

Abstract
The high frequency, recent origin, and geographic distribution of the CCR5-Δ32 deletion allele together indicate that it has been intensely selected in Europe. Although the allele confers resistance against HIV-1, HIV has not existed in the human population long enough to account for this selective pressure. The prevailing hypothesis is that the selective rise of CCR5-Δ32 to its current frequency can be attributed to bubonic plague. By using a population genetic framework that takes into account the temporal pattern and age-dependent nature of specific diseases, we find that smallpox is more consistent with this historical role.
Keywords: selection, epidemic, disease resistance, population genetics
The CCR5 chemokine receptor is fundamental to establishing HIV-1 infection. The receptor is exploited by HIV strains that predominate during the primary phase of infection to gain entry into immune system cells, including macrophages and CD4+ T cells (1-3). The CCR5-Δ32 deletion confers resistance to HIV-1 by preventing expression of the receptor on the cell surface (4-11). This allele provides almost complete resistance to HIV-1 in the homozygous state (4, 7, 12) and partial resistance with slower disease progression in the heterozygous state (4, 5, 8, 12). Interference with the expression of the CCR5 receptor also appears to have promise in the treatment of HIV (13).
The CCR5-Δ32 deletion allele is currently under intense selection in populations with a high prevalence of HIV-1 (14). However, HIV has not infected humans long enough to account for the selective rise of this resistance allele, the frequency of which is estimated at an average of ≈10% in European populations (4, 12, 15-18). The allele is virtually absent in African, Asian, Middle Eastern, and American Indian populations, suggesting a recent origin, specifically estimated at 700 years based on coalescent theory (16). The assertion that the high frequency of the variant in Europe arose through strong selection from bubonic plague (16) has become known as the classic example of the signature of historical selection on a clinically important locus. This hypothesis has been gaining widespread acceptance in both population genetic and medical literature, despite the absence of quantitative assessment. Here, we evaluate the feasibility of the hypothesis that bubonic plague provided the selective pressure that brought CCR5-Δ32 to high frequencies in Caucasian populations. Three lines of evidence indicate that the smallpox Variola major virus is a more likely candidate: predictions from a population genetic model, the geographical distribution of the allele, and the clinical effect of the deletion.
The enormous impact of bubonic plague on human mortality during the infamous Black Death pandemic led to the suggestion that plague had selected for CCR5-Δ32 (16). Indeed, from 1346 to 1352, bubonic plague killed an estimated 25-40% of Europeans of all ages (20-23). After the Black Death pandemic, plague struck in a series of less severe intermittent epidemics (20, 21, 24, 25). Although individual cities were sporadically hit hard by these intermittent epidemics, total mortality of plague epidemics amounted to only a few percent of Europe's overall population (20, 21, 25). In 1665 and 1666, there was a second pandemic, known as the “Great Plague,” that killed 15-20% of Europe's population (20, 24). After this pandemic, plague declined in Europe. France did not experience bubonic plague after 1722 and England was not afflicted by plague after 1667 (20). By 1750, bubonic plague had virtually disappeared from Europe (20, 26).
The intermittent nature of plague epidemics is caused, at least in part, by indirect transmission from a rodent reservoir via fleas (27). By comparison, smallpox was transmitted directly between humans, resulting in more continuous transmission. In populations where smallpox epidemics occurred frequently, children were the only immunologically naïve individuals, making smallpox a childhood disease. Indeed, smallpox infected the vast majority of Europeans before the age of 10 (25, 28) until its recent eradication. In addition, smallpox had a high case fatality rate of ≈30% (29, 30). Thus, the Black Death and Great Plague pandemics in Europe represent strong bouts of episodic selection, whereas ongoing smallpox epidemics represent weaker, but more continuous selection. Here, we investigate which disease is most likely to have generated the high frequency of CCR5-Δ32.
