding were normal puppies (Table 1). This is slightly higher than the expected 50%. Carrier offspring accounted for about 23% puppies, slightly lower than the expected 25%. Sixteen percent puppies were affected. Although this appears lower than the expected 25%, it is not statistically significant (p = 0.2278). Seven cases of neonatal death were observed from six rounds of carrier breeding. Six deaths occurred during the first two rounds of breeding. They included three wild type puppies, two affected puppies and one carrier (Table 1). Among these, four puppies were stillborn.
Western blot and immunofluorescence staining were performed on muscles obtained from stillborn puppies. On western blot, full-length dystrophin was not detected in limb muscles obtained from the affected puppy. Compared to that of the normal puppy, dystrophin level was apparently reduced in the carrier (Figure 6d). On immunofluorescence staining, we observed strong uniform dystrophin expression in skeletal muscle and the heart in the normal puppy. Dystrophin was not detected in an affected puppy. In skeletal muscle of a carrier puppy, dystrophin staining was much weaker. The signal became easily visible after prolonged exposure (Figure 6e top middle panel, insert). In the heart of the carrier puppy we found mosaic expression consisting of broad dystrophin positive regions adjacent to large dystrophin negative regions (Figure 6e bottom middle panel).
We also compared the serum CK levels in newborn puppies (Figure 6f). The average CK level in carrier puppies was about twice that of normal puppies. However, it did not reach statistical significance. As expected, the affected puppies had a significantly higher serum CK level. Nevertheless, caution should be taken when using the CK level for diagnosis. The lowest CK in affected puppies was 9,670 unit/liter. This is approaching the highest value in carriers (8,609 unit/liter) and normal (6,719 unit/liter) puppies. At 1 month of age, the serum CK level dropped to the baseline in normal puppies (361 ± 66 unit/liter) while it remained elevated in affected puppies (19,773 ± 7,754 unit/liter).
We also analyzed the growth curve (Figure 6g and h, Table 2). While the average birth body weights of affected males and carriers were similar to those of normal males and normal females, respectively, a dramatic difference in the growth rate was noted in the first 5 days (Table 2). Normal male puppies grew at an average rate of 34.4 g/day, while the average growth rate of affected male puppies was less than one-third of normal. A similar trend was seen in female puppies. The growth rate of a normal female was ~2.4-fold higher than that of a carrier. From day 6 to day 20, a higher growth rate was still noted in normal puppies but the relative difference to that of affected and carrier puppies became smaller. From day 21 to day 45, affected males continued to show a slower growth rate compared to that of normal male. Interestingly, the growth rate of carrier puppies was increased. Overall, the growth curve of normal males was significantly different from that of affected males (Figure 6G, p < 0.0001). The growth curve of normal females was also significantly different from that of carrier females (Figure 6H, p < 0.0001). Nevertheless, by the age of 45 days, growth delay was apparent only in affected males as the body weight of carriers had now caught up with that of normal female dogs.
Discussion
DMD has been a great therapeutic challenge for modern medicine. The discovery of the dystrophin gene in the 1980s has sparked more than two decades of intense research. Yet, a cure remains distant. Over the years, investigators have turned to dystrophin-deficient mdx mice as the primary workhorse to study disease pathogenesis and to develop therapy. Intermediate animal models with a body size between mice and humans may provide critical insights into translational research in DMD. Here, we described a symptomatic canine DMD model in the corgi breed. We identified the gene mutation, characterized histopathology and most importantly, established a research colony.
Among many mutations reported in human DMD patients, transposon-like repetitive element insertion constitutes a unique category. Transposons are mobile elements in the eukaryotic genomes and they represent a major source of genomic diversity (48). Their insertion has been frequently observed in the human dystrophin gene. Specifically, insertions have been found in intron 2 (31), intron 7 (32), intron 11 (33, 34), intron 43 (35), exon 44 (36, 37) and exon 48 (38). While the insertion in intron 2 appears to be a polymorphic change, all other reported insertions result in clinical disease. In general, the inserted repetitive sequences disrupt normal dystrophin splicing. This either leads to pathological exon skipping or formation of a new exon containing an in-frame stop codon (33, 34, 36–38). In affected corgi dogs, we detected a LINE-1 element in intron 13. It contained the expected polyA tract and in-frame premature stop codon but not target site duplication (49) (Figure 5, Supplementary Figure 2). Based on genomic and cDNA sequence analysis, we postulate that the LINE-1 element insertion activated a cryptic splicing signal in intron 13. The first 167 nucleotides of the inserted LINE-1 sequence were spliced into the mature mRNA. Dystrophin translation is truncated by the nonsense codon in the LINE-1 element (Figure 5, Supplementary Figure 2).
