

Summary
Background
Hypersensitivity reactions towards non-steroidal anti-inflammatory drugs (NSAID) are common, although true allergies are detectable only in a subgroup of patients. The current study was prompted by a case observation, where a patient experienced generalized urticaria following his second course of diclofenac and proton pump inhibitor medication, and was found to have diclofenac-specific IgE. During recent years, our group has been investigating the importance of gastric digestion in the development of food allergies, demonstrating anti-acid medication as a risk factor for sensitization against food proteins.
Objective
Here, we aimed to investigate whether the mechanism of food allergy induction described can also be causative in NSAID allergy, using diclofenac as a paradigm.
Methods
We subjected BALB/c mice to several oral immunization regimens modelled after the patient’s medication intake. Diclofenac was applied with or without gastric acid suppression, in various doses, alone or covalently coupled to albumin, a protein abundant in gastric juices. Immune responses were assessed on the antibody level, and functionally examined by in vitro and in vivo crosslinking assays.
Results
Only mice receiving albumin-coupled diclofenac under gastric acid suppression developed anti-diclofenac IgG1 and IgE, whereas no immune responses were induced by the drug alone or without gastric acid suppression. Antibody induction was dose dependent with the group receiving the higher dose of the drug showing sustained anti-diclofenac titres. The antibodies induced triggered basophil degranulation in vitro and positive skin tests in vivo.
Conclusion
Gastric acid suppression was found to be a causative mechanism in the induction of IgE-mediated diclofenac allergy.
Keywords: acid suppression, adverse drug reactions, allergy, anti-ulcer drugs, diclofenac, gastric hypoacidity, hypersensitivity, IgE, non-steroidal anti-inflammatory drugs (NSAID)
Introduction
Non-steroidal anti-inflammatory drugs (NSAID) are among the most widely prescribed classes of medication [1]. In some countries, they are even available as over-the-counter pain killers. Mostly, patients are advised to combine NSAID with gastric acid suppressive drugs, to prevent upper gastrointestinal bleeding. Hypersensitivity reactions against NSAID are a known problem, and have been reported to increase in industrialized countries [2, 3]. With this type of reactions, it is essential for future patient management to differentiate between non-allergic drug hypersensitivity and true IgE-mediated allergies.
Diclofenac is one of the most commonly used anti-inflammatory drugs, and often causes immediate-type reactions [4]. It is also one of the NSAID for which true allergies, i.e. presence of specific IgE antibodies, have been reported [5, 6] – and in some cases even nearly fatal or fatal anaphylactic reactions [7-11]. It is generally thought that for a drug to elicit an immune response, it must first be covalently bound to a carrier protein. This is certainly the case with diclofenac, which is in itself too small to be immunogenic. Diclofenac has been demonstrated to be metabolized to acyl glucuronides, which then covalently bind to proteins, thereby generating antigenic determinants capable of stimulating immune cells [12-15].
The present study has been prompted by a case observation, where an otherwise healthy male patient experienced generalized urticaria following his second course of diclofenac and proton pump inhibitor medication. He had no previous history of allergy or urticaria, but had been using proton pump inhibitors repeatedly for recurrent reflux oesophagitis. This history bore marked similarities with many adult food allergy patients, who also have no prior history of atopy, but develop IgE-mediated hypersensitivities to food proteins while being under gastric acid-suppressive medication [16]. In recent years, we have been studying this correlation, and found that normally innocuous, easily degradable food proteins can lead to IgE formation and sensitization when they persist undegraded, in the gastric passage [17]. Gastric digestion is impaired by hypoacidity, as induced by anti-ulcer treatments or prophylactic acid-suppressive therapy during NSAID medication. We hypothesized that this mechanism could also be causative in the sensitization phase of IgE-mediated NSAID hypersensitivity. Therefore, the first aim of the present study was to examine if the urticaria patient was suffering from an intolerance reaction or a true IgE-mediated allergy to diclofenac. In case of an IgE-mediated hypersensitivity, we further aimed to investigate if the suspected sensitization mechanism could be experimentally reproduced in a mouse model.
Methods
Patient characteristics
The urticaria patient is an otherwise healthy 60-year-old male subject with no previous history of allergy or urticaria. He is intermittently under proton pump inhibitor medication for recurrent reflux oesophagitis. He was under an ongoing proton pump inhibitor medication [esomeprazole (Nexium®, AstraZeneca, London, UK), 20 mg, once daily] for this condition when he was first taking diclofenac (Voltaren®, 50 mg coated tablets, twice daily, for 1 week). When he was prescribed diclofenac for the second time approximately a year later, he was not currently taking acid-suppressive drugs. Therefore, he was given a proton pump inhibitor [pantoprazole (Pantoloc®), 20 mg] for mucosal protection, to be taken 1 h before the diclofenac intake. Urticaria started to develop 30 min after he ingested 1 tablet of diclofenac (Diclofenac Genericon®, 50 mg coated tablet), and was generalized after 1 h. Diclofenac medication was ceased immediately, and the urticarial reaction abated after approximately 4 h. The patient was taking no other medication at the time. Blood samples were obtained 2 weeks after the reaction, and skin testing performed a month later. The patient subsequently tolerated aspirin (acetylsalicylic acid), but did not try other NSAID while under our care.