Model Description
We previously showed that age structuring of a host population can affect the selection of a resistance allele when the corresponding disease is responsible for significant mortality, particularly if disease virulence depends on host age and even more so if disease dynamics are episodic (31). Thus, here we use a population genetic framework that takes into account the temporal pattern and age-specific nature of different diseases. We divided the population into 55 age classes, each of 1 year. The annual probability of survival at age x is μx, which represents survival from background mortality caused by sources other than plague and smallpox. The number of female offspring born to a female of age x is mx. The survival (μx) and fecundity (mx) parameters were based on estimates from 19th-century Europe (32, 33). The number of females in an age class x at time t is nx,t. The systems of deterministic equations below were initiated at the stable age distribution determined by these parameters. Stochastic effects can be ignored in a population as large as the human population in Europe several hundred years ago (34).
We parameterized our model with upper estimates of plague mortality in Europe. Thus, we assumed that the Black Death (from 1346 to 1352) and Great Plague (from 1665 to 1666) wiped out 40% and 20% of Europe's population, respectively. Additionally, intermittent plague epidemics were assumed to kill 10% of the population every 10 years over a period of 400 years (21, 22, 24, 25). Thus, we assumed that plague did not disappear until 1750, an upper estimate of the duration of the plague era. Plague mortality was also assumed to affect all ages equally, consistent with historical accounts (22, 23). To assess selection generated by smallpox, we used the age distribution of smallpox burials in York between 1770 and 1812 (25) to parameterize smallpox mortality in the different age classes. The case fatality rate of smallpox was ≈30% (29, 30).
Our model incorporated the temporal patterns of smallpox and plague, respectively. Thus, the proportion of susceptible hosts within an age class x that is killed by disease, σx,t, depends on the temporal pattern of disease transmission dynamics. Consistent with disease time series data, we assumed that smallpox mortality peaked every 5 years, although disease mortality between these peaks was still 25% of that during the peak years (25, 28). In contrast, plague epidemics were more sporadic, with interepidemic periods of virtually no plague in most of Europe (21, 22, 24, 25).
Our diploid model is based on a single locus with two alleles: a common allele, at a frequency q, that confers susceptibility to disease, and a rare resistance allele, at a frequency p = 1 - q. We assumed that offspring were produced according to Hardy-Weinberg ratios based on p and q in each age class. Thus, the population dynamics of the system are defined by difference equations, where changes in number of susceptible homozygotes (z), heterozygotes (h), and resistant homozygotes (r) in age class 1 during a single time step are given by:
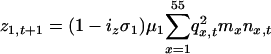 |
[1] |
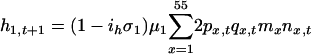 |
[2] |
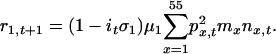 |
[3] |
Dynamics of z, h, and r in age classes 2-55 are given by:
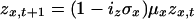 |
[4] |
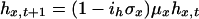 |
[5] |
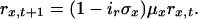 |
[6] |
The degree to which genetic resistance is conferred by each genotype is represented by iz, ih, and ir, respectively. This parameter decreases linearly with increasing protection against disease, such that a value of 1 corresponds to genetic susceptibility to infection and 0 represents full protection against disease mortality. We compared the case where the resistance allele is completely dominant with the case where the allele is incompletely dominant and has an additive effect on resistance. In the case of genetic dominance, iz = 1, while ih = ir = 0. In the case of additive resistance, iz = 1, ih = 0.5, and ir = 0.
We considered the rise in frequency of the resistance allele from an initial frequency, p0, of 5 × 10-5 over 700 years, an estimate of the age of CCR5-Δ32 (16). A lower frequency is equivalent to an earlier origin, which is quite possible given the 95% confidence intervals of this estimate (275-1,875 years) (16) and earlier estimates from other studies, including 1,400 (17) and 1,000-1,200 years (18). The crucial point for this comparative analysis is that p0 is the same for both bubonic plague and smallpox.