To establish the corgi model for translational study, we characterized dystrophin, utrophin and DGC expression, histopathology (general morphology, fibrosis and calcification) and inflammation (B cell, T cell, neutrophil and macrophage) (Figures 2 to 4, Supplementary Figure 1). Besides rare revertant fibers (Supplementary Figure 1), dystrophin was absent from the sarcolemma. As a consequence of utrophin up-regulation, we detected low-level expression of several DGC components (Figure 2). In contrast to dystrophin, utrophin cannot restore nNOS to the sarcolemma (50). We did not see nNOS at the membrane (Figure 2).
Dystrophic muscle histopathology has been noticed in all dystrophin-deficient dogs irrespective of the breed. In a 2-year-old affected weimaraner, Baltzer et al. showed endomysial fibrosis in the diaphragm and extremely high central nucleation (~ 90%) in the infraspinatus msucle (26). In a 10-mold affected rat terrier, Wetterman et al. found pronounced fiber size variation, moderate central nucleation (~15%) but no calcium deposition in limb muscles (24). In a 3-m-old affected old English sheepdog, Wieczorek et al. reported focal degeneration and apparent myofiber calcification in the cranial tibialis muscle (23). In three affected Cavalier King Charles spaniels (6 to 10-m-old), Walmsley et al. found fiber size variation, multifocal necrosis and small basophilic regenerated fibers in several limb muscles (14). However, muscle calcification was rarely observed (14). In seven affected Japanese spitz dogs (4 to 12-month-old), Jones et al. showed segmental muscle necrosis, frequent calcification and occasional regeneration (~3% central nucleation) from multiple muscles collected at the necropsy (20). In a 3.5-m-old affected labrador retriever, Bergman et al. noticed myofiber size variation, some fibrosis, necrosis, regeneration and numerous calcific deposits in the vastus lateralis muscle (21). Klarenbeek et al. examined multiple muscles from three ~2-m-old affected grand basset griffon vendeen dogs. The histological lesions were multifocal and included fiber size variation, clusters of small basophilic regenerative fibers, and occasional necrosis and mineralization (18). Presthus and Nordstoga reported fiber size difference, hyaline degeneration and macrophage infiltration in three 12-week-old affected Samoyed dogs (25). In a 7-m-old affected miniature schnauzer, Paola et al. found small foci of inflammatory cell infiltration and myofiber calcification in the gastrocnemius muscle (22). In a 7-m-old affected Brittany spaniel, van Ham et al. observed fiber size variation, focal lymphocyte infiltration, endomysial fibrosis and calcification (13). They also found increased fatty deposition in muscle tissues obtained from necropsy (13). van Ham et al. also studied three 5-m-old affected Belgian groenendaeler shepherds and found fiber size variation, hyaline degeneration, hypercontraction, and centrally nucleated regenerative fiber (12). We performed necropsy in three affected dogs that were 1.8-, 8.7- and 16-m-old (Supplementary Table 1). We observed a high level of central nucleation (~23%), extensive fibrosis, calcification and fiber size variation in skeletal muscle (Figure 3) (17, 51–54). Recently, Urasawa et al. showed selective vacuolar degeneration in the hearts of dystrophin-deficient beagles (52). We also observed vacuolar degeneration in the heart of a 16-m-old affected corgi dog (Figure 3).
Cozzi et al. examined the types of inflammatory cells in dystrophic muscle in the GRMD model (51). They found that ~65% of infiltrated mononuclear cells were macrophages. Remaining were CD4 positive (~15%) and CD8 positive (~16%) T cells. In affected corgis, macrophages and neutrophils were the predominant infiltrates. Surprisingly, T cells were rarely observed (Figure 4).