Controls were two volunteers with no history of allergy or urticaria, nor history of gastric acid-suppressive treatment. Blood sampling and skin testing was performed at the allergy outpatient clinic of the Department of Dermatology, Medical University of Vienna, Austria.
Diclofenac-specific enzyme-linked immunosorbent assay
To establish a diclofenac-specific ELISA, we covalently coupled diclofenac to the carrier molecule keyhole limpet hemocyanin (KLH; Sigma, St Louis, MO, USA) via the N-hydroxy-succinimide–1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (NHS-EDAC) method, which results in a covalent (amide) bond between the carboxylic acid group of diclofenac and a primary amine group of KLH. The reaction took place at room temperature at pH 7.3. In contrast to diclofenac alone, the conjugate can be coated onto ELISA plates. We incubated 96-well ELISA plates (Nunc, Roskilde, Denmark) with 1 μg/mL diclofenac–KLH in carbonate/bicarbonate coating buffer (pH 9.6) at 4 °C overnight. Thereafter, plates were washed with Tris-buffered saline (TBS)/0.05% Tween 20 and non-specific binding was blocked with TBS containing 1% dry milk. Validity of the assay was assessed with a highly sensitive and specific rabbit anti-diclofenac antibody, that recognizes diclofenac to a limit of detection of 6 ng/mL [18] (kindly provided by PD Dr Dietmar Knopp, Technische Universitä München, Munich, Germany) as a positive control, and a panel of rabbit antibodies to irrelevant antigens as negative controls. Inhibition experiments were performed with the positive control antibody and diclofenac to ensure specificity of the assay (data not shown). For control purposes, plates were coated in the same manner with 1 μg/mL KLH.
For detection of diclofenac-specific serum IgE antibodies, plates were incubated with patient sera, diluted 1 : 5 in TBS containing 0.1% dry milk. IgE was directly detected by a peroxidase-coupled mouse anti-human IgE antibody (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA). For quantification, a human IgE standard (Southern Biotech, Birmingham, AL, USA) was coated onto the same ELISA plates in serial dilutions. The reaction was developed with 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (Sigma) as substrate. Optical density was measured at 405 nm (reference wavelength 490 nm). Quality control experiments to rule out potential cross-reactivity between the anti-IgE antibody and serum IgG were performed on coated human IgG standards (Southern Biotech), applying up to a 1000-fold more IgG than IgE. All ELISA experiments were performed in duplicate, and repeated for confirmation of reproducibility.
Skin prick testing
Skin prick tests (SPTs) were performed according to the guidelines of the European Academy of Allergology and Clinical Immunology [19] in the urticaria patient and two healthy volunteers, after obtaining written informed consent. A commercially available diclofenac preparation for intravenous (i.v.) application (Diclobene®, Ratiopharm, Vienna, Austria) was used at 37.5 mg/mL (undiluted), and at 500, 100, 30, and 10 μg/mL; the diclofenac–KLH conjugate and KLH alone were used at 500, 100, 30, and 10 μg/mL. Histamine dihydrochloride at 10 mg/mL (ALK-Abelló, Hørsholm, Denmark) served as positive and 0.9% NaCl as negative control. The test was read after 20 min. The weal size was measured using the formula: (D+d)/2, where D is the maximum diameter, and d its perpendicular diameter. A weal size ≥3 mm was defined as a positive reaction.
Oral immunization of BALB/c mice
Six-week-old female BALB/c mice (n = 10 per group) were obtained from the Institute for Laboratory Animal Science and Genetics (Medical University of Vienna, Vienna, Austria) and were treated according to European Union rules of animal care. Mice were immunized according to the oral immunization regimen for IgE induction developed by our group [20], with modifications accounting for the common intake of anti-inflammatory drugs not only once, but for a period of time. In brief, each animal received 11.6 μg esomeprazole (Nexium®) i.v. on days 1 and 2 of each immunization cycle. On days 3–5, esomeprazole was given i.v. twice, 2 h and 15 min before intragastric immunization. This regimen has been shown to cause a consistent elevation of the gastric pH [21]. One hundred and thirty-five microlitres of the respective antigen preparation was mixed with 2 mg (15 μL) sucralfate (Ulcogant, Merck, Vienna, Austria) and administered intragastrically in a final volume of 150 μL. Control mice received diclofenac alone (without esomeprazol and sucralfate). Immunization cycles were repeated every 3 weeks to a total of four; i.e. immunizations were performed on days 3–5, 24–26, 45–47, and 66–68. Blood was taken from the tail vein on day 0 [pre-immune serum (PIS)], and 10 days after each immunization cycle on days 17, 38, 59, and 80 [1.–4. mouse immune serum (MIS), respectively; also see Fig. 1a].
