

Abstract
Haematopoiesis is crucial for immunity because it results in the production of leucocytes. Bacterial and viral infections alter leucocyte production by promoting granulopoiesis or lymphopoiesis. Recent studies suggest that changes in leucocyte production may be caused by the effects of inflammatory responses on the differentiation of haematopoietic progenitors in the bone marrow. We investigated the mechanisms through which infection regulates the formation of bone marrow-derived dendritic cells (BMDCs) in vitro. We mimicked infection by stimulating developing cells with molecules associated with bacteria and viruses and with inactivated influenza viruses. We showed that toll-like receptor (TLR) ligands act as modulators of haematopoiesis, and that signalling through different TLRs results in differing effects on the production of BMDCs. We demonstrated that ligands for TLR3 and influenza viruses reduce the production of BMDCs, resulting in increased neutrophil numbers, and that ligands for TLR4 and TLR9 drive the production of plasmacytoid dendritic cells. Furthermore, there are distinct signalling mechanisms involved in these effects. Signalling pathways triggered by TLR4 and TLR9 involve MyD88 and are partially mediated by the cytokine tumour necrosis factor-α (TNF-α). Mechanisms activated by TLR3 were Tir-domain-containing adaptor-inducing interferon dependent. Haematopoietic modulation induced by inactivated influenza viruses was associated with the activation of an antiviral pathway mediated by type-1 interferons.
Keywords: dendritic cells, flow cytometry/fluorescence-activated cell sorter (FACS), haematopoiesis, toll receptors/toll-like receptors
Introduction
Toll-like receptors (TLRs) are a family of pattern recognition receptors (PRRs) which are involved in the recognition of pathogen-related molecular patterns (PAMPs) associated with bacteria, viruses and fungi. Although the importance of TLRs for innate and adaptive immunity has been well documented, recent studies have suggested that they may also have a role in tissue homeostasis. Rakoff-Nahoum et al.1 demonstrated that signalling through TLR4 plays a role in the maintenance of epithelial homeostasis in the gut. They found that commensal bacteria are recognized by TLRs under normal steady-state conditions and that this interaction plays a role in maintaining gut epithelial cells and protecting the epithelium from injury.
Inflammation has been shown to alter leucocyte production by reducing lymphopoiesis and promoting granulopoiesis in vivo; this bias towards granulopoiesis is generated by inflammation-induced tumour necrosis factor (TNF)-α initiating a reduction in the level of chemokines such as CXCL12.2,3 Borrow et al.4 demonstrated that influenza virus infection leads to a depletion of early B-lineage cells in the bone marrow. This depletion was mediated by a TNF receptor (TNFR)-dependent mechanism and involved the cytokines TNF-α and lymphotoxin (LT)-α. Taken together, these data show that infection and inflammation can influence the production of haematopoietic cells in vivo.
On ligand binding, TLRs initiate signalling cascades that result ultimately in the production of cytokines and chemokines. These signalling cascades are mediated by the adaptor molecules MyD88 (all TLRs excluding TLR3)5 and Tir-domain-containing adaptor-inducing interferon (TRIF) (TLR3 and TLR4).6 Although most studies have focussed on their expression on immune cells, TLRs are widely distributed and have been shown to be expressed on haematopoietic progenitor cells7 and their ligation has been demonstrated to influence haematopoiesis.8
More recently it has also been suggested that TLRs may have a role to play in directing haematopoiesis at the progenitor cell level. TLRs have been shown to be expressed on haematopoietic stem cells (HSCs) and early progenitors in the bone marrow. Stimulation with ligands for TLR2 and TLR4 induced proliferation and increased the production of mature progeny.7 Furthermore, stimulation of granulocyte/monocyte progenitor (GMP) and common myeloid progenitor (CMP) cultures with lipopolysaccharide (LPS) resulted in a loss of dependence on the growth factors macrophage colony-stimulating factor (M-CSF) and granulocyte–macrophage colony-stimulating factor (GM-CSF) for cell survival and differentiation in vitro. Ligands for TLR2 and TLR4 thus appear to act on haemopoietic progenitor cells to bias haemopoiesis towards monocyte and macrophage production. McGettrick and O'Neill8 reviewed this role for TLRs in haematopoiesis, suggesting that TLRs can supply initiation, survival and proliferation cues in a way similar to that of endogenous cytokines.