Consistent with a previous study in GRMD dogs (51), we also noted mild myopathy in corgi carrier muscle (Figures 3B and 4B). Heterogeneous dystrophin expression may explain this observation. In fact, Cooper et al have demonstrated mosaic dystrophin expression in both skeletal muscle and the heart in GRMD carriers (55). We also found mosaic dystrophin expression in newborn carrier heart. However, in newborn carrier skeletal muscle, we did not see large areas of dystrophin negative fibers (Figure 6e). While we cannot exclude non-uniform expression in individual myofibers in carrier skeletal muscle, our results (western blot and immunostaining) clearly demonstrated a reduced dystrophin expression in corgi carrier skeletal muscle (Figure 6d, e).
There is no doubt that an important accomplishment of our study is the establishment of a corgi DMD colony. In this regard, we have developed a robust genotyping protocol (Figure 6B). Breeding with female carriers did not alter the litter size and sex ratio. The number of affected puppies appeared lower than expected but it did not reach statistical significance (Table 1).
Neonatal death has been reported in approximately one-third to nearly half of affected puppies in the beagle and golden retriever background (10, 11, 56). Except for the first two rounds of breeding, we have not observed significant death in our colony. Since we only have a relatively small sample size, additional studies are needed to determine whether the corgi model shows a different neonatal mortality.
An intriguing question revolves around the timing of visible clinical signs of the disease. The high CK level suggests the presence of the disease at birth (Figure 6f). There is also anecdotal evidence suggesting a weaker suckling ability of affected puppies. However, controversy exists as to whether there is a significant growth delay at an early age. From weekly body weight data, Valentine et al. concluded that affected puppies had a significantly low body weight in the first two weeks and the trend persisted until 8 weeks of age (the end of the study) (11). Kornegay et al. detected a lower body weight in GRMD dogs at three months of age and the growth delay persisted up to one year (57). Interestingly, a recent study by Shimatsu et al. showed no body weight difference between normal and affected male dogs from birth to 6 months of age (10). We analyzed daily body weight change in the corgi colony (Figure 6g, h; Table 2). Interestingly, the greatest growth rate difference was found in the first 5 days. During this period, we provided supplemental nutrition support to all puppies ad libitum. However, the growth rates of normal males and females were 3.4-fold and 2.4-fold higher than these of affected males and carrier females, respectively (Table 2). Our results suggest that the absence of dystrophin (affected dogs) or reduced dystrophin expression (carriers) may influence early growth. Since we only analyzed body weight in 3 affected males and 5 carrier females, future studies are needed to determine whether growth retardation represents an early clinical sign.
Unlike golden retrievers, corgi is a chondrodysplastic (short-legged) breed. A recent study suggested that the stunted legs are associated with atypical expression of fibroblast growth factor 4 (FGF4) from a retrogene (58). Several members of the FGF family have been implicated in myogenesis (59, 60). Interestingly, FGF2 expression is increased in DMD patients (61, 62). Since the colony has been outbred, it now consists of a mixture of chondrodysplastic and non-chondroplastic dogs. To date, no difference in the disease process has been noted. Nevertheless, the sample size remains small for each category (chondrodysplastic or non-chondroplastic). Future comparative studies between these two categories may offer critical insight on whether (and how) FGF4 up-regulation may influence the DMD phenotype.
Large animal models hold the key in translational biomedical research. The successful establishment of the corgi model provides important alternatives to the current models. The vast majority of current DMD models are due to point mutations in the dystrophin gene. The corgi model reported here represents the first insertion mutation model. Comparative studies between the corgi model and other existing models will offer new insight on how the genetic background and dystrophin mutation influence disease progression and therapeutic outcome.
Supplementary Material
Immunofluorescence staining was performed on 8 μm muscle sections with antibodies specific for dystrophin spectrin-like repeats R6-8 (Dys-1), R10/11 (Mandys1), R15 (Mandys101), R14–18 (Mandys105), and the C-terminal domain (Dys-2). Dys-1 and Dys-2 were obtained from Novocastra, Newcastle, UK. Remaining antibodies were gifts from Dr. Glenn Morris, The Robert Jones and Agnes Hunt Orthopaedic Hospital, UK. Arrow, revertant fiber.