Fig. 1.
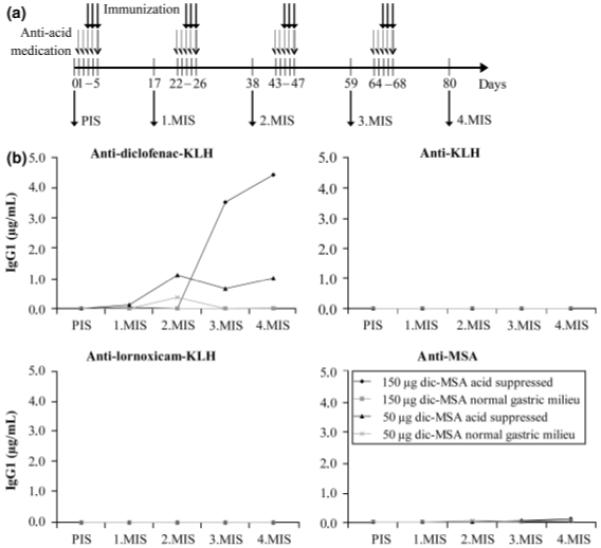
(a) Timeline of the oral immunization regimen (PIS, pre-immune serum; MIS, mouse immune serum). (b) Antibody titres against diclofenac–keyhole limpet hemocyanin (KLH), KLH, lornoxicam-KLH, and murine serum albumin (MSA) in pooled sera of all four treatment groups during the course of immunization. Only diclofenac–MSA immunizations under gastric acid suppression induced antibodies against diclofenac, with the group receiving the higher dose (150 μg/gavage) developing higher titres. In no group any reactivity to the control conjugate lornoxicam-KLH, the carrier KLH, or MSA was observed at any time-point.
A first set of mice received 30 μg diclofenac per immunization, calculated from human dosage and body weight. A second set of mice was given 300 μg diclofenac per immunization, taking into account the higher rate of murine metabolism. For a third set of experiments, diclofenac was covalently coupled to murine serum albumin (MSA; Sigma), via the NHS–EDAC coupling method described above. As the stomachs of laboratory mice are continuously filled with food pellet material, we decided to perform this binding reaction extracorporeally. Mice were gavaged either 50 or 150 μg of the diclofenac–MSA conjugate.
Measurement of murine diclofenac-specific antibodies in enzyme-linked immunosorbent assay
For detection of murine diclofenac-specific serum antibodies, plates were coated with 1 μg/mL diclofenac–KLH as described above. As an irrelevant hapten conjugate control, the NSAID lornoxicam (Nycomed, Linz, Austria) was coupled to KLH as described for diclofenac. Control plates were coated with 1 μg/mL lornoxicam-KLH, 1 μg/mL KLH, or 1 μg/mL MSA, respectively. Plates were then incubated with murine sera, diluted in TBS containing 0.1% dry milk. Dilution was 1 : 5 for IgE detection and 1 : 50 for IgG1 detection. For the inhibition experiment, pooled 4.MIS of the 150 μg acid-suppressed mouse group was incubated for 2 h with 5, 50, 250, or 500 μg/mL of diclofenac–KLH, KLH, diclofenac–MSA, MSA, and diclofenac alone, before addition to the coated ELISA plates. Murine IgE and IgG1 were detected by respective rat anti-mouse Ig subclass antibodies (BD Pharmingen, Heidelberg, Germany), followed by incubation with a peroxidase-coupled goat anti-rat IgG antibody (GE Healthcare, Little Chalfont, UK). For quantification, mouse Ig standards (Southern Biotech) were coated onto the same ELISA plates in serial dilutions. The reaction was developed and measured as described above. Quality control experiments to rule out potential cross-reactivity between anti-IgE and serum IgG, or anti-IgG and serum IgE, were performed on coated murine IgG or IgE standards (Southern Biotech), respectively. All ELISA experiments were performed in duplicates, and repeated for confirmation of reproducibility.
β-hexosaminidase release assay from rat basophilic leukaemia cells
Rat basophilic leukaemia (RBL)-2H3 cells, which express rodent FcεRI as their only antibody receptor, were used to determine functional diclofenac-specific IgE. Cells were incubated for 2 h with pooled 4.MIS of the acid-suppressed or the control mice to allow for binding of serum IgE to FcεRI. Passively sensitized RBL-2H3 cells were then incubated for 30 min with the diclofenac–KLH conjugate, or with KLH, MSA, or diclofenac alone as controls (each antigen at 5 μg/mL). For dose–response experiments, 5, 1, 0.5, and 0.1 μg/mL of diclofenac–KLH were used. IgE recognition of its target antigen leads to cross-linking of FcεRI, RBL cell degranulation, and thus β-hexosaminidase release. Released β-hexosaminidase was detected by 4-methyl-umbelliferyl-N-acetyl-β-d-glucosaminide (4-MUG) and the resulting fluorescence was measured at 465 nm (excitation wavelength 360 nm). All antigens were also incubated with non-sensitized RBL-2H3 cells to assess possible non-IgE-mediated effects.