The cytokine TNF-α is a potential product of TLR signalling and has been found to affect the generation of dendritic cells (DCs) from haematopoietic progenitors in the bone marrow. Studies have shown that TNF-α, along with GM-CSF, is involved in the in vitro differentiation of CD34+ cells into cells displaying a DC phenotype,9 while interleukin (IL)-6 has been shown to suppress monocyte differentiation into DCs and to promote the development of macrophages.10 In addition there are also reports that IL-6, in conjunction with GM-CSF or Flt-3,11 can initiate in vivo DC differentiation from CD34+ progenitors. Type-1 interferons (IFN-αβ) are produced following TLR signalling initiated by viral PAMPs and in response to viral infection, and there is also evidence to suggest that IFN-αβ is involved in the generation and maturation of DCs. The capacity of type 1 IFNs to induce DC maturation has been well documented; they have been shown to increase the capacity of DCs to stimulate T lymphocytes through the upregulated expression of specific costimulatory molecules, including CD86.12–14 Reports have also suggested that DCs generated in vitro from monocyte precursors display enhanced maturation and function in response to IFN-α. Santini et al.14 showed that treatment of monocytes with IFN-α led to the rapid acquisition of high levels of CD40, CD80 and CD86, whereas Radvanyi et al.13 demonstrated that the addition of IFN-α to cultures of human peripheral blood mononuclear cells cultured with GM-CSF and TNF-α greatly increased the expression of CD86 on developing DCs.
The hypothesis of this study was that TLR-mediated signalling initiated by bacterial and viral products would lead to changes in mature leucocyte production from murine bone marrow in vitro. To examine this hypothesis we investigated the effect of TLR ligands (representing bacterial and viral PAMPs) and inactivated influenza viruses on the production of bone marrow-derived dendritic cells (BMDCs) in vitro.
Materials and methods
Animals
BALB/c mice were bred and maintained in the animal facility at the University of Liverpool. C57Bl/6 mice were purchased from Banting and Kingman Universal Ltd (North Humberside, UK) and maintained in the animal facility at the University of Liverpool. 129Ev mice and type 1 IFN receptor (IFNAR)-deficient mice on the 129 background were originally purchased from Banting and Kingman Universal Ltd and bred and maintained in the specific pathogen-free unit at the Institute for Animal Health (Compton, UK). Bone marrow was supplied by Dr P. Borrow. MyD88−/− mice on a C57Bl/6 background, TRIF−/− mice and their TRIF+/+ littermates were made available by Prof. R. K. Grencis (Faculty of Life Sciences, University of Manchester) with the generous permission of Prof. S. Akira (Department of Host Defense, Osaka University). All mice were used at > 8 weeks of age. All animal studies were carried out in accordance with local and UK Home Office regulations for animal care and use.
Media and reagents
RPMI-1640 medium (Sigma, Gillingham, UK) supplemented with 2 mm l-glutamine, 100 U/ml of penicillin, 100 U/ml of streptomycin, 5 × 10−5m 2-mercaptoethanol and 5% (v/v) fetal calf serum (Biosera, Ringmer, UK) was used throughout these experiments. Medium from P3-X63 cells transfected with the murine GM-CSF vector was used as a source of GM-CSF. The medium was titrated for potency to induce DC generation from murine bone marrow. The cells were originally made by Dr Brigitta Stockinger (Division of Molecular Immunology, National Institute for Medical Research) and were a gift from Prof. David Gray (Institute of Immunology and Infection, The University of Edinburgh). LPS from Escherichia coli, Poly I and Poly I:C were purchased from Sigma, and cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN) 1826 was purchased from MWG (London, UK). Influenza viruses Jap (A/Jap/1/57), PR8 (A/Puerto Rico/8/34) and the recombinant virus X31 (A/Aichi/2/68 × A/Puerto Rico/8/34), grown in the allantoic cavity of hen eggs, were a gift from Dr B. Thomas (Sir William Dunn School of Pathology, University of Oxford). Viruses were inactivated by exposure for 3-min to ultraviolet (UV) light from a 60 W source at a distance of 20 cm and treated with polymyxin-B (Sigma) to eliminate possible contamination with LPS. CpG ODN, LPS, Jap, X31 and PR8 were used at 1 μg/ml in all experiments; Poly I and Poly I:C were used at 25 μg/ml. These doses were selected as they have been shown to be effective at eliciting an innate immune response in vitro. Recombinant TNF-α was purchased from Hycult Biotechnology (Eindhoven, Netherlands) and neutralizing antibody to TNF-α was purchased from Sigma. Recombinant TNF-α was used at a concentration of 5 ng/ml. Monoclonal antibody (mAb) to TNF-α was titrated for potency in neutralizing recombinant TNF-α and subsequently used at a concentration of 20 μg/ml.
