

Abstract
Introduction
Propofol's lipophilicity has required dispersion in a soybean macroemulsion. We hypothesized that the anesthetic properties of propofol are preserved when reformulated as a transparent microemulsion rather than as a turbid macroemulsion and that the dose-response relationship can be selectively modified by altering the microemulsion's surfactant type and concentration.
Methods
Microemulsions of propofol were formulated using purified poloxamer 188 (3%, 5%, 7%), and sodium salt of fatty acids (C8, C10, C12) in saline and characterized using ternary/binary diagrams, particle sizing, and stability upon dilution. Rats received propofol (10 mg/kg/min) as either a microemulsion or conventional macroemulsion to determine these endpoints: induction (dose; stunned; loss of lash reflex, righting reflex, withdrawal to toe pinch) and recovery (recovery of lash, righting, withdrawal reflexes). After a 14 day recovery period, rats were crossed over into the opposite experimental limb.
Results
Forty-eight microemulsions (diameter: 11.9-47.7 nm) were formulated. Longer carbon chain length led to a marked increase in the volume of diluent necessary to break these microemulsions. All rats experienced anesthetic induction with successful recovery although significantly greater doses of propofol were required to induce anesthesia with microemulsions irrespective of surfactant concentration or type than with macroemulsions. The sodium salt of C10 fatty acid microemulsion required the greatest dose and longest time for anesthetic induction.
Conclusion
Propofol microemulsions cause induction in rat similar to that from macroemulsions. The surfactant concentration and type markedly affects the spontaneous destabilization and anesthetic properties of microemulsions, a phenomenon suggesting a mechanism whereby dose-response relationship can be selectively modified.
Introduction
Propofol possesses several favorable characteristics including an anti-emetic effect and rapid emergence from unconsciousness with minimal residual drowsiness.1-4 However, a primary drawback of propofol (2,6-diisopropylphenol) is this drug's extreme lipophilicity that necessitates dispersion in soybean macroemulsions to produce white, opaque formulations. This solvent requirement potentially causes several adverse drug outcomes including bacterial growth leading to postoperative infection, pain in a significant number of patients with peripheral intravenous injection, inclusion of egg products, and others.5-8 Alternative formulations yielding similar pharmacodynamic characteristics as the conventional formulations, but without the associated liabilities, would be useful to enhance patient safety and comfort. Recently, efforts have been made to achieve these goals using other lipid solvents and concentrations, cyclodextrin formulations, microemulsions technology, and prodrug techniques that depend on native enzymes such as alkaline phosphatase to metabolize a parent compound (i.e., phosphono-2,6-diisopropylphenol) to the active drug molecule (i.e., propofol).9-14
To address these drawbacks of conventional formulations, we hypothesized that propofol could be associated with biocompatible surfactants to form transparent, colorless, thermodynamically stable, low viscosity, oil-in-water microemulsions with droplets having a 10-50 nm diameter (fig. 1). Thus, instead of regarding propofol's extreme lipophilicity as a hindrance to be overcome, the lipophilicity of propofol (which exists as an oil at room and physiological temperatures) was leveraged to construct the physical core of the microemulsions. Therefore, propofol served dual roles as both the lipid oil core of the microemulsion and the pharmacologically active agent to induce unconsciousness. In addition, we hypothesized that the spontaneous destabilization of the microemulsion nanodroplets to release propofol could be selectively altered by modifying the concentration and nature of the associated surfactants with resultant changes in latency to anesthetic induction. That is, we hypothesized that the dose-response relationship of propofol microemulsions could be modified by varying the nature and concentration of surfactants. To examine these hypotheses, we first determined the physical limitations of possible propofol microemulsions using a variety of propofol and surfactant concentrations to construct pseudo-ternary phase diagrams of these systems. Pseudoternary diagrams are equilateral triangles that describe the compositional phase behavior of microemulsions wherein the apices represent pure components, the boundaries denote two component systems, and the interior demonstrates all three components in the microemulsion system.15 These diagrams can be used to note the physical boundary conditions of oils and surfactants necessary to formulate thermodynamically stable microemulsions. The diagram is defined as pseudoternary, vis-à-vis ternary, because the fourth component of the microemulsion system, 0.9% NaCl in water, is not plotted, but did not change throughout the studies. Second, we measured several parameters of anesthetic induction and emergence in rats receiving the experimental and conventional formulations of propofol using a randomized, crossover design.
Figure 1.
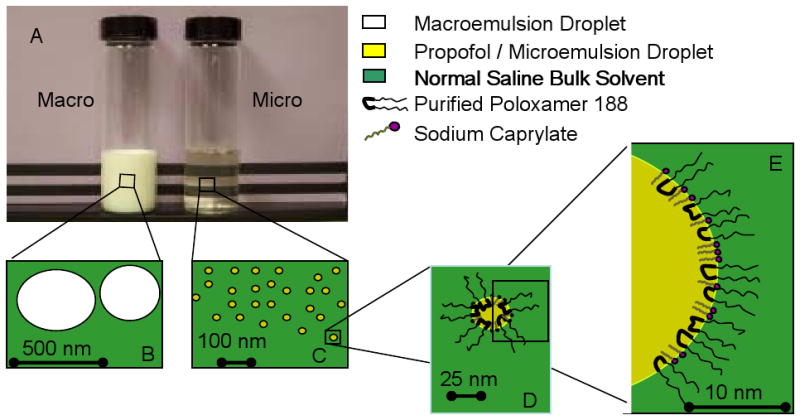
Photograph and cartoons of a macroemulsion and microemulsion of propofol. Panel A. Shown in the photograph is an opaque, white macroemulsion (Macro) and a clear, colorless microemulsion (Micro) of propofol. Panels B and C: cartoon demonstrating the relative particle sizes of the macroemulsion (Panel B) and microemulsion (Panel C). Panel D. Cartoon of a microemulsion in cross-section. Panel E: Magnified view of cross section of a microemulsion particle demonstrating propofol surrounded by a corona of purified poloxamer 188 and sodium caprylate surfactant in a bulk media of normal saline. Calibration bars apply only to their respective panels. Color legend is applies to all panels.