For 100% release, RBL-2H3 cells were treated with the unspecific degranulation mediator ionomycin (Sigma) for 30 min, and the relative experimental releases determined as follows:
All RBL assay experiments were performed in triplicates, and repeated for confirmation of reproducibility.
Skin tests for cutaneous anaphylaxis in mice
Eight weeks after the last immunization, type I hypersensitivity skin testing was performed. On the day of killing, Evan’s blue (100 μL at 5 mg/mL) was injected into the tail vein of mice. Subsequently, 30 μL of diclofenac–KLH, KLH, MSA, diclofenac alone, mast cell degranulation compound 48/80 as positive control (all at 20 μg/mL), or 0.9% NaCl as negative control were administered intradermally into the shaved abdominal skin. After 20 min, mice were killed and skinned. A positive response is seen as a blue colour reaction on the inside of the abdominal skin due to IgE-mediated effector cell degranulation, mediator release, and subsequent vascular leakage.
Statistical analysis
Statistical analysis was performed using the two-tailed Student’s t-test. P-values <0.05 were considered statistically significant.
Results
Characterization of diclofenac reactivity in the urticaria patient
As a first step, we examined if the patient was suffering from a true IgE-mediated allergy or a non-specific intolerance reaction. To this end, we established a diclofenac-specific ELISA, and analysed the patient’s serum (obtained 2 weeks after the urticaria reaction) for IgE. Indeed, he was found to have high levels of diclofenac-specific IgE (1.19±0.34 μg/mL; mean±SD of two independent experiments), whereas sera from two healthy volunteer controls did not contain diclofenac-specific IgE (0.10±0.04 and 0.12±0.04 μg/mL, respectively). No binding was observed to the carrier molecule KLH.
To assess the clinical relevance of this serological finding, SPT was performed. We used serial dilutions of a commercially available soluble diclofenac preparation, and of the diclofenac–KLH conjugate generated for ELISA coating purposes. KLH alone was included as control. The patient demonstrated dose-dependent positive SPT reactions to the diclofenac conjugate only. Neither diclofenac alone nor KLH alone led to skin reactions. Two healthy volunteers showed no skin reactivity to any test substance, applied at the highest concentration (Table 1).
Table 1.
Skin prick test results
| Patient | Control 1 | Control 2 | |
|---|---|---|---|
| Diclofenac–KLH | |||
| 500 μg/mL | 17 mm* | 0 mm | 0 mm |
| 100 μg/mL | 15 mm | ND | ND |
| 30 μg/mL | 7 mm | ND | ND |
| 10 μg/mL | 0 mm | ND | ND |
| Diclofenac | |||
| 37.5 mg/mL† | 0 mm | 0 mm | 0 mm |
| 500 μg/mL | 0 mm | ND | ND |
| 100 μg/mL | 0 mm | ND | ND |
| 30 μg/mL | 0 mm | ND | ND |
| 10 μg/mL | 0 mm | ND | ND |
| KLH | |||
| 500 μg/mL | 0 mm | 0 mm | 0 mm |
| 100 μg/mL | 0 mm | ND | ND |
| 30 μg/mL | 0 mm | ND | ND |
| 10 μg/mL | 0 mm | ND | ND |
Diameter of weal reaction.
Undiluted concentration of commercially available i.v. diclofenac preparation.
i.v., intravenous; KLH, keyhole limpet hemocyanin; ND, not done.
Antibody levels after oral immunization of BALB/c mice
To investigate if the suspected mechanism of sensitization could be reproduced experimentally, BALB/c mice were immunized intragastrically with diclofenac under concomitant suppression of gastric acid (Fig. 1a). The first two sets of experiments, with either 30 μg diclofenac/gavage or 300 μg diclofenac/gavage, did not result in any antibody titres (data not shown). In humans, orally applied diclofenac is metabolized to a reactive intermediate, an acyl glucuronide, which then forms covalent bonds with surrounding proteins. This covalently bound diclofenac is considered to be the immunogenic form [12-14]. To ensure that the experimental animals were exposed to covalently bound diclofenac, we immunized them with a diclofenac–MSA conjugate in the second set of experiments. Indeed, now high anti-diclofenac antibody titres were observed in the acid-suppressed groups. We immunized four groups of mice, applying two diclofenac–MSA concentrations, 150 and 50 μg/gavage. The control groups received the same amount of antigen without concomitant gastric acid suppression, i.e. they had a normal gastric milieu.