Cell culture
Bone marrow was harvested from mouse femurs by flushing through with complete RPMI medium and the cells were treated with 1 ml of 0·83% NH4Cl for 3 min to lyse the red blood cells. The cell suspension was plated out at 5 × 105 cells/ml (1 ml/well) in the wells of 24-well plates, in RPMI-1640 medium containing 20% (v/v) GM-CSF. Cultures were stimulated with Poly I, Poly I:C, LPS, CpG ODN, Jap, X31 or PR8, as indicated above, and incubated at 37° for 6 days. Experiments over a time course from 6 to 9 days were initially undertaken, and a culture period of 6 days was selected because the cultures demonstrated an effect that was not increased over longer time-periods of culture.
Flow cytometry
Surface antigen staining was performed using either directly conjugated mAb or biotinylated mAb followed by staining with phycoerythrin (PE) or Cy-chrome-conjugated streptavidin (both from BD Biosciences Pharmingen, Oxford, UK). The following antibody conjugates were used: mouse CD11c–PE or CD11c–biotin, Gr1–fluorescein isothiocyanate (FITC) or Gr1–PE (Caltag, Buckingham, UK), B220–allophycocyanin (APC), CD19–biotin (BD Biosciences Pharmingen) and PDCA–biotin (Miltenyi Bergisch Gladbach, Germany), and a mAb to major histocompatibility complex class II (MHCII) (KB6, a gift from Dr M. Parkhouse, Department of Infection and Immunity, Instituto Gulbenkian de Ciencia) was purified and coupled to FITC using standard methods. Fluorescence was analysed using a FACSCalibur flow cytometer (Becton Dickinson, Oxford, UK).
Morphological analysis
Cells were stained with haematoxylin and eosin, and morphological analysis was performed under a light microscope at 400× magnification. Cells were counted in five fields of view and the numbers of different cell types were assessed. Neutrophil-like cells were defined as cells with cytoplasm that stained neutral pink and a multilobed nucleus. Cells containing a large oval nucleus surrounded by a voluminous cytoplasm were classed as monocytes, and cells containing a large, dark nucleus, with little or no cytoplasm, were classed as lymphoid. Photographs were taken at 200× magnification using a Canon Powershot G3 mounted onto a Nikon TMS-F inverted microscope.
Results
Influenza A viruses and TLR ligands reduce generation of bone marrow-derived dendritic cells in vitro
To examine the effects of TLR ligands (representing bacterial and viral PAMPs) and inactivated influenza viruses on the generation of BMDCs, BALB/c bone marrow was cultured in the presence of GM-CSF, with or without the inactivated influenza A viruses Jap (H2N2), X31 (H3N1), or PR8 (H1N1), the TLR3 ligands Poly I and Poly I:C, the TLR4 ligand LPS or the TLR9 ligand CpG ODN. The production of BMDCs was determined by assessing the surface expression of CD11c and MHCII by flow cytometry. The results (Fig. 1a) showed that the addition of influenza virus to BMDC-generating cultures resulted in a marked reduction in the proportion of cells expressing the expected CD11c+/MHCII+phenotype. The addition of ligands for TLR3, TLR4 and TLR9 (Fig. 1b) also resulted in reduced production of cells of this phenotype. Analysis of cell numbers (Fig. 1c) revealed that cultures treated with LPS or CpG ODN had a greater number of total cells present after 6 days than were present in control cultures. By contrast, cultures to which Poly I or Poly I:C had been added showed reduced cell numbers. The increase in cell numbers induced by LPS and CpG ODN could mainly be attributed to a significant increase in the number of cells expressing a CD11clo/MHCIIlo phenotype (Fig. 1c), while the number of these cells present in cultures stimulated with influenza virus, Poly I or Poly I:C was reduced in comparison to unstimulated cultures (Fig. 1c). Therefore, we suggest that the reduction in cell numbers observed in response to influenza virus, Poly I or Poly I:C was caused by reduced BMDC production, whereas the increased cell number observed in response to LPS or CpG ODN was caused by the production of cells other than BMDCs.