Methods and Materials
Synthesis of Propofol Microemulsions
Propofol was obtained from Albemarle Corporation (Baton Rouge, Louisiana, U.S.A.). Purified poloxamer 188, a nonionic coblock polymer consisting of polyethylene and polypropylene monomers, was purchased from the BASF Corporation (Florham Park, New Jersey, U.S.A.). Sodium salts of fatty acid (C8, C10, and C12) were supplied by Sigma Chemical Co. (St. Louis, Missouri, U.S.A.). Microemulsions were prepared by combining propofol (0-100 mg/ml) with purified poloxamer 188 (0-70 mg/ml), and a fatty acid salt (0-12.5 mg/ml) in normal saline (0.90 mg/ml NaCl) bulk media. Water was ultra-purified using a water purification system (Nanopure, Barnstead/Thermolyne, Dubuque, Iowa, U.S.A.) to provide a minimal electrical resistance of 18.2 MΩ. Following agitation with a magnetic stirrer, these components combined to form clear, colorless microemulsions with adjustment of pH to 7.40 using either HCl or NaOH. All experimental formulations were stored under a nitrogen headspace. To characterize the dimension of the individual droplets, the effective droplet size of the nanoparticles was measured by the dynamic light scattering method using a submicron particle sizer analyzer (90Plus, Brookhaven Instruments Corporation, Holtsville, New York, U.S.A.) as previously described.16
Animal Preparation
All experimental protocols involving the use of animals were reviewed and approved by the University of Florida Institutional Animal Care and Use Committee (Gainesville, Florida, U.S.A.). Sprague-Dawley rats (350-500 g; either sex) were purchased from Charles Rivers Laboratories (Wilmington, Massachusetts, U.S.A.). Before delivery of animals to the investigators by the vendor, rats underwent catheterization of the left femoral vein with subcutaneous tunneling to an exit site between the scapulae. Thus, rats were supplied by the vendor with pre-implanted, heparin-bonded, femoral vein catheters and did not require additional instrumentation at the time of the experiments that might require sedation or anesthesia that could confound interpretation of results. Rats were caged singly in order to avoid damage to the catheter by cage mates. They were allowed unlimited access to food and water with a 12:12 hour light:dark cycle. On the day of experimentation, each rat was weighed. Thereafter, the central venous line of each rat was easily accessed in conscious, unrestrained rats. All catheters were aspirated until blood was observed and then flushed with 0.5 ml of normal saline. Subsequently, rats entered the animal experimental protocol.
Animal Experimental Protocol
Rats were randomized using a random number generator to receive either 1) an experimental propofol microemulsion (n=6 per microemulsion) after filtration through a 200 nm pore filter in order to ensure sterility or 2) a conventional macroemulsion of propofol in a soybean-based solvent (Diprivan, AstraZeneca Pharmaceuticals, Wilmington, Delaware, U.S.A.). In both cases, the formulation was infused at a rate of 10 mg/kg/min propofol via a microprocessor-controlled syringe pump (sp2000i, World Precision Instruments, Sarasota, Florida, U.S.A.) in order to avoid varying rates of infusion associated with manual injection that could potentially confound anesthetic induction time parameters. The endpoints of anesthetic induction were total drug dose, time to loss of exploratory behavior (i.e., stunned), time to loss of righting reflex, time to loss of lash reflex to gentle stroking (i.e., canthal reflex), and time to loss of reflexive withdrawal of the leg following a great toe pinch every 10 s by a metal clamp. In these experiments, the metal clamp was rubber shod to avoid tissue damage during the experiment. Following loss of withdrawal to the toe pinch, the drug infusion was discontinued. Endpoints of anesthetic recovery were return of the withdrawal response to a toe pinch, recovery of spontaneous eye blinking, recovery of a sustained head lift, and recovery of the righting reflex. Following the first anesthetic, each rat was recovered for at least 14 days before enrollment in the experimental limb opposite the original assignment. Thus, each rat was anesthetized with both experimental and conventional propofol formulations with random initial assignment and with a 14 day recovery interval before crossover between the formulations. Following a 14 day period of observation after the participation in the crossover limb, the experiment was ended.
Statistical Analysis
Measurements are reported as mean±SD. Statistical analysis was performed with SAS 9.1 (SAS Institute, Inc., Cary, N.C., U.S.A.). Linear mixed models for incomplete randomized block design were used to analyze the data from this cross-over study.17 Outcome variables were evaluated for normality assumptions using the Box-Cox procedure, with transformations (i.e., square root, inverse square root) if indicated.18 Initial models included terms for order effect and cross-over effect. These were not included in final models if not significant, based on Akaike's Information Criterion. Models were parameterized to include treatment versus control differences, purified poloxamer 188 concentration category, fatty acid salt carbon chain length category, and interactions between treatment difference and propofol formulation variables. Pre-specified contrasts were used to compare treatment differences at each level of the formulation variables and between levels of the formulation variables. A P-value of less than 0.05 was considered statistically significant.