In a first ELISA experiment, we investigated the overall development of antibodies against diclofenac using pooled sera of each mouse group (Fig. 1b). For control purposes, both anti-KLH and anti-MSA titres were assessed, as well as titres against an irrelevant hapten conjugate control, lornoxicam-KLH. As can be seen from Fig. 1, induced antibodies only reacted with diclofenac. No reactivity against the carrier molecule KLH could be detected, nor did mice develop antibodies against the hapten conjugate control or their self-protein MSA. Because of the usage of pooled sera, statistical analysis could not be performed, but antibody titres were found to be markedly higher in the group receiving the higher dose of the antigen (150 μg/gavage). Therefore, this group was chosen for further experiments. We performed an inhibition ELISA as an additional test for diclofenac-specificity of the induced antibodies (Fig. 2). The interaction of anti-diclofenac antibodies (4.MIS) and coated diclofenac could be inhibited by diclofenac alone, and to an even greater extent by both the diclofenac–KLH and the diclofenac–MSA conjugates. KLH and MSA did not lead to any inhibition, consistent with the first ELISA results. The antibodies induced were thus shown to be specific for diclofenac.
Fig. 2.

Specificity test of induced antibodies by inhibition ELISA. The interaction of anti-diclofenac antibodies (4.MIS, higher dosage group) with coated diclofenac could be inhibited in a dose-dependent fashion by diclofenac alone (Dic), and by the diclofenac-keyhole limpet hemocyanin (KLH) and the diclofenac–murine serum albumin (MSA) conjugates. KLH or MSA alone did not cause any inhibition. MIS, mouse immune serum.
Subsequently, anti-diclofenac IgG1 and IgE responses were examined at the individual serum level. When comparing IgG1 of the acid-suppressed group to the control group with normal gastric milieu, the difference was statistically significant (3.MIS: P<0.05; 4.MIS: P<0.01). With IgE, statistical significance was not reached, but the P-values showed a definite trend towards significance with time (Fig. 3).
Fig. 3.
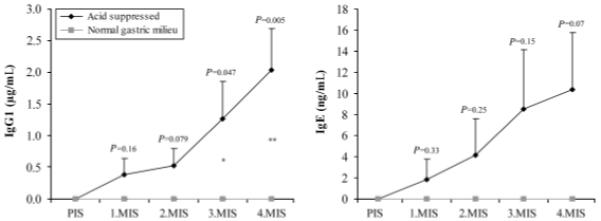
Single serum analysis of diclofenac-specific antibodies from the higher dosage groups (150 μg diclofenac–MSA/gavage, ± acid suppression). Only gastric acid-suppressed mice show both IgG1 (left panel) and IgE (right panel) antibody induction. Mean values+SEM are shown. Statistical comparisons between the groups are indicated above error bars. *P<0.05; **P<0.01. MIS, mouse immune serum; MSA, murine serum albumin
Analysis of the cross-linking capability of induced immunoglobulin E by rat basophilic leukaemia assay
To assess the biological relevance of the induced IgE antibodies, we performed a RBL cell release assay. Anti-diclofenac IgE from the acid-suppressed mouse group resulted in a marked degranulation of RBL cells upon triggering with diclofenac–KLH, with relative specific release reaching 53%. Only very low background release was observed upon triggering with KLH, MSA, or diclofenac alone, and upon all triggers in the control group. To exclude toxic effects, trigger substances were incubated with non-sensitized RBL cells, and did not cause any degranulation (Fig. 4a). As an additional proof of specificity, anti-diclofenac antibody-mediated degranulation could be shown to be antigen dose dependent (Fig. 4b).
Fig. 4.
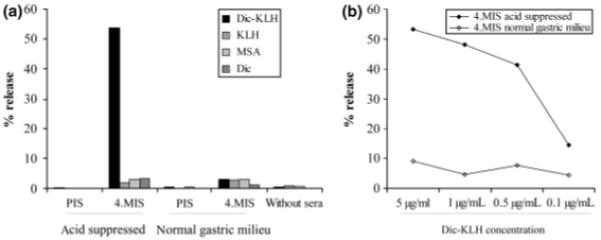
Functional analysis of diclofenac-specific IgE from the higher dosage groups by an in vitro cross-linking rat basophilic leukaemia assay. Only immune sera from gastric acid suppressed mice, but not from the control group, contained diclofenac-specific IgE triggering degranulation after cross-linking with multivalent diclofenac–keyhole limpet hemocyanin (KLH). No degranulation was triggered by KLH, murine serum albumin (MSA), or monovalent diclofenac (Dic), and no antibody-independent degranulation was caused by the tested substances alone (a). Diclofenac-specific degranulation was found to be antigen dose dependent (b).
In vivo skin test for cutaneous anaphylaxis in mice
To assess the in vivo relevance of the induced anti-diclofenac antibodies, skin testing for cutaneous anaphylaxis was performed. Corresponding to the RBL assay results, the acid-suppressed group showed specific type I skin reactivity upon challenge with the diclofenac–KLH conjugate, but not with the control substances or monovalent diclofenac alone. In the group with normal gastric milieu, no reactivity to any test substance was observed (data not shown).