Figure 1.
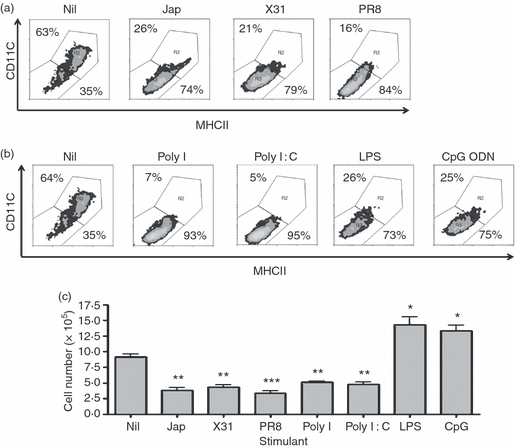
Influenza viruses and bacterial and viral ligands reduce the production of bone marrow-derived dendritic cells (DCs). BALB/c bone marrow cells (5 × 105 cells) were cultured with granulocyte–macrophage colony-stimulating factor (GM-CSF), with or without (a) Jap, X31 or PR8 or (b) Poly I, Poly I:C, lipopolysaccharide (LPS) or cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN) for 6 days. Surface markers were analysed by flow cytometry. The results are based on data for 10 000 gated events. The numbers shown in the density plots indicate the percentage of cells falling into two defined regions representing CD11c+ major histocompatibility complex class II (MHCII)+ cells (upper right) or CD11clo MHCIIlo cells (lower left). The data shown are the representative results of three similar experiments. (c) Cell numbers were determined by counting on a Neubauer haemocytometer. The counts represent the mean values of triplicate wells for three similar experiments. *P < 0·05, **P < 0·001 and ***P < 0·0001, determined using the Student's t-test, with respect to Nil.
Influenza A viruses and TLR ligands induce production of different cell types
To characterize the cells generated in response to stimulation of bone marrow cultures with influenza viruses or TLR ligands, bone marrow cells were cultured in the presence of GM-CSF, with or without stimulation, for 6 days and the cellular morphology was assessed by staining cells with haematoxylin and eosin. Differential counts were performed to assess the cell populations present. The results presented in Fig. 2 show that cells grown in the presence of GM-CSF alone were predominantly (50–60%) large cells displaying the described morphology of DCs. The proportion of cells of this type was clearly reduced with all tested stimuli. However, the predominant cell types produced depended on the nature of the added stimulus. In cultures treated with influenza viruses, Poly I or Poly I:C there was a marked increase in the number of cells with a neutrophil-like morphology (Fig. 2a). Conversely, in the presence of LPS or CpG ODN, most of the cells generated displayed a lymphoid morphology (Fig. 2b). Differential counts (Fig. 2c) clearly showed this change in the type of cells generated.
Figure 2.