Results
Microemulsion Synthesis and Characterization
Forty-eight microemulsions of propofol were formulated using varying concentrations of propofol, purified poloxamer 188, and C8 fatty acid salt in a normal saline bulk media in order to construct a pseudoternary diagram of these systems (fig. 2). Depending on the concentrations of surfactants, propofol concentrations of 12-15 mg/ml or less produced microemulsions. In contrast, propofol concentrations exceeding 20 mg/ml with these concentrations of surfactants formed opaque, white macroemulsions similar to conventional propofol formulations. For purposes of testing in the rat, the final concentration of propofol in the microemulsions was selected to be 1% (10 mg/ml), a concentration equal to that in the conventional commercially available macroemulsion formulation.
Figure 2.

Pseudoternary diagram of an oil-in-water propofol microemulsion of propofol (oil), purified poloxamer 188 (nonionic surfactant), and sodium caprylate (ionic co-surfactant; C8 fatty acid salt). (Note: the diagram is defined as pseudoternary, vis-à-vis ternary, because the fourth component of the microemulsion system is the bulk media, 0.9%w/v NaCl in water, and is not plotted, but did not change throughout the studies). The region of the phase diagram below and above the dashed line represents microemulsion (Microemulsion) and macroemulsion (Macroemulsion) regions, respectively. As is typical of these diagrams, the axes are normalized with values representing a fraction of the maximal concentration of components used (propofol: 100 mg/ml; purified poloxamer 188: 70 mg/ml; sodium caprylate: 12.5 mg/ml) and are, therefore, without units of measure.
Subsequently, we more closely investigated the interaction of the concentration and types of surfactants and co-surfactants necessary to form microemulsions (or macroemulsions) while holding the concentration of propofol constant at 10 mg/ml. At the greatest concentration of purified poloxamer 188 (70 mg/ml), no fatty acid co-surfactant was required to create microemulsions of propofol as shown in the binary diagram (fig. 3). However, as the concentration of purified poloxamer 188 decreased, the concentration of the fatty acid surfactant necessary to form a microemulsion increased. In addition, the nature of the fatty acid significantly affected its concentration of fatty acid salt necessary to create a microemulsion. Thus, the concentration of fatty acid salt required to formulate propofol as the oil core of a microemulsion was markedly diminished by lengthening the carbon chain of the co-surfactant fatty acid from 8 to 10 or 12 carbon atoms at any concentration of purified poloxamer 188.
Figure 3.

Binary diagram noting the concentrations of purified poloxamer 188 and the co-surfactant fatty acids necessary to form propofol microemulsions in a bulk media of normal saline. The concentration of propofol was constant at 10 mg/ml. For a given fatty acid co-surfactant, the region below the line is a microemulsion whereas the area above the line represents a macroemulsion formulation.
The measured particle sizes for various formulations of the microemulsions varied from 11.9 to 47.7 nm (fig. 4). In general, as the concentration of purified poloxamer 188 was increased, the diameter of the particles also increased irrespective of the carbon length of the fatty acid co-surfactant. No change was noted in the dimension of the microemulsion droplets containing C8 fatty acid salt after four months of storage at room temperature.
Figure 4.

Effects of purified poloxamer 188 concentration and fatty acid chain length on nanodroplet diameter for propofol microemulsions. In these systems, the concentration of propofol was held constant at 1% (10 mg/ml) and the minimum necessary amount of fatty acid salt was added to formulate propofol microemulsions.
We also investigated the relative stability of these propofol microemulsions to dilution with normal saline. That is, we determined the volume of the saline diluent necessary to break the microemulsion into a macroemulsion (fig. 5). The volume of diluent required to cause these changes increased with lengthening of the carbon chain in the fatty acid salt. This increase in the diluent volume was most noticeable for the C12 propofol microemulsions that required approximately a six-fold dilution to break the microemulsion.
Figure 5.

Effect of dilution on propofol microemulsions formulated using various concentrations of purified poloxamer 188 (see abscissa) and several fatty acid salts with variable carbon chain length (see figure legend). Shown are the relative volumes of 0.9% NaCl diluents that caused the microemulsions to break into macroemulsions (i.e., a transition from a transparent to a turbid state). The concentration of propofol in the undiluted microemulsions was constant at 1% (10 mg/ml).
Anesthetic Properties in the Rat
Thirty-eight rats were enrolled in this protocol, but eight rats completed only one limb of this protocol due to retraction of the catheter under the skin during the recovery interval from the first limb of the study. Data from these eight rats (n=5 in microemulsion groups, n=3 macroemulsion group) were removed from the study. The remaining rats (n=30) completed the crossover study and were anesthetized with these microemulsions (n=6 per experimental group; 5 experimental groups) and with the macroemulsion control (n=30). All rats experienced rapid induction without apnea and recovered for at least 14 days without apparent injury. Following the 14 day observation period after crossover, the experiment was terminated. The summary data from these experiments are presented in Table 1.
Table 1.