Discussion
The first observation that peptic ulcer patients receiving multiple medications show increased incidences of food and drug allergies dates back to 1984 [22]. In recent years, we were repeatedly able to show that gastric acid suppression is an important risk factor for food allergy development [16, 17, 20, 23, 24]. The underlying mechanism is thought to be a hindrance of peptic digestion, which requires a low gastric pH for pepsin activation. Thus persisting undegraded food proteins can reach the small intestine, where sensitization occurs [25]. Prompted by a case observation, where an otherwise healthy male patient suffered from generalized urticaria following his second course of diclofenac and proton pump inhibitor medication, the aim of this study was to investigate whether this mechanism of food allergy induction may also be causative in IgE-mediated hypersensitivities against NSAID.
The patient was found to have diclofenac-specific IgE antibodies in his serum and dose-dependent diclofenac reactivity in SPT, and could thus be diagnosed with IgE-mediated diclofenac allergy. He was known to having been under an ongoing proton pump inhibitor medication for reflux oesophagitis during his first intake of diclofenac, and received proton pump inhibitors again as a mucosal protection concomitantly with his second prescription of this NSAID.
To investigate if the suspected sensitization mechanism could be reproduced experimentally, BALB/c mice were subjected to an acid-suppressive treatment regimen effectively increasing the gastric pH [21], and fed with different amounts of diclofenac. Interestingly, when feeding the drug alone, no antibody induction could be observed. However, when we covalently coupled the drug to murine albumin, both of the murine anaphylactogenic antibody classes, IgG1 and IgE, were induced. The coupling step was introduced to mirror the human situation as closely as possible, where the immunogenic form of diclofenac is described to be covalently bound to serum and surrounding proteins, such as albumin [26], enzymes of the small intestine (aminopeptidase, sucrose-isomaltase, etc.) [13], other enterocyte macromolecules [27], or several liver proteins [14, 28]. In this study, we chose to work with albumin as coupling partner, as this is the most common binding partner described for diclofenac in humans [26], and also most readily available. Clinical relevance of the antibodies induced was further assessed in in vitro and in vivo functionality tests, which both showed strong reactivity and confirmed the highly specific immune response.
Taken together, these murine data indicate that gastric acid suppression can indeed lead to true IgE-mediated allergies to certain drugs. The fact that the mice did not become sensitized after gavage of diclofenac alone, but only after extra-corporeal coupling, is in contrast to the observation in the patient. We speculate that the continuous presence of the food pellet material in the stomachs of laboratory mice inhibits reactions with surrounding proteins that would occur more easily in the relatively empty and liquid conditions of the human stomach. On the other hand, the necessary introduction of a covalent coupling to albumin highlights the importance of a protein component. This is in accordance with the assumed sensitization mechanism, where gastric protein digestion is the crucial checkpoint for tolerance or allergy induction [17]. It also is in line with the established hapten concept for most drug sensitivities, stating that small molecules only become immunogenic after being bound to a protein carrier [29]. We are aware of the fact that this study uses a mouse model, and murine sensitization to diclofenac may not be the same as human. It also only analyses diclofenac allergy induction, but we believe that the above described mechanism of hypoacidity-associated sensitization could be pertinent to all IgE-mediated NSAID hypersensitivities, if not even a wider array of drugs that form covalent bonds with proteins. Further studies into these questions are warranted.
If murine experiments are relevant to humans, the use of acid-suppressive drugs may be a mechanism of sensitization. However, as millions of patients throughout the world are taking both acid suppressants and NSAID, and the incidence of single drug reactions are quite low (1–3% in most population studies) [1], there would have to be other variables that single out the occasional reactors from the mainstream human experience. One of these variables could be the extent of gastric acid suppression upon first contact with the potential allergen. The urticaria patient already was under an ongoing proton pump inhibitor treatment when he first received diclofenac. This observation is also true for the very first patient in which we observed the acid suppression/allergy correlation: this Beluga caviar-allergic individual had also been under an ongoing treatment for dyspeptic disorders when he first ingested caviar [30]. It might thus be speculated that for sensitization to occur, gastric acid has to be effectively suppressed. Most patients who are prescribed acid-suppressive medication together with their NSAID will begin to take them simultaneously, and hypoacidity will not be complete in the first days of treatment. Other factors may be general genetic susceptibility to allergies, or different activity levels of enzymes that catalyse protein conjugation.
A last observation from this study relates to SPT for diagnostic purposes in patients. SPTs are often negative in drug hypersensitivity testing, and we suspected this to be due to the drug molecules being monovalent, and thus unable to cross-link receptor-bound antibodies on the surface of effector cells [31]. We therefore included the diclofenac–KLH conjugate that we had prepared for ELISA coating purposes (which carries multiple diclofenac molecules on one KLH carrier) in the SPT of the urticaria patient; and in the RBL assay with the murine sera and the murine skin tests. Indeed, we found that only the conjugate was able to elicit degranulation and mediator release in all these assays. A follow-up study, which examines the significance of this observation in routine clinical diagnostic procedures, is currently being conducted.
In conclusion, in this study we provide evidence in mice that gastric acid suppression can be a causative mechanism in the induction of true diclofenac allergy. This mechanism may also account for other IgE-mediated NSAID/drug hypersensitivities, in case the compound or a metabolite is covalently bound to a protein. For some patients, it may thus be risky to automatically combine NSAID prescriptions with acid-suppressive medication.