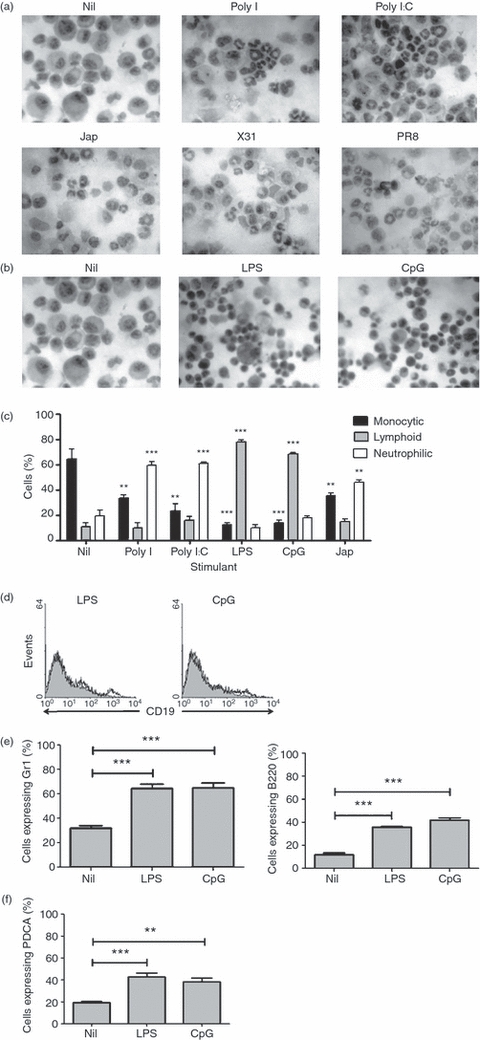
Influenza viruses and bacterial and viral agents induce the production of different cell types. BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with granulocyte–macrophage colony-stimulating factor (GM-CSF) in the presence or absence of (a) Jap, X31, PR8, or the toll-like receptor 3 (TLR3) ligands Poly I or Poly I:C and (b) lipopolysaccharide (LPS) or cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN). Cells were spun and fixed onto slides and then stained with haematoxylin and eosin. Photographs were taken using a Nikon light microscope at 200× magnification. Data shown are representative of three experiments. (c) Cell numbers were assessed under a Nikon light microscope at 400× magnification. Data shown represent the average cell counts from five fields of view for three similar experiments. **P < 0·001 and ***P < 0·0001, determined using the Student's t-test with respect to Nil. (d) BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with GM-CSF in the presence or absence of LPS or CpG ODN. CD19 expression was analysed by flow cytometry. Histograms represent unstimulated cultures (shaded histograms) and stimulated cultures (white histograms). Data shown are representative of two experiments. (e) CD11c, major histocompatibility complex class II (MHCII), Gr1 and B220 expression was analysed by flow cytometry. Cells were gated to low expression of CD11c and MHCII and the expression of Gr1 and B220 in this population was assessed. ***P < 0·0001, determined using the Student's t-test with respect to Nil. (f) CD11c, MHCII and PDCA expression was analysed using flow cytometry. Cells were gated to low expression of CD11c and MHCII and the expression of PDCA in this population was assessed. Results are based on 10 000 gated events. The data shown are representative of two similar experiments. **P < 0·001 and ***P < 0·0001, determined using the Student's t-test with respect to Nil.
LPS and CpG ODN induce the production of plasmacytoid DCs in vitro
The lymphoid appearance of cells generated in cultures containing LPS or CpG ODN suggested that they could belong to the B lineage, or might possibly be plasmacytoid DCs (pDCs). To explore the possibility of these cells belonging to the B lineage, we analysed the expression of the B-lineage marker CD19. The resulting data (Fig. 2d) showed that CD19 was not expressed by the cells generated under these conditions, excluding the possibility that they belong to the B-cell lineage.
pDCs are reported to have a morphology similar to that of lymphocytes and have been found to have a CD11clo/CD11b−/MHCIIlo/B220+/Gr1+ phenotype.15 We therefore examined the expression of these markers using flow cytometry. The data (Fig. 2e) showed that, as described above, cells generated in cultures containing LPS or CpG ODN predominantly express low levels of CD11c and MHCII. However, they were found to express both B220 and Gr1, markers not expressed by cells generated in untreated cultures. Cells from LPS- and CpG ODN-containing cultures were also analysed for the expression of the pDC marker PDCA. The majority of cells displaying the CD11clo/MHCIIlo phenotype also expressed PDCA (Fig. 2f). Taken together, these data suggest that under the influence of LPS and CpG ODN, progenitor cells preferentially differentiate towards the production of pDC and away from producing conventional DCs (cDCs).