Effects of Purified Poloxamer 188 and Fatty Acid Chain Length Concentrations on Anesthetic Induction and Emergence.
| PARAMETER | PROPOFOL FORMULATION | |||||
|---|---|---|---|---|---|---|
| Macroemulsion | Macroemulsions | |||||
| Purified Poloxamer 188 (%) | Not applicable | 3% | 5% | 7% | 5% | 5% |
| Fatty Acid Salt Length | Not applicable | C8 | C8 | C8 | C10 | C12 |
| N | 30 | 6 | 6 | 6 | 6 | 6 |
| Anesthetic Induction | ||||||
| Dose (mg/kg) | 20.5 ± 4.4 | 26.6 ± 4.0* | 30.4 ± 4.5* | 30.2 ± 4.5* | 41.9 ± 3.4*† | 33.9 ± 1.6*†¶ |
| Stunned (s) | 40 ± 9 | 48 ± 8*† | 69 ± 13* | 64 ± 12* | 69 ± 10* | 83 ± 12*¶ |
| Loss of Righting Reflex (s) | 55 ± 12 | 68 ± 11* | 91 ± 12* | 78 ± 15* | 110 ± 13* | 100 ± 13* |
| Loss of Lash Reflex (s) | 89 ± 17 | 93 ± 17 | 129 ± 24* | 106 ± 16 | 156 ± 11* | 142 ± 33* |
| Loss of Leg Withdrawal (s) | 123 ± 26 | 160 ± 23* | 182 ± 26* | 181 ± 28* | 251 ± 20*† | 203 ± 10*†¶ |
| Anesthetic Emergence | ||||||
| Return of Leg Withdrawal (s) | 312 ± 163 | 368 ± 77 | 405 ± 193 | 274 ± 105*†‡ | 318 ± 63 | 516 ± 176*†¶ |
| Return of Lash Reflex (s) | 341 ± 191 | 347 ± 84 | 362 ± 195 | 230 ± 107‡ | 334 ± 57 | 528 ± 240 |
| Return of Righting Reflex (s) | 658 ± 119 | 460 ± 64* | 629 ± 126 | 581 ± 155 | 712 ± 192 | 889 ± 175*†¶ |
| Return of Sustained Headlift (s) | 684 ± 113 | 495 ± 83* | 644 ± 133 | 596 ± 143* | 725 ± 197 | 850 ± 119*†¶ |
Notes: Dose and latency intervals for anesthetic induction and emergence in rat following intravenous infusion of a propofol macroemulsion formulation and several propofol microemulsion formulations with differing fatty acid salt carbon chain length and differing purified poloxamer 188 concentrations. Data are expressed as mean±SD of N number of experiments. Propofol was given as a macroemulsion or a microemulsion in a rat femoral vein catheter using a randomized, crossover design (see Methods and Materials) separated by 14 days rest. The anesthetic emergence intervals started when the propofol infusion was discontinued.
P<0.05:
microemulsion formulation compared to the macroemulsion formulation;
compared to microemulsion formulated with 5% purified poloxamer and C8;
compared to microemulsion formulation with 3% purified poloxamer and C8;
compared to microemulsion formulation with 5% purified poloxamer and C10.
Discussion
Propofol is an organic liquid that exists as an oil at room temperature with minimal aqueous solubility. This insolubility is both a fundamental property of propofol and a significant problem for purposes of drug delivery. To address this issue, propofol was formulated in a castor bean oil macroemulsion in the early 1980s, but this solvent was subsequently abandoned due to its propensity to cause anaphylaxis.19,20 Instead, a soy bean oil macroemulsion was selected as an alternative emulsification agent to deliver propofol.21 Currently, these white, opaque macroemulsions available from different pharmaceutical companies are formulated with the following components: propofol (10 mg/ml), soybean oil (100 mg/ml), glycerol (22.5 mg/ml), egg lecithin (12 mg/ml), and a preservative (0.05 mg/ml disodium edetate or 0.25 mg/ml sodium metabisulfite). The requirement that propofol be dissolved in 10% soybean oil with associated surfactant has caused a number of liabilities including support of bacterial growth, addition of egg products that is objectionable to vegans, and the necessity of preservatives.5,6,8
The need for a suitable alternative formulation is evidenced by past and ongoing research into other types of propofol delivery systems. Several investigators have reported favorable results using formulations composed of medium- and long-chain triglyceride macroemulsions to deliver propofol at varying concentrations of 10-60 mg/ml.11,22,23 In addition, propofol has been complexed with sulfobutylether 7-β-cyclodextrin molecules to form clear, colorless formulations that are stable at a wide range of temperatures (4-50°C), a clear benefit compared to macroemulsion technology, and that have pharmacokinetic properties similar to the conventional drug delivery system.12,24 Finally, pro-drug methods have been exploited to develop phosphono-2,6-diisopropylphenol, an agent that is metabolized by native alkaline phosphatase to generate the active agent, propofol, and two byproducts, formaldehyde and phosphate.25 In this report, we demonstrate that microemulsion methods represent another suitable technology to deliver propofol intravenously with anesthetic parameters similar to the commercially available macroemulsions.
Microemulsion Technology for Drug Delivery
Microemulsions have been previously used to deliver several drugs by the oral and transcutaneous routes. Currently, an oral microemulsion of cyclosporine is available for prevention and treatment of transplant rejection of solid organs.26 In addition, a number of other drugs have been formulated in microemulsions for oral (e.g., paclitaxel, heparin) and transdermal (e.g., apomorphine, estrogen) delivery.27-30 Fewer drugs have been delivered using an intravenous route, but include trans-retinoic acid, and flurbiprofen.31,32 Unlike other oil-in-water microemulsions wherein the active drug is dissolved in an oil excipient, propofol acted in two different, complementary roles in the present investigation. That is, the need for an excipients oil for propofol dispersal was obviated by the fact that propofol is itself an oil at room and physiological temperatures. Therefore, propofol could serve not only as the active pharmacological agent, but also could exist as the physical platform for the preparation of these microemulsions. By doing so, the need for additional excipients oils (e.g., soybean or castor bean oil) is eliminated along with the potential for these excipients to nourish bacteria. Although the present authors also hypothesize that propofol microemulsions will not support bacterial growth to the same extent as macroemulsions, additional investigations are needed to test this thesis.