Acknowledgements
The authors would like to thank Prof. Dr Walter Jäger (Department of Clinical Pharmacy and Diagnostics, University of Vienna, Austria for the expert pharmacological counselling, Dr Fritz Andreae and Dr Werner Sallegger, piCHEM, Graz, Austria for consultance in conjugation chemistry, Harald Kurz for the excellent technical assistance, and PD Dr Dietmar Knopp (Technische Universität München, Munich, Germany) for his kind gift of the positive control anti-diclofenac antibody. This study was funded by grants L467-B05 and (in part) SFB-F1808-B13 of the Austrian Science Fund (FWF); I. Pali-Schöll is a recipient of the Hertha Firnberg stipend T283-B13 of the Austrian Science Fund; and E. Untersmayr of grant 11375 of the Austrian National Bank ‘Jubiläumsfonds’.
Footnotes
Cite this as: A. B. Riemer, S. Gruber, I. Pali-Schöll, T. Kinaciyan, E. Untersmayr and E. Jensen-Jarolim, Clinical & Experimental Allergy,2010 (40) 486–493.
References
- 1.Nettis E, Colanardi MC, Ferrannini A, Tursi A. Update on sensitivity to nonsteroidal antiinflammatory drugs. Curr Drug Targets Immune Endocr Metabol Disord. 2001;1:233–40. doi: 10.2174/1568008013341235. [DOI] [PubMed] [Google Scholar]
- 2.Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998;279:1200–5. doi: 10.1001/jama.279.15.1200. [DOI] [PubMed] [Google Scholar]
- 3.Sanchez-Borges M, Capriles-Hulett A, Caballero-Fonseca F. NSAID-induced urticaria and angioedema: a reappraisal of its clinical management. Am J Clin Dermatol. 2002;3:599–607. doi: 10.2165/00128071-200203090-00002. [DOI] [PubMed] [Google Scholar]
- 4.Leone R, Conforti A, Venegoni M, et al. Drug-induced anaphylaxis: case/non-case study based on an italian pharmacovigilance database. Drug Saf. 2005;28:547–56. doi: 10.2165/00002018-200528060-00006. [DOI] [PubMed] [Google Scholar]
- 5.Gamboa P, Sanz ML, Caballero MR, et al. The flow-cytometric determination of basophil activation induced by aspirin and other non-steroidal anti-inflammatory drugs (NSAIDs) is useful for in vitro diagnosis of the NSAID hypersensitivity syndrome. Clin Exp Allergy. 2004;34:1448–57. doi: 10.1111/j.1365-2222.2004.02050.x. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez-Trabado A, Camara-Hijon C, Ramos-Cantarino A, et al. Basophil activation test for the in vitro diagnosis of nonsteroidal anti-inflammatory drug hypersensitivity. Allergy Asthma Proc. 2008;29:241–9. doi: 10.2500/aap.2008.29.3114. [DOI] [PubMed] [Google Scholar]
- 7.Alkhawajah AM, Eifawal M, Mahmoud SF. Fatal anaphylactic reaction to diclofenac. Forensic Sci Int. 1993;60:107–10. doi: 10.1016/0379-0738(93)90098-u. [DOI] [PubMed] [Google Scholar]
- 8.Gupta D, Aggarwal AN, Aggarwal PN, Jindal SK. Near fatal asthma following ingestion of diclofenac sodium tablet. J Assoc Physicians India. 2000;48:258–9. [PubMed] [Google Scholar]
- 9.Hadar A, Holcberg G, Mazor M. Anaphylactic shock after diclofenac sodium (Voltaren) Harefuah. 2000;138:211–212. 270. [PubMed] [Google Scholar]
- 10.Ray M, Mitra S, Parmar V. Diclofenac induced fatal anaphylactic reaction. Indian Pediatr. 1999;36:1067–9. [PubMed] [Google Scholar]
- 11.Sen I, Mitra S, Gombar KK. Fatal anaphylactic reaction to oral diclofenac sodium. Can J Anaesth. 2001;48:421. doi: 10.1007/BF03014979. [DOI] [PubMed] [Google Scholar]
- 12.Riley RJ, Leeder JS. In vitro analysis of metabolic predisposition to drug hypersensitivity reactions. Clin Exp Immunol. 1995;99:1–6. doi: 10.1111/j.1365-2249.1995.tb03463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ware JA, Graf ML, Martin BM, Lustberg LR, Pohl LR. Immunochemical detection and identification of protein adducts of diclofenac in the small intestine of rats: possible role in allergic reactions. Chem Res Toxicol. 1998;11:164–71. doi: 10.1021/tx970182j. [DOI] [PubMed] [Google Scholar]
- 14.Tang W. The metabolism of diclofenac-enzymology and toxicology perspectives. Curr Drug Metab. 2003;4:319–29. doi: 10.2174/1389200033489398. [DOI] [PubMed] [Google Scholar]