{kind=link}
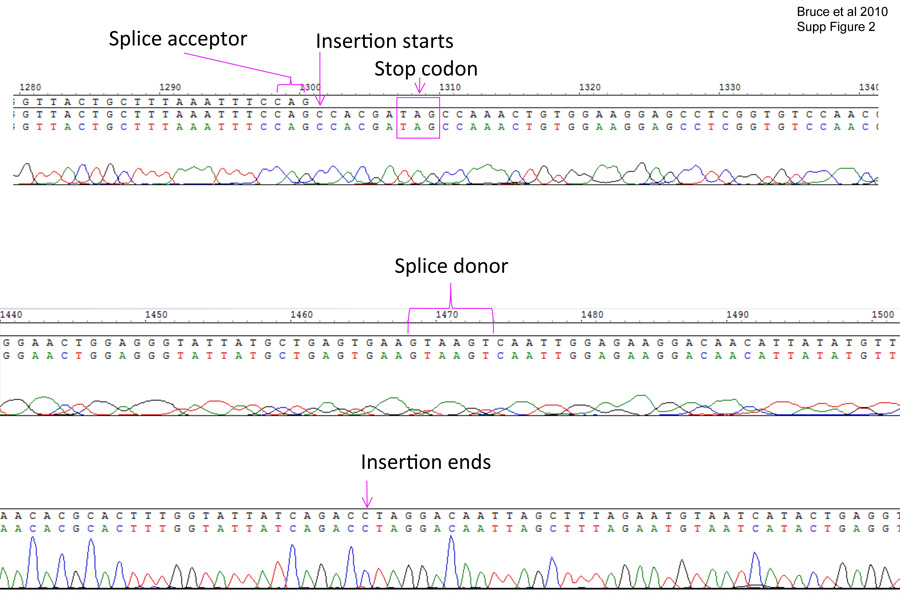
The upper chromatogram shows where the insertion begins within the intron and the CAG splicing acceptor site just upstream of the insertion. The in-frame stop codon is marked. The middle chromatogram shows the conserved splicing donor signal GTAAGT where the “pseudo-intron” is spliced. The bottom chromatogram shows the end of the insertion and resumption of the normal intron sequence.
{kind=link}
Supplementary Table 1. Summary of muscle tissues used in the study
Supplementary Video 1. Video documentation of daily activity of an affected (the first dog appeared in the video) and a normal corgi. The affected corgi became fatigued readily and had to sit on the floor while the normal corgi walked away.
Acknowledgments
This work was supported by grants from the National Institutes of Health AR-49419 and AR-57209 (DD and BFS), AR-48650 (BFS), the Muscular Dystrophy Association (DD and BFS). The authors (YY, JS, and DD) thank Robert J McDonald, Jr., M.D. for the generous support to Duchenne muscular dystrophy research in the Duan lab. We thank Dr. Jianguo Sun for the help with SAS statistic analysis. We thank Drs Glenn Morris and Lam Le for providing dystrophin monoclonal antibodies. We also thank Dr. Lucia Alvarez and Ms. Kerriann Sparks for excellent technical help.
Grant: NIH AR-49418 and AR-57209 (BFS and DD), NIH AR-48650 (BFS), MDA (BFS and DD)
Footnotes
Conflict of interest:
none declared
Supplementary information is available on Laboratory Investigation’s web site.
References
- 1.Hoffman EP, Brown RH, Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51(6):919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Romitti P, Mathews K, Zamba G, Andrews J, Druschel C, Ciafaloni E, et al. Prevalence of Duchenne/Becker muscular dystrophy among males aged 5–24 years - four states, 2007. MMWR Morb Mortal Wkly Rep. 2009;58(40):1119–1122. [PubMed] [Google Scholar]
- 3.Jeppesen J, Green A, Steffensen BF, Rahbek J. The Duchenne muscular dystrophy population in Denmark, 1977–2001: prevalence, incidence and survival in relation to the introduction of ventilator use. Neuromuscul Disord. 2003;13(10):804–812. doi: 10.1016/s0960-8966(03)00162-7. [DOI] [PubMed] [Google Scholar]
- 4.Dooley J, Gordon KE, Dodds L, MacSween J. Duchenne muscular dystrophy: a 30-year population-based incidence study. Clin Pediatr (Phila) 2010;49(2):177–179. doi: 10.1177/0009922809347777. [DOI] [PubMed] [Google Scholar]
- 5.Bradley DM, Parsons EP, Clarke AJ. Experience with screening newborns for Duchenne muscular dystrophy in Wales. Bmj. 1993;306(6874):357–360. doi: 10.1136/bmj.306.6874.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins CA, Morgan JE. Duchenne’s muscular dystrophy: animal models used to investigate pathogenesis and develop therapeutic strategies. Int J Exp Pathol. 2003;84(4):165–172. doi: 10.1046/j.1365-2613.2003.00354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nonaka I. Animal models of muscular dystrophies. Lab Anim Sci. 1998;48(1):8–17. [PubMed] [Google Scholar]
- 8.Willmann R, Possekel S, Dubach-Powell J, Meier T, Ruegg MA. Mammalian animal models for Duchenne muscular dystrophy. Neuromuscul Disord. 2009;19(4):241–249. doi: 10.1016/j.nmd.2008.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Shelton GD, Engvall E. Canine and feline models of human inherited muscle diseases. Neuromuscul Disord. 2005;15(2):127–138. doi: 10.1016/j.nmd.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 10.Shimatsu Y, Yoshimura M, Yuasa K, Urasawa N, Tomohiro M, Nakura M, et al. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMD. J Acta Myol. 2005;24(2):145–154. [PubMed] [Google Scholar]