The onset of induction with the propofol microemulsion was similar to that caused by the commercially available propofol macroemulsion. Whereas the time to achieve induction (defined by the protocol as loss of leg withdrawal to a pinch) during macroemulsion injection was 123 s, the latency periods during microemulsion treatment were 160-251 s. This delay is favorable when compared to pro-drug technologies that rely on metabolism, a phenomenon that may vary within any patient population.33 The differences in induction times between experimental groups reported herein may be caused by differential release of propofol from the individual droplets into the blood. That is, the different propofol nanoparticles have markedly different stabilities against dilution by blood based on the emulsifier structure and concentration selected for the formulation. In general, a microemulsion is thermodynamically stable at equilibrium. One can destabilize a microemulsion by significantly changing pressure, temperature, or chemical compositions.15 The last variable can be changed simply by diluting a microemulsion with saline or blood. It is well recognized that the formation of microemulsions requires an ultra low interfacial tension (e.g., ∼10-3 mNewton/m) at the oil/water interface.15 Upon dilution with saline, the interfacial tension will increase substantially as the emulsifier molecules (i.e., both poloxamer 188 and fatty acid salt molecules) desorb from the droplet surface. This event will markedly increase the interfacial tension at the droplet surface and ultimately destabilize the microemulsion, with release of the active pharmacological core (i.e., propofol). However, each emulsifier film around the microemulsion droplet has inherent molecular packing and, hence, stability. The extent of dilution required to destabilize a microemulsion represents its inherent stability. Thus, a microemulsion requiring greater dilution for destabilization indicates that its emulsifier film has a greater stability. The data in fig. 5 suggests that the C12 fatty acid salt microemulsions are the most stable as they require the greatest dilution to become a turbid macroemulsion. It is likely, however, that microemulsion destabilization is affected by more than just dilution in vivo as the C10 fatty acid microemulsion required the longest time to cause anesthetic induction whereas the C12 fatty acid microemulsion was most stable to dilution in vitro. Further understanding and selectively modifying these destabilization rates by adapting surfactant type and concentrations alludes to the possibility of controlling release times of active pharmaceuticals from nearly immediate (e.g., propofol) to longer times (e.g., chemotherapeutic or antifungal agents). Moreover, although an understanding of the parameters governing release rate allows further refinement of these microemulsions, only a fraction of the formulations presented herein could be considered for additional study with an aim towards clinical use. That is, the induction times for some microemulsions (e.g., 5% purified poloxamer 188 with C10) were more than double that caused by the macroemulsion. Whether this observation is species-specific or a clinically significant result remains to be determined.
Selection of Surfactants
The nonionic and ionic surfactants used to formulate propofol microemulsions were carefully selected not only for their ability to combine with the drug to form stable microemulsions, but also because both surfactants have been previously injected intravenously into humans without reported adverse incidents. Justification for choosing these surfactants is presented subsequently.
Nonionic Surfactant
The selection of purified poloxamer 188, an ethylene oxide / propylene oxide block copolymer, was justified based on a number of characteristics related to chemical and medical requirements. First, this surfactant allowed rapid and reproducible preparation of propofol microemulsions that possessed good stability over time. In general, microemulsions are thermodynamically stable and hence exhibit infinite shelf life. These formulations are destabilized upon dilution with blood or plasma or saline beyond its tolerance limit. Second, purified poloxamer 188 has been previously administered intravenously in large doses to humans without any apparent adverse effect. For example, this surfactant was infused to patients suffering chest crisis due to sickle cell disease in an attempt to reduce the duration and severity of the crisis.34,35 Although not efficacious to treat chest crisis at large doses of purified poloxamer 188 (e.g., 88 g), no apparent untoward effects were noted in the subjects. Third, this surfactant is primarily (>95%) excreted by kidneys and does not stimulate or inhibit human cytochrome P450 systems.36 In fact, others have hailed this coblock surfactant as a model surfactant for pharmacological use although additional toxicological evaluation may be needed before more widespread use in the clinical arena.36 For example, the present investigators have also demonstrated that poloxamer surfactants may reduce platelet function in vitro as assessed by thromboelastography.37 Additional work is needed to determine if this effect on platelets translates into functional defects in thrombosis.
For the experiments examining microemulsions formulated with C8, C10, or C12, the authors held the purified poloxamer 188 concentration constant at 5% (vis-à-vis 3% or 7%). The 5% concentration of purified poloxamer 188 was selected for further study because microemulsions formulated with 7% purified poloxamer 188 required minimal-to-no fatty acid surfactant (see fig 2) whereas microemulsions with 3% purified poloxamer 188 required concentrations of fatty acid necessary to form stable propofol microemulsion likely to be unacceptable for in vivo use in humans.