We calculated selection coefficients averaged over the total number of generations since the origin of the resistance allele (i.e., over a total of 28 generations, T). Each generation was ≈25 years. Thus, we define s as the average selection coefficient per generation acting on the resistance allele, calculated from the change in p:
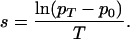 |
[7] |
The final frequency of the resistant allele reached by the end of the simulation is pr.
Results
Dominance. Even when we assumed that the resistance allele was dominant, we found that bubonic plague could not generate sufficient selective pressure to account for current CCR5-Δ32 frequencies, despite the periods of unprecedented disease mortality. The 400-year period of plague epidemics in Europe did not remove enough individuals of high reproductive potential to generate a sufficient selection coefficient. Our results suggest that plague could not even have driven the resistance allele to 1% during the period that it existed in Europe (Fig. 1). However, we found that the more continuous smallpox mortality that afflicted European children since the origin of the allele could have provided the necessary selective pressure to generate the rise of CCR5-Δ32 deletion to current frequencies of 10% (Fig. 2).
Fig. 1.

Change in p, the frequency of a dominant resistance allele, arising from plague mortality. The era of bubonic plague in Europe drives p to <0.8%.
Fig. 2.

Change in p, the frequency of a dominant resistance allele, generated by smallpox mortality. A total of 680 years of smallpox are required for p to reach 10%.
When the resistance allele was assumed to be dominant, s was calculated to be 0.18 for plague and 0.28 for smallpox. Although the difference in the magnitude of these selection coefficients is less than a factor of 2, it translates into a difference in allele frequency of >10-fold after 700 years of selection. This derives from Eq. 7, which demonstrates that the final frequency of the resistance allele increases exponentially with s.
Incomplete Dominance. In the case of incomplete dominance, we assumed that the resistance allele in the heterozygous state conferred 50% protection against disease mortality, relative to a susceptible homozygote, e.g., the case fatality rate for smallpox was 15% instead of the full 30%. Additionally, a resistant homozygote was completely protected against disease mortality. These conditions resulted in a selection coefficient (s) of 0.09 for plague and 0.17 for smallpox averaged over the total 700 years.
When incomplete dominance was assumed, 1,135 years of smallpox epidemics are required to reach a CCR5-Δ32 frequency of 10%. This is within the 95% confidence interval of the allele age estimate by Stephens et al. (275-1,875 years) (16) and within the range of other estimates of an earlier origin ≈1,400 years (17) or 1,000-1,200 years (18). In contrast, extending the age of the allele to 1,135 actually reduces the average selection coefficient for plague to 0.06 (in the additive case), because plague epidemics only affected Europe for a restricted period, which is not extended by an earlier origin of CCR5-Δ32.
Comparing Incomplete and Complete Dominance. The ratio of the selection coefficients for incomplete versus complete dominance was 50% for plague and 61% for smallpox, arising from reduction in heterozygote fitness by 50% for incomplete relative to complete dominance. That is, reduction in fitness of the heterozygote generates a decrease in the selection coefficient by a similar magnitude. Heterozygote fitness is important to the speed of the evolution because most of the selection on the resistance allele occurs via heterozygotes when the frequency of the resistance allele is initially very low.
Discussion
No other disease has killed such a large proportion of people in Europe in such a short period as the bubonic plague during the Black Death. Nonetheless, the cumulative number of deaths during the last 700 years from smallpox was greater than that from plague (26, 35). Additionally, smallpox disproportionately affected younger people. Consequently, a typical smallpox death removed greater reproductive potential than an average plague victim. While a number of other diseases, including measles, poliomyletis, whooping cough, rubella, scarlet fever, chicken pox, and influenza, have also been sources of mortality in Europe, typical case fatality rates of these diseases were only a few percent. Although a combination of diseases may constitute sufficient selection, our results suggest that smallpox alone can account for current frequencies of the HIV-1 resistance allele.