- 11.Valentine BA, Cooper BJ, de Lahunta A, O’Quinn R, Blue JT. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: clinical studies. J Neurol Sci. 1988;88(1–3):69–81. doi: 10.1016/0022-510x(88)90206-7. [DOI] [PubMed] [Google Scholar]
- 12.van Ham LML, Desmidt M, Tshamala M, Hoorens JK, Mattheeuws DRG. Canine X-linked muscular dystrophy in Belgian Groenendaeler shepherds. J Am Anim Hosp Assoc. 1993;29:570–574. [Google Scholar]
- 13.van Ham LML, Roels SLMF, Hoorens JK. Congital dystrophy-like myopathy in a Brittany Spaniel puppy. Prog Vet Neurol. 1995;6(4):135–138. [Google Scholar]
- 14.Walmsley GL, Arechavala-Gomeza V, Fernandez-Fuente M, Burke MM, Nagel N, Holder A, et al. A duchenne muscular dystrophy gene hot spot mutation in dystrophin-deficient cavalier king charles spaniels is amenable to exon 51 skipping. PLoS One. 2010;5(1):e8647. doi: 10.1371/journal.pone.0008647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schatzberg SJ, Olby NJ, Breen M, Anderson LV, Langford CF, Dickens HF, et al. Molecular analysis of a spontaneous dystrophin ‘knockout’ dog. Neuromuscul Disord. 1999;9(5):289–295. doi: 10.1016/s0960-8966(99)00011-5. [DOI] [PubMed] [Google Scholar]
- 16.Cooper BJ, Winand NJ, Stedman H, Valentine BA, Hoffman EP, Kunkel LM, et al. The homologue of the Duchenne locus is defective in X-linked muscular dystrophy of dogs. Nature. 1988;334(6178):154–156. doi: 10.1038/334154a0. [DOI] [PubMed] [Google Scholar]
- 17.Kornegay JN, Tuler SM, Miller DM, Levesque DC. Muscular dystrophy in a litter of golden retriever dogs. Muscle Nerve. 1988;11(10):1056–1064. doi: 10.1002/mus.880111008. [DOI] [PubMed] [Google Scholar]
- 18.Klarenbeek S, Gerritzen-Bruning MJ, Rozemuller AJ, van der Lugt JJ. Canine X-linked muscular dystrophy in a family of Grand Basset Griffon Vendeen dogs. J Comp Pathol. 2007;137(4):249–252. doi: 10.1016/j.jcpa.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 19.Wentink GH, van der Linde-sipman JS, Meijer AEFH, Kamphuisen HAC, van Vorstenbosch CJAHV, Hartman W, et al. Myopathy with a possible recessive X-linked inheritance in a litter of Irish Terriers. Vet Path. 1972;9:328–349. doi: 10.1177/030098587200900504. [DOI] [PubMed] [Google Scholar]
- 20.Jones BR, Brennan S, Mooney CT, Callanan JJ, McAllister H, Guo LT, et al. Muscular dystrophy with truncated dystrophin in a family of Japanese Spitz dogs. J Neurol Sci. 2004;217(2):143–149. doi: 10.1016/j.jns.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 21.Bergman RL, Inzana KD, Monroe WE, Shell LG, Liu LA, Engvall E, et al. Dystrophin-deficient muscular dystrophy in a Labrador retriever. J Am Anim Hosp Assoc. 2002;38(3):255–261. doi: 10.5326/0380255. [DOI] [PubMed] [Google Scholar]
- 22.Paola JP, Podell M, Shelton GD. Muscular dystrophy in a miniature Schnauzer. Prog Vet Neurol. 1993;4(1):14–18. [Google Scholar]
- 23.Wieczorek LA, Garosi LS, Shelton GD. Dystrophin-deficient muscular dystrophy in an old English sheepdog. Vet Rec. 2006;158(8):270–273. doi: 10.1136/vr.158.8.270. [DOI] [PubMed] [Google Scholar]
- 24.Wetterman CA, Harkin KR, Cash WC, Nietfield JC, Shelton GD. Hypertorphic muscular dystrophy in a young dog. J Am Vet Med Assoc. 2000;216(6):878–881. doi: 10.2460/javma.2000.216.878. [DOI] [PubMed] [Google Scholar]
- 25.Presthus J, Nordstoga K. Congenital myopathy in a litter of Samoyed dogs. Prog Vet Neurol. 1993;4(2):37–40. [Google Scholar]