Ionic Surfactant
Sodium caprylate was chosen as the best ionic surfactant for the following reasons. First, the surfactant allows reliable synthesis of propofol microemulsions with particle diameters of <50 nm range. Second, sodium caprylate is already present in many blood products (e.g., human albumin) currently in clinical use to stabilize proteins.38,39 Third, the smaller length of the carbon backbone reduces the risks of cellular injury associated with fatty acids with longer carbon chains such as sodium oleate that is used to cause lung injury as a model of acute respiratory distress syndrome.40 For these reasons, sodium caprylate is the best choice currently available for additional investigations using microemulsions of propofol, although the information from use of C10 and C12 allowed us to understand how surfactant selection can affect the dose-response of propofol microemulsions. In addition to the selection of surfactant type, however, the concentration of this ionic surfactant must be carefully considered. In the future, the present investigators hope to determine if the concentrations of sodium caprylate used in this study interact with erythrocyte membranes or cause hemolysis as can occur in vitro with higher concentrations (EC50 of 213 mM) of sodium caprylate.37
In conclusion, we report the successful preparation, characterization, and use of propofol microemulsions to induce anesthesia in rat similar to that caused by propofol macroemulsions. Formulation of propofol microemulsions was achieved although no excipient oil (e.g., soybean or castor bean oil) was used because propofol exists as an oil and can serve as its own core of an oil-in-water microemulsion. Thus, propofol was used both to solubilize itself and as an active pharmaceutical. The differential diluent volumes in vitro and destabilization times in vivo based on changing the concentration and type of surfactant alludes to the possibility of selectively modifying the dose-response properties of propofol, and potentially other lipophilic drugs. Future work will be aimed towards exploring these differences for propofol and other lipophilic agents and to determining if these propofol microemulsions reduce the liabilities associated with soybean oil-based macroemulsions.
Summary Statement.
Transparent propofol microemulsions were prepared and characterized because these formulations may offer advantages over conventional turbid macroemulsions. These microemulsion formulations caused induction and emergence in rat similar to that caused by a macroemulsion of propofol, but with a larger dose and longer time of onset with some microemulsion formulations.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (Bethesda, Maryland, U.S.A.) via a Small Business Innovation Research Phase I award (1R43GM072142-01) and by NanoMedex, Inc. (Alachua, Florida, U.S.A.). The methodology for preparation of the propofol microemulsion used in this study is protected by United States Patents 6,623,765 and 6,638,537, which are assigned to University of Florida, Research Foundation, Incorporated (Gainesville, FL, U.S.A.) and licensed to NanoMedex, Inc. Drs. Morey, Modell, Shah, Gravenstein, and Dennis own stock in NanoMedex, Inc. and as such may benefit financially as a result of the outcomes of the research reported in this publication.
Footnotes
Institutional Attribution: Departments of Anesthesiology and Chemical Engineering, University of Florida, Gainesville, FL and NanoMedex, Inc., Alachua, FL
Contributor Information
Timothy E. Morey, Associate Professor, Department of Anesthesiology, University of Florida, Gainesville, FL
Jerome H. Modell, Professor Emeritus, Department of Anesthesiology, University of Florida, Gainesville, FL and, Chief Scientific Officer, NanoMedex, Inc. Alachua, FL, Professor and Chairman, Department of Anesthesiology, Professor of Neurosurgery, University of Florida, Gainesville, FL.
Dushyant Shekhawat, Department of Chemical Engineering, University of Florida, Gainesville, FL
Todd Grand, Biological Scientist, NanoMedex, Inc., Alachua, FL
Dinesh O. Shah, Professor Emeritus, Departments of Chemical Engineering and Anesthesiology, University of Florida, Gainesville, FL and Vice President for Research, NanoMedex, Inc., Alachua, FL
Nikolaus Gravenstein, Department of Anesthesiology, Professor of Neurosurgery, University of Florida, Gainesville, FL
Susan P. McGorray, Research Assistant Professor, Division of Biostatistics, Department of Epidemiology and Health Policy Research, University of Florida, Gainesville, FL
Donn M. Dennis, The J.S. Gravenstein, M.D. Professor of Anesthesiology, Department of Anesthesiology, Professor of Pharmacology & Experimental Therapeutics, and Psychiatry, University of Florida, Gainesville, FL
References
- 1.Chhabra A, Pandey R, Khandelwal M, Subramaniam R, Gupta S. Anesthetic techniques and postoperative emesis in pediatric strabismus surgery. Reg Anesth Pain Med. 2005;30:43–7. doi: 10.1016/j.rapm.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 2.Song D, Whitten CW, White PF, Yu SY, Zarate E. Antiemetic activity of propofol after sevoflurane and desflurane anesthesia for outpatient laparoscopic cholecystectomy. Anesthesiology. 1998;89:838–43. doi: 10.1097/00000542-199810000-00007. [DOI] [PubMed] [Google Scholar]