- 15.Naisbitt DJ, Sanderson LS, Meng X, Stachulski AV, Clarke SE, Park BK. Investigation of the immunogenicity of diclofenac and diclofenac metabolites. Toxicol Lett. 2007;168:45–50. doi: 10.1016/j.toxlet.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 16.Untersmayr E, Bakos N, Schöll I, et al. Anti-ulcer drugs promote IgE formation toward dietary antigens in adult patients. FASEB J. 2005;19:656–8. doi: 10.1096/fj.04-3170fje. [DOI] [PubMed] [Google Scholar]
- 17.Untersmayr E, Jensen-Jarolim E. The effect of gastric digestion on food allergy. Curr Opin Allergy Clin Immunol. 2006;6:214–9. doi: 10.1097/01.all.0000225163.06016.93. [DOI] [PubMed] [Google Scholar]
- 18.Deng A, Himmelsbach M, Zhu QZ, et al. Residue analysis of the pharmaceutical diclofenac in different water types using ELISA and GC-MS. Environ Sci Technol. 2003;37:3422–9. doi: 10.1021/es0341945. [DOI] [PubMed] [Google Scholar]
- 19.Position paper: allergen standardization and skin tests. The European Academy of Allergology and Clinical Immunology. Allergy. 1993;4:48–82. [PubMed] [Google Scholar]
- 20.Untersmayr E, Schöll I, Swoboda I, et al. Antacid medication inhibits digestion of dietary proteins and causes food allergy: a fish allergy model in BALB/c mice. J Allergy Clin Immunol. 2003;112:616–23. doi: 10.1016/s0091-6749(03)01719-6. [DOI] [PubMed] [Google Scholar]
- 21.Diesner SC, Knittelfelder R, Krishnamurthy D, et al. Dose-dependent food allergy induction against ovalbumin under acid-suppression: a murine food allergy model. Immunol Lett. 2008;121:45–51. doi: 10.1016/j.imlet.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Budagovskaia VN, Voitko NE. Allergic reactions in patients with peptic ulcer; incidence of food and drug allergy. Vopr Pitan. 1984;3:30–3. [PubMed] [Google Scholar]
- 23.Schöll I, Untersmayr E, Bakos N, et al. Antiulcer drugs promote oral sensitization and hypersensitivity to hazelnut allergens in BALB/c mice and humans. Am J Clin Nutr. 2005;81:154–60. doi: 10.1093/ajcn/81.1.154. [DOI] [PubMed] [Google Scholar]
- 24.Schöll I, Ackermann U, Ozdemir C, et al. Anti-ulcer treatment during pregnancy induces food allergy in mouse mothers and a Th2-bias in their offspring. FASEB J. 2007;21:1264–70. doi: 10.1096/fj.06-7223com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brandtzaeg PE. Current understanding of gastrointestinal immunoregulation and its relation to food allergy. Ann NY Acad Sci. 2002;964:13–45. doi: 10.1111/j.1749-6632.2002.tb04131.x. [DOI] [PubMed] [Google Scholar]
- 26.Chan KK, Vyas KH, Brandt KD. In vitro protein binding of diclofenac sodium in plasma and synovial fluid. J Pharm Sci. 1987;76:105–8. doi: 10.1002/jps.2600760204. [DOI] [PubMed] [Google Scholar]
- 27.Atchison CR, West AB, Balakumaran A, et al. Drug enterocyte adducts: possible causal factor for diclofenac enteropathy in rats. Gastroenterology. 2000;119:1537–47. doi: 10.1053/gast.2000.20186. [DOI] [PubMed] [Google Scholar]
- 28.Kretz-Rommel A, Boelsterli UA. Diclofenac covalent protein binding is dependent on acyl glucuronide formation and is inversely related to P450-mediated acute cell injury in cultured rat hepatocytes. Toxicol Appl Pharmacol. 1993;120:155–61. doi: 10.1006/taap.1993.1097. [DOI] [PubMed] [Google Scholar]
- 29.Naisbitt DJ, Gordon SF, Pirmohamed M, Park BK. Immunological principles of adverse drug reactions: the initiation and propagation of immune responses elicited by drug treatment. Drug Saf. 2000;23:483–507. doi: 10.2165/00002018-200023060-00002. [DOI] [PubMed] [Google Scholar]
- 30.Untersmayr E, Focke M, Kinaciyan T, et al. Anaphylaxis to Russian Beluga caviar. J Allergy Clin Immunol. 2002;109:1034–5. doi: 10.1067/mai.2002.124893. [DOI] [PubMed] [Google Scholar]
- 31.Schöll I, Kalkura N, Shedziankova Y, et al. Dimerization of the major birch pollen allergen Bet v 1 is important for its in vivo IgE-cross-linking potential in mice. J Immunol. 2005;175:6645–50. doi: 10.4049/jimmunol.175.10.6645. [DOI] [PubMed] [Google Scholar]