Our results suggest that an origin for the CCR5-Δ32 deletion of 700 years ago is consistent with the allele conferring dominant resistance against smallpox mortality. This would mean that both heterozygotes and resistant homozygotes are fully protected against smallpox mortality (although not necessarily infection). The 95% confidence interval of the 700-year estimate for the origin of the CCR5-Δ32 deletion (275-1,875 years) (16) indicates that the deletion may have arisen earlier. Indeed, another study used the frequency of microsatellite mutations to estimate an age of 1,400 (with 95% confidence interval of 375-3,675) (17). In addition, correlation between the frequencies of the CCR5-Δ32 deletion and European geography has been taken to suggest that the mutation occurred in the northern Europe 1,000-1,200 years ago before spreading in a continuous gradient down to the south and the Mediterranean coasts via Viking dispersal (18). Our results demonstrate that incomplete dominance with an additive effect of the CCR5-Δ32 deletion for smallpox resistance is most consistent with an age of ≈1,100 years. Under the assumption of incomplete dominance with an additive effect of the resistance allele, case fatality rates of heterozygotes were half those of a susceptible homozygote. That is, the intensity of selection is reduced if dominance is incomplete, requiring more generations of exposure to smallpox epidemics to reach an allele frequency of 10%. In contrast, we found that plague cannot generate sufficient selective pressure regardless of whether the resistance allele is dominant or has an additive effect.
Irrespective of the date at which CCR5-Δ32 originated, plague could not have provided sufficient selective pressure to drive CCR5-Δ32 to current frequencies. Plague epidemics only occurred in Europe during a limited window of ≈400 years. Therefore, an earlier origin for CCR5-Δ32 simply reduces the average selection coefficient caused by plague and does not increase the frequency of the allele. In contrast, historical evidence suggests that smallpox epidemics occurred in Europe >2,000 years ago (35). Indeed, an earlier origin for CCR5-Δ32 would give smallpox more generations during which to drive the allele to current frequencies. Thus, an earlier origin would further strengthen the case for smallpox (relative to plague) as the historical selective pressure of the resistance allele, because the era of heavy plague mortality in Europe started with the Black Death in 1346, making the time before that largely irrelevant for plague-mediated selection.
Although no ill effects associated with the CCR5-Δ32 deletion have been documented, the central role that chemokine receptors play in the inflammatory immune response makes it seem probable that the obliteration of CCR5 has negative fitness repercussions in the absence of a compensatory protective effect. Any such cost of resistance further argues against the hypothesis that bubonic plague selected for CCR5-Δ32. Plague has not been an important source of mortality for at least the last 250 years in Europe. In the absence of plague-mediated selection during this time, the postulated cost of resistance would be expected to drive down the frequency of CCR5-Δ32. In contrast, smallpox was not eradicated until 1978, coincidental with the start of the AIDS pandemic. Consequently, under the smallpox scenario, there has not been a long period without positive selection since the origin of CCR5-Δ32.
The geographic distribution of the resistance allele also implicates smallpox as the most likely historical source of selection on the CCR5-Δ32 deletion. Although bubonic plague was more intense within central Europe than in Scandinavian populations, the latter were hit particularly hard by intense smallpox epidemics (35). Indeed, the frequency of the CCR5-Δ32 variant forms a north-to-south cline from 0 to 14% across Eurasia (15-18, 36), with greatest prevalence in Scandinavian countries (17, 18, 36, 37).
HIV and poxviruses exhibit disease similarities in terms of viral infection of leukocytes associated with the dysfunction of cellular immunity. Moreover, poxviruses, like HIV, enter leukocytes by using chemokine receptors (19). Thus, it is biologically plausible that the obliteration of the CCR5 chemokine receptor could confer resistance against both HIV and smallpox. In contrast, clinical characterization of HIV and the plague-causing bacillus Yersinia pestis are quite distinct.
In summary, results from our age-structured model indicate that even the heavy mortality during the Black Death and Great Plague pandemics, combined with a series of intermittent epidemics, does not generate sufficient selective pressure to drive a resistance allele to 10% frequency. Instead, a disease with relatively high case fatality rates that persisted more continuously since the origin of the allele was likely to have been responsible responsible. Smallpox is such a disease. No single smallpox pandemic was as devastating as the Black Death, but the cumulative toll of human life caused by smallpox constituted an even stronger selection pressure than the episodic decimation of bubonic plague.