- 3.Jellish WS, Leonetti JP, Murdoch JR, Fowles S. Propofol-based anesthesia as compared with standard anesthetic techniques for middle ear surgery. J Clin Anesth. 1995;7:292–6. doi: 10.1016/0952-8180(95)00030-l. [DOI] [PubMed] [Google Scholar]
- 4.Chan VW, Chung FF. Propofol infusion for induction and maintenance of anesthesia in elderly patients: recovery and hemodynamic profiles. J Clin Anesth. 1996;8:317–23. doi: 10.1016/0952-8180(96)00041-4. [DOI] [PubMed] [Google Scholar]
- 5.Centers for Disease Control and Prevention. Postsurgical Infections Associated with an Extrinsically Contaminated Intravenous Anesthetic Agent -- California, Illinois, Maine, and Michigan, 1990. MMWR. 1990;39:426–7. [PubMed] [Google Scholar]
- 6.Yu HP, Tang GJ, Liaw WJ, Yien HW, Lee TY. Pseudomonas cepacia induced septic shock after propofol--a case report. Acta Anaesthesiol Sin. 2000;38:53–6. [PubMed] [Google Scholar]
- 7.Ho CM, Tsou MY, Sun MS, Chu CC, Lee TY. The optimal effective concentration of lidocaine to reduce pain on injection of propofol. J Clin Anesth. 1999;11:296–300. doi: 10.1016/s0952-8180(99)00040-9. [DOI] [PubMed] [Google Scholar]
- 8.Hofer KN, McCarthy MW, Buck ML, Hendrick AE. Possible anaphylaxis after propofol in a child with food allergy. Ann Pharmacother. 2003;37:398–401. doi: 10.1345/aph.1C227. [DOI] [PubMed] [Google Scholar]
- 9.Doenicke AW, Roizen MF, Rau J, O'Connor M, Kugler J, Klotz U, Babl J. Pharmacokinetics and pharmacodynamics of propofol in a new solvent. Anesth Analg. 1997;85:1399–403. doi: 10.1097/00000539-199712000-00040. [DOI] [PubMed] [Google Scholar]
- 10.Morey TE, Shah DO, Gravenstein N, Modell JH, Dennis DM. Novel microemulsion-based nanoparticles for propofol delivery. Anesthesiology. 2004;101:A506. [Google Scholar]
- 11.Knibbe CA, Voortman HJ, Aarts LP, Kuks PF, Lange R, Langemeijer HJ, Danhof M. Pharmacokinetics, induction of anaesthesia and safety characteristics of propofol 6% SAZN vs propofol 1% SAZN and Diprivan-10 after bolus injection. Br J Clin Pharmacol. 1999;47:653–60. doi: 10.1046/j.1365-2125.1999.00942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Egan TD, Kern SE, Johnson KB, Pace NL. The pharmacokinetics and pharmacodynamics of propofol in a modified cyclodextrin formulation (Captisol) versus propofol in a lipid formulation (Diprivan): an electroencephalographic and hemodynamic study in a porcine model. Anesth Analg. 2003;97:72–9. doi: 10.1213/01.ane.0000066019.42467.7a. [DOI] [PubMed] [Google Scholar]
- 13.Fechner J, Ihmsen H, Hatterscheid D, Jeleazcov C, Schiessl C, Vornov JJ, Schwilden H, Schuttler J. Comparative pharmacokinetics and pharmacodynamics of the new propofol prodrug GPI 15715 and propofol emulsion. Anesthesiology. 2004;101:626–39. doi: 10.1097/00000542-200409000-00011. [DOI] [PubMed] [Google Scholar]
- 14.Fechner J, Ihmsen H, Hatterscheid D, Schiessl C, Vornov JJ, Burak E, Schwilden H, Schuttler J. Pharmacokinetics and clinical pharmacodynamics of the new propofol prodrug GPI 15715 in volunteers. Anesthesiology. 2003;99:303–13. doi: 10.1097/00000542-200308000-00012. [DOI] [PubMed] [Google Scholar]
- 15.Holmberg K. In: Quarter Century Progress and New Horizons in Microemulsions, Micelles, Microemulsions, and Monolayers: Science and Technology. Shah DO, editor. New York: Marcel Dekker Inc.; 1998. pp. 161–92. [Google Scholar]
- 16.Morey TE, Varshney M, Flint JA, Rajasekaran S, Shah DO, Dennis DM. Treatment of local anesthetic-induced cardiotoxicity using drug scavenging nanoparticles. Nano Letters. 2004;4:757–9. doi: 10.1021/nl049880w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Littell RC, Milliken GA, Stroup WW, Wolfinger RD. SAS System for Mixed Models. Cart, N.C.: SAS Institute; 1996. [Google Scholar]
- 18.Cook RD, Weisberg S. Residuals and Influence in Regression. New York, N,Y: Chapman and Hall; 1982. [Google Scholar]
- 19.Glen JB, Hunter SC. Pharmacology of an emulsion formulation of ICI 35 868. Br J Anaesth. 1984;56:617–26. doi: 10.1093/bja/56.6.617. [DOI] [PubMed] [Google Scholar]
- 20.Huttel MS, Schou OA, Stoffersen E. Complement-mediated reactions to diazepam with Cremophor as solvent (Stesolid MR) Br J Anaesth. 1980;52:77–9. doi: 10.1093/bja/52.1.77. [DOI] [PubMed] [Google Scholar]
- 21.Dutta S, Matsumoto Y, Ebling WF. Propofol pharmacokinetics and pharmacodynamics assessed from a cremophor EL formulation. J Pharm Sci. 1997;86:967–9. doi: 10.1021/js970118m. [DOI] [PubMed] [Google Scholar]
- 22.Doenicke AW, Roizen MF, Rau J, Kellermann W, Babl J. Reducing pain during propofol injection: the role of the solvent. Anesth Analg. 1996;82:472–4. doi: 10.1097/00000539-199603000-00007. [DOI] [PubMed] [Google Scholar]
- 23.Suchner U, Katz DP, Furst P, Beck K, Felbinger TW, Thiel M, Senftleben U, Goetz AE, Peter K. Impact of sepsis, lung injury, and the role of lipid infusion on circulating prostacyclin and thromboxane A(2) Intensive Care Med. 2002;28:122–9. doi: 10.1007/s00134-001-1192-3. [DOI] [PubMed] [Google Scholar]