- 26.Baltzer WI, Calise DV, Levine JM, Shelton GD, Edwards JF, Steiner JM. Dystrophin-deficient muscular dystrophy in a Weimaraner. J Am Anim Hosp Assoc. 2007;43(4):227–232. doi: 10.5326/0430227. [DOI] [PubMed] [Google Scholar]
- 27.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34(2):135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- 28.Flanigan KM, Dunn DM, von Niederhausern A, Soltanzadeh P, Gappmaier E, Howard MT, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat. 2009;30(12):1657–1666. doi: 10.1002/humu.21114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuffery-Giraud S, Beroud C, Leturcq F, Yaou RB, Hamroun D, Michel-Calemard L, et al. Genotype-phenotype analysis in 2,405 patients with a dystrophinopathy using the UMD-DMD database: a model of nationwide knowledgebase. Hum Mutat. 2009;30(6):934–945. doi: 10.1002/humu.20976. [DOI] [PubMed] [Google Scholar]
- 30.Heydemann A, Doherty KR, McNally EM. Genetic modifiers of muscular dystrophy: Implications for therapy. Biochim Biophys Acta. 2007;1772(2):216–228. doi: 10.1016/j.bbadis.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Muntoni F, Cau M, Congiu R, Congia M, Cao A, Melis MA. Identification of a novel T-insertion polymorphism at the DMD locus. Hum Genet. 1993;92(1):103. doi: 10.1007/BF00216157. [DOI] [PubMed] [Google Scholar]
- 32.McNaughton JC, Broom JE, Hill DF, Jones WA, Marshall CJ, Renwick NM, et al. A cluster of transposon-like repetitive sequences in intron 7 of the human dystrophin gene. J Mol Biol. 1993;232(1):314–321. doi: 10.1006/jmbi.1993.1389. [DOI] [PubMed] [Google Scholar]
- 33.Ferlini A, Galie N, Merlini L, Sewry C, Branzi A, Muntoni F. A novel Alu-like element rearranged in the dystrophin gene causes a splicing mutation in a family with X-linked dilated cardiomyopathy. Am J Hum Genet. 1998;63(2):436–446. doi: 10.1086/301952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferlini A, Muntoni F. The 5’ region of intron 11 of the dystrophin gene contains target sequences for mobile elements and three overlapping ORFs. Biochem Biophys Res Commun. 1998;242(2):401–406. doi: 10.1006/bbrc.1997.7976. [DOI] [PubMed] [Google Scholar]
- 35.Pizzuti A, Pieretti M, Fenwick RG, Gibbs RA, Caskey CT. A transposon-like element in the deletion-prone region of the dystrophin gene. Genomics. 1992;13(3):594–600. doi: 10.1016/0888-7543(92)90129-g. [DOI] [PubMed] [Google Scholar]
- 36.Musova Z, Hedvicakova P, Mohrmann M, Tesarova M, Krepelova A, Zeman J, et al. A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: further evidence for possible bias in retroposon integration. Biochem Biophys Res Commun. 2006;347(1):145–149. doi: 10.1016/j.bbrc.2006.06.071. [DOI] [PubMed] [Google Scholar]
- 37.Narita N, Nishio H, Kitoh Y, Ishikawa Y, Minami R, Nakamura H, et al. Insertion of a 5’ truncated L1 element into the 3’ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J Clin Invest. 1993;91(5):1862–1867. doi: 10.1172/JCI116402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holmes SE, Dombroski BA, Krebs CM, Boehm CD, Kazazian HH., Jr A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat Genet. 1994;7(2):143–148. doi: 10.1038/ng0694-143. [DOI] [PubMed] [Google Scholar]
- 39.Woods P, Sharp NJ, Schatzberg S. Muscular dystrophy in Pembroke Corgis and other dogs. In: Stiff M, Sellon DC, editors. American College of Veterinary Internal Medicine; Proceedings of 16th American College of Veterinary Internal Medicine Forum; 1998; San Diego, CA. 1998. pp. 301–303. [Google Scholar]