Acknowledgments
We are grateful for valuable discussions with Jeffrey Townsend and comments on the manuscript from Paul Harvey. A.P.G. was funded by a Miller Research Fellowship. M.S. was funded by National Institutes for Health Grant GM40282.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Alkhatib, G., Combadiere, C., Broder, C. C., Feng, Y., Kennedy, P. E., Murphy, P. M. & Berger, E. A. (1996) Science 272, 1955-1958. [DOI] [PubMed] [Google Scholar]
- 2.Dragic, T., Litwin, V., Allaway, G. P., Martin, S. R., Huang, Y., Nagashima, K. A., Cayanan, C., Maddon, P. J., Koup, R. A., Moore, J. P. & Paxton, W. A. (1996) Nature 381, 667-673. [DOI] [PubMed] [Google Scholar]
- 3.Deng, H., Liu, R., Ellmeier, W., Choe, S., Unutmaz, D., Burkhart, M., Di Marzio, P., Marmon, S., Sutton, R. E., Hill, C. M., et al. (1996) Nature 381, 661-666. [DOI] [PubMed] [Google Scholar]
- 4.Dean, M., Carrington, M., Winkler, C., Huttley, G. A., Smith, M. W., Allikmets, R., Goedert, J. J., Buchbinder, S. P., Vittinghoff, E., Gomperts, E., et al. (1996) Science 273, 1856-1862. [DOI] [PubMed] [Google Scholar]
- 5.Huang, Y., Paxton, W. A., Wolinsky, S. M., Neumann, A. U., Zhang, L., He, T., Kang, S., Ceradini, D., Zhanqun, J., Yazdanbakhsh, K., et al. (1996) Nat. Med. 2, 1240-1243. [DOI] [PubMed] [Google Scholar]
- 6.Biti, R., French, R., Young, J., Bennetts, B. & Stewart, G. (1997) Nat. Med. 3, 252-253. [DOI] [PubMed] [Google Scholar]
- 7.Liu, R., Paxton, W. A., Choe, S., Ceradini, D., Martin, S. R., Horuk, R., MacDonald, M. E., Stuhlmann, H., Koup, R. A. & Landau, N. R. (1996) Cell 86, 367-377. [DOI] [PubMed] [Google Scholar]
- 8.Michael, N. L., Chang, G., Louie, L. G., Mascola, J. R., Dondero, D., Brix, D. L. & Sheppard, H. W. (1997) Nat. Med. 3, 338-340. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien, T., Winkler, C., Dean, M., Nelson, J. A. E., Carrington, M., Michael, N. L. & White, G. C. I. (1997) Lancet 349, 1219. [DOI] [PubMed] [Google Scholar]
- 10.Theodorou, I., Meyer, L., Magierowska, M., Katlama, C., Rouzious, C. & Group, S. S. (1997) Lancet 349, 1219-1220. [PubMed] [Google Scholar]
- 11.Zimmerman, P. A., Buckler-White, A., Alkhatib, G., Spalding, T., Kubofcik, J., Combadiere, C., Weissman, D., Cohen, O., Rubbert, A., Lam, G., et al. (1997) Mol. Med. 3, 23-26. [PMC free article] [PubMed] [Google Scholar]
- 12.Samson, M., Libert, F., Doranz, B. J., Rucker, J., Liesnard, C., Farber, C.-M., Saragosti, S., Lapoumeroulie, C., Cognaux, J., Forceille, C., et al. (1996) Nature 382, 722-725. [DOI] [PubMed] [Google Scholar]
- 13.Qin, X. F., An, D. S., Chen, I. S. Y. & Baltimore, D. (2003) Proc. Natl. Acad. Sci. USA 100, 183-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schliekelman, P., Garner, C. & Slatkin, M. (2001) Nature 411, 545-546. [DOI] [PubMed] [Google Scholar]