- 24.Babu MK, Godiwala TN. Toward the development of an injectable dosage form of propofol: preparation and evaluation of propofol-sulfobutyl ether 7-beta-cyclodextrin complex. Pharm Dev Technol. 2004;9:265–75. doi: 10.1081/pdt-200031428. [DOI] [PubMed] [Google Scholar]
- 25.Fechner J, Ihmsen H, Schiessl C, Jeleazcov C, Vornov JJ, Schwilden H, Schuttler J. Sedation with GPI 15715, a water-soluble prodrug of propofol, using target-controlled infusion in volunteers. Anesth Analg. 2005;100:701–6. doi: 10.1213/01.ANE.0000144772.13372.F4. [DOI] [PubMed] [Google Scholar]
- 26.Ferraresso M, Ghio L, Zacchello G, Murer L, Ginevri F, Perfumo F, Zanon GF, Fontana I, Amore A, Edefonti A, Vigano S, Cardillo M, Scalamogna M. Pharmacokinetic of Cyclosporine Microemulsion in Pediatric Kidney Recipients Receiving A Quadruple Immunosuppressive Regimen: The Value of C2 Blood Levels. Transplantation. 2005;79:1164–8. doi: 10.1097/01.tp.0000160762.37225.2b. [DOI] [PubMed] [Google Scholar]
- 27.Priano L, Albani G, Brioschi A, Calderoni S, Lopiano L, Rizzone M, Cavalli R, Gasco MR, Scaglione F, Fraschini F, Bergamasco B, Mauro A. Transdermal apomorphine permeation from microemulsions: a new treatment in Parkinson's disease. Mov Disord. 2004;19:937–42. doi: 10.1002/mds.20054. [DOI] [PubMed] [Google Scholar]
- 28.Andersson M, Lofroth JE. Small particles of a heparin/chitosan complex prepared from a pharmaceutically acceptable microemulsion. Int J Pharm. 2003;257:305–9. doi: 10.1016/s0378-5173(03)00131-5. [DOI] [PubMed] [Google Scholar]
- 29.Gao P, Rush BD, Pfund WP, Huang T, Bauer JM, Morozowich W, Kuo MS, Hageman MJ. Development of a supersaturable SEDDS (S-SEDDS) formulation of paclitaxel with improved oral bioavailability. J Pharm Sci. 2003;92:2386–98. doi: 10.1002/jps.10511. [DOI] [PubMed] [Google Scholar]
- 30.Peltola S, Saarinen-Savolainen P, Kiesvaara J, Suhonen TM, Urtti A. Microemulsions for topical delivery of estradiol. Int J Pharm. 2003;254:99–107. doi: 10.1016/s0378-5173(02)00632-4. [DOI] [PubMed] [Google Scholar]
- 31.Hwang SR, Lim SJ, Park JS, Kim CK. Phospholipid-based microemulsion formulation of all-trans-retinoic acid for parenteral administration. Int J Pharm. 2004;276:175–83. doi: 10.1016/j.ijpharm.2004.02.025. [DOI] [PubMed] [Google Scholar]
- 32.Park KM, Kim CK. Preparation and evaluation of flurbiprofen-loaded microemulsion for parenteral delivery. Int J Pharm. 1999;181:173–9. doi: 10.1016/s0378-5173(99)00029-0. [DOI] [PubMed] [Google Scholar]
- 33.Blesch KS, Gieschke R, Tsukamoto Y, Reigner BG, Burger HU, Steimer JL. Clinical pharmacokinetic/pharmacodynamic and physiologically based pharmacokinetic modeling in new drug development: the capecitabine experience. Invest New Drugs. 2003;21:195–223. doi: 10.1023/a:1023525513696. [DOI] [PubMed] [Google Scholar]
- 34.Orringer EP, Casella JF, Ataga KI, Koshy M, Adams-Graves P, Luchtman-Jones L, Wun T, Watanabe M, Shafer F, Kutlar A, Abboud M, Steinberg M, Adler B, Swerdlow P, Terregino C, Saccente S, Files B, Ballas S, Brown R, Wojtowicz-Praga S, Grindel JM. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: A randomized controlled trial. JAMA. 2001;286:2099–106. doi: 10.1001/jama.286.17.2099. [DOI] [PubMed] [Google Scholar]
- 35.Grindel JM, Jaworski T, Emanuele RM, Culbreth P. Pharmacokinetics of a novel surface-active agent, purified poloxamer 188, in rat, rabbit, dog and man. Biopharm Drug Dispos. 2002;23:87–103. doi: 10.1002/bdd.297. [DOI] [PubMed] [Google Scholar]
- 36.Grindel JM, Jaworski T, Piraner O, Emanuele RM, Balasubramanian M. Distribution, metabolism, and excretion of a novel surface-active agent, purified poloxamer 188, in rats, dogs, and humans. J Pharm Sci. 2002;91:1936–47. doi: 10.1002/jps.10190. [DOI] [PubMed] [Google Scholar]
- 37.Morey TE, Varshney M, Flint JA, Seubert CN, Smith WB, Bjoraker DG, Shah DO, Dennis DM. Activity of microemulsion-based nanoparticles at the human bio-nano interface: concentration-dependent effects on thrombosis and hemolysis in whole blood. Journal of Nanoparticle Research. 2004;6:159–70. [Google Scholar]
- 38.Johnston A, Uren E, Johnstone D, Wu J. Low pH, caprylate incubation as a second viral inactivation step in the manufacture of albumin. Parametric and validation studies. Biologicals. 2003;31:213–21. doi: 10.1016/s1045-1056(03)00062-9. [DOI] [PubMed] [Google Scholar]
- 39.Anraku M, Tsurusaki Y, Watanabe H, Maruyama T, Kragh-Hansen U, Otagiri M. Stabilizing mechanisms in commercial albumin preparations: octanoate and N-acetyl-L-tryptophanate protect human serum albumin against heat and oxidative stress. Biochim Biophys Acta. 2004;1702:9–17. doi: 10.1016/j.bbapap.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 40.Hubloue I, Biarent D, Abdel KS, Bejjani G, Melot C, Naeije R, Leeman M. Endothelin receptor blockade in canine oleic acid-induced lung injury. Intensive Care Med. 2003;29:1003–6. doi: 10.1007/s00134-003-1683-5. [DOI] [PubMed] [Google Scholar]