- 40.Li D, Long C, Yue Y, Duan D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum Mol Genet. 2009;18(7):1209–1220. doi: 10.1093/hmg/ddp015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bostick B, Yue Y, Long C, Duan D. Prevention of Dystrophin-Deficient Cardiomyopathy in Twenty-One-Month-Old Carrier Mice by Mosaic Dystrophin Expression or Complementary Dystrophin/Utrophin Expression. Circ Res. 2008;102(1):121–130. doi: 10.1161/CIRCRESAHA.107.162982. [DOI] [PubMed] [Google Scholar]
- 42.Lai Y, Yue Y, Liu M, Ghosh A, Engelhardt JF, Chamberlain JS, et al. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat Biotechnol. 2005;23(11):1435–1439. doi: 10.1038/nbt1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yue Y, Liu M, Duan D. C-terminal truncated microdystrophin recruits dystrobrevin and syntrophin to the dystrophin-associated glycoprotein complex and reduces muscular dystrophy in symptomatic utrophin/dystrophin double knock-out mice. Mol Ther. 2006;14(1):79–87. doi: 10.1016/j.ymthe.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yue Y, Ghosh A, Long C, Bostick B, Smith BF, Kornegay JN, et al. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol Ther. 2008;16(12):1944–1952. doi: 10.1038/mt.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thanh LT, Nguyen TM, Helliwell TR, Morris GE. Characterization of revertant muscle fibers in Duchenne muscular dystrophy, using exon-specific monoclonal antibodies against dystrophin. Am J Hum Genet. 1995;56(3):725–731. [PMC free article] [PubMed] [Google Scholar]
- 46.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin Gene Therapy of Cardiomyopathy Restores Dystrophin-Glycoprotein Complex and Improves Sarcolemma Integrity in the Mdx Mouse Heart. Circulation. 2003;108(13):1626–1632. doi: 10.1161/01.CIR.0000089371.11664.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lai Y, Thomas GD, Yue Y, Yang HT, Li D, Long C, et al. Dystrophins carrying spectrin-like repeats 16 and 17 anchor nNOS to the sarcolemma and enhance exercise performance in a mouse model of muscular dystrophy. J Clin Invest. 2009;119(3):624–635. doi: 10.1172/JCI36612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303(5664):1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 49.Han JS, Boeke JD. LINE-1 retrotransposons: modulators of quantity and quality of mammalian gene expression? Bioessays. 2005;27(8):775–784. doi: 10.1002/bies.20257. [DOI] [PubMed] [Google Scholar]
- 50.Li D, Bareja A, Judge L, Yue Y, Lai Y, Fairclough R, et al. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci. 2010;123(Pt 12):2008–2013. doi: 10.1242/jcs.064808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cozzi F, Cerletti M, Luvoni GC, Lombardo R, Brambilla PG, Faverzani S, et al. Development of muscle pathology in canine X-linked muscular dystrophy. II. Quantitative characterization of histopathological progression during postnatal skeletal muscle development. Acta Neuropathol (Berl) 2001;101(5):469–478. doi: 10.1007/s004010000308. [DOI] [PubMed] [Google Scholar]
- 52.Urasawa N, Wada MR, Machida N, Yuasa K, Shimatsu Y, Wakao Y, et al. Selective vacuolar degeneration in dystrophin-deficient canine Purkinje fibers despite preservation of dystrophin-associated proteins with overexpression of Dp71. Circulation. 2008;117(19):2437–2448. doi: 10.1161/CIRCULATIONAHA.107.739326. [DOI] [PubMed] [Google Scholar]