- 15.Martinson, J. J., Chapman, N. H., Rees, D. C., Liu, Y.-T. & Clegg, J. B. (1997) Nat. Genet. 16, 100-102. [DOI] [PubMed] [Google Scholar]
- 16.Stephens, J. C., Reich, D. E., Goldstein, D. B., Shin, H. D., Smith, M. W., Carrington, M., Winkler, C., Huttley, G. A., Allikmets, R., Schriml, L., et al. (1998) Am. J. Hum. Genet. 62, 1507-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Libert, F., Cochaux, P., Beckman, G., Samson, M., Askenova, M., Cao, A., Czeizel, A., Claustres, M., de la Rua, C., Ferrari, M., et al. (1998) Hum. Mol. Genet. 7, 399-406. [DOI] [PubMed] [Google Scholar]
- 18.Lucotte, G. (2001) Hum. Immunol. 62, 933-936. [DOI] [PubMed] [Google Scholar]
- 19.Lalani, A. S., Masters, J., Zeng, W., Barrett, J., Pannu, R., Everett, H., Arendt, C. W. & McFadden, G. (1999) Science 286, 1968-1971. [DOI] [PubMed] [Google Scholar]
- 20.McEvedy, C. (1988) Sci. Am. 258, 118-123. [DOI] [PubMed] [Google Scholar]
- 21.Hatcher, J. (1977) Plague, Population, and the English Economy 1348-1530 (MacMillan, London).
- 22.Russell, J. C. (1948) British Mediaeval Population (University of New Mexico Press, Albuquerque).
- 23.Twigg, G. (1984) The Black Death: A Biological Reappraisal (Batsford, London).
- 24.Gottfried, R. S. (1983) The Black Death: Natural and Human Disaster in Medieval Europe (Free Press, New York).
- 25.Galley, C. (1998) The Demography of Early Modern Towns: York in the Sixteenth and Seventeenth Centuries (Liverpool Univ. Press, Liverpool, U.K.).
- 26.Giblin, J. C. (1995) When Plague Strikes: The Black Death, Smallpox, AIDS (Harper Trophy, New York).
- 27.Keeling, M. J. & Gilligan, C. A. (2000) Nature 407, 903-906. [DOI] [PubMed] [Google Scholar]
- 28.Anderson, R. M. & May, R. M. (1991) Infectious Diseases of Humans: Dynamics and Control (Oxford Univ. Press, Oxford).
- 29.Fenner, F., Henderson, D. A., Arita, I., Jezek, Z. & Ladnyi, I. D. (1988) Smallpox and Its Eradication (World Health Organization, Geneva).
- 30.Nathanson, N. (2001) in Fields Virology, ed. Howley, P. M. (Lippincott Williams & Wilkins, Philadelphia), Vol. 1, pp. 371-392. [Google Scholar]
- 31.Galvani, A. P. & Slatkin, M. W. (2003) Proc. R. Soc. London Ser. B., in press.
- 32.Preston, S. H., Heuveline, P. & Guillot, M. (2001) Demography: Measuring and Modeling Population Processes (Blackwell, Oxford, U.K.).
- 33.Coale, A. J. & Demeny, P. (1983) Regional Model Life Tables and Stable Populations (Academic, New York).
- 34.Ewens, W. J. (1979) Mathematical Population Genetics (Springer, New York).
- 35.Hopkins, D. R. (2002) The Greatest Killer in History: Smallpox (Univ. of Chicago Press, Chicago).
- 36.Lucotte, G. & Mercier, G. (1998) J. Acquired Immune Defic. Syndr. Hum. Retrovirol. 19, 174-177. [DOI] [PubMed] [Google Scholar]
- 37.Kalev, I., Mikelsaar, A. V., Beckman, L., Tasa, G. & Parlist, P. (2000) Eur. J. Epidemiol. 16, 1107-1109. [DOI] [PubMed] [Google Scholar]