- 53.Valentine BA, Cooper BJ, Cummings JF, de Lahunta A. Canine X-linked muscular dystrophy: morphologic lesions. J Neurol Sci. 1990;97(1):1–23. doi: 10.1016/0022-510x(90)90095-5. [DOI] [PubMed] [Google Scholar]
- 54.Nguyen F, Cherel Y, Guigand L, Goubault-Leroux I, Wyers M. Muscle lesions associated with dystrophin deficiency in neonatal golden retriever puppies. J Comp Pathol. 2002;126(2–3):100–108. doi: 10.1053/jcpa.2001.0526. [DOI] [PubMed] [Google Scholar]
- 55.Cooper BJ, Gallagher EA, Smith CA, Valentine BA, Winand NJ. Mosaic expression of dystrophin in carriers of canine X-linked muscular dystrophy. Lab Invest. 1990;62(2):171–178. [PubMed] [Google Scholar]
- 56.Valentine BA, Cooper BJ. Canine X-linked muscular dystrophy: selective involvement of muscles in neonatal dogs. Neuromuscul Disord. 1991;1(1):31–38. doi: 10.1016/0960-8966(91)90040-y. [DOI] [PubMed] [Google Scholar]
- 57.Kornegay JN, Bogan DJ, Bogan JR, Childers MK, Cundiff DD, Petroski GF, et al. Contraction force generated by tarsal joint flexion and extension in dogs with golden retriever muscular dystrophy. J Neurol Sci. 1999;166(2):115–121. doi: 10.1016/s0022-510x(99)00118-5. [DOI] [PubMed] [Google Scholar]
- 58.Parker HG, VonHoldt BM, Quignon P, Margulies EH, Shao S, Mosher DS, et al. An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science. 2009;325(5943):995–998. doi: 10.1126/science.1173275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Armand AS, Laziz I, Chanoine C. FGF6 in myogenesis. Biochim Biophys Acta. 2006;1763(8):773–778. doi: 10.1016/j.bbamcr.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 60.Olwin BB, Hannon K, Kudla AJ. Are fibroblast growth factors regulators of myogenesis in vivo? Prog Growth Factor Res. 1994;5(2):145–158. doi: 10.1016/0955-2235(94)90002-7. [DOI] [PubMed] [Google Scholar]
- 61.D’Amore PA, Brown RH, Jr, Ku PT, Hoffman EP, Watanabe H, Arahata K, et al. Elevated basic fibroblast growth factor in the serum of patients with Duchenne muscular dystrophy. Ann Neurol. 1994;35(3):362–365. doi: 10.1002/ana.410350320. [DOI] [PubMed] [Google Scholar]
- 62.Saito A, Higuchi I, Nakagawa M, Saito M, Uchida Y, Inose M, et al. An overexpression of fibroblast growth factor (FGF) and FGF receptor 4 in a severe clinical phenotype of facioscapulohumeral muscular dystrophy. Muscle Nerve. 2000;23(4):490–497. doi: 10.1002/(sici)1097-4598(200004)23:4<490::aid-mus6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Immunofluorescence staining was performed on 8 μm muscle sections with antibodies specific for dystrophin spectrin-like repeats R6-8 (Dys-1), R10/11 (Mandys1), R15 (Mandys101), R14–18 (Mandys105), and the C-terminal domain (Dys-2). Dys-1 and Dys-2 were obtained from Novocastra, Newcastle, UK. Remaining antibodies were gifts from Dr. Glenn Morris, The Robert Jones and Agnes Hunt Orthopaedic Hospital, UK. Arrow, revertant fiber.
The upper chromatogram shows where the insertion begins within the intron and the CAG splicing acceptor site just upstream of the insertion. The in-frame stop codon is marked. The middle chromatogram shows the conserved splicing donor signal GTAAGT where the “pseudo-intron” is spliced. The bottom chromatogram shows the end of the insertion and resumption of the normal intron sequence.
Supplementary Table 1. Summary of muscle tissues used in the study
Supplementary Video 1. Video documentation of daily activity of an affected (the first dog appeared in the video) and a normal corgi. The affected corgi became fatigued readily and had to sit on the floor while the normal corgi walked away.