MyD88 and TRIF are differentially required to modulate changes in BMDC production
Because we had shown that TLR ligands were able to modulate the differentiation of DCs from murine bone marrow in vitro, it seemed likely that signalling via TLRs would be implicated in these effects. It is well known that, with the exception of TLR3, TLRs use the adaptor molecule MyD88 to initiate signalling cascades;5 it was therefore important to establish whether MyD88 was required for the induction of changes in haematopoiesis observed in vitro. To assess this, bone marrow from MyD88+/+ and MyD88−/− mice was cultured in the presence of GM-CSF, with or without LPS or the influenza viruses Jap, X31 or PR8. The production of BMDC was determined by assessing the surface expression of CD11c and MHCII using flow cytometry. The results (Fig. 3a) show that the presence of LPS and the influenza viruses reduced the production of BMDCs in cultures of MyD88+/+ bone marrow, as observed previously for BALB/c bone marrow cells. The same reduction in BMDC production was observed when bone marrow cells from MyD88−/− mice were stimulated with influenza viruses. By contrast, when MyD88−/− bone marrow cells were stimulated with LPS, a large proportion of cells expressed a CD11c+/MHCII+ phenotype.
Figure 3.
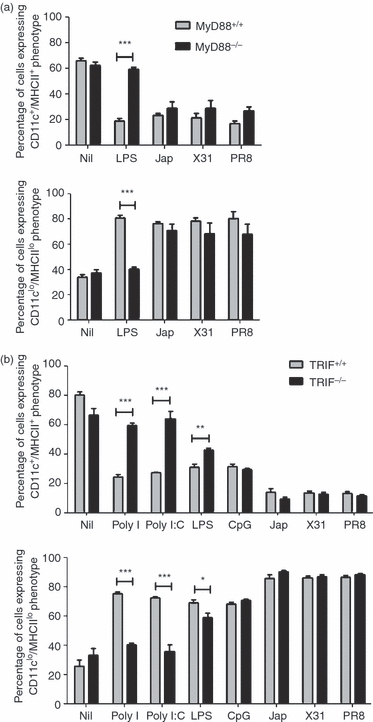
Effects on bone marrow-derived dendritic cells (BMDC) production are differentially dependent on MyD88 and Tir-domain-containing adaptor-inducing interferon (TRIF). (a) MyD88+/+ and MyD88−/− bone marrow cells (5 × 105 cells) were cultured for 6 days with granulocyte–macrophage colony-stimulating factor (GM-CSF) in the presence of lipopolysaccharide (LPS), Jap, X31 or PR8. ***P < 0·0001, determined using the Student's t-test. (b) TRIF+/+ and TRIF−/− bone marrow cells (5 × 105) were cultured for 6 days with GM-CSF in the presence or absence of Poly I, Poly I:C, LPS, cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN), Jap, X31 or PR8. Surface markers were analysed by flow cytometry. The results are based on data for 10 000 gated events. The data shown are representative of two similar experiments. *P < 0·05, **P < 0·001 and ***P < 0·0001, determined using the Student's t-test.
Signalling via TLR3 is known to involve a second adaptor molecule, TRIF, and TLR4 signalling can also involve this adaptor.6 The involvement of TRIF in the modulation of BMDC production was therefore investigated. To achieve this, bone marrow from TRIF-deficient mice and their wild-type littermates was cultured in the presence of GM-CSF, with or without Poly I, Poly I:C, LPS, CpG ODN, Jap, X31 or PR8, and the generation of CD11c+/MHCII+ BMDCs was monitored. The results (Fig. 3b) demonstrate that treatment of TRIF+/+ bone marrow cultures with these agents resulted in a dramatic reduction in the production of CD11c+/MHCII+ BMDCs, as observed for BALB/c bone marrow cells. A similar reduction in BMDC production was observed when TRIF−/− bone marrow cultures were stimulated with CpG ODN or influenza viruses, indicating that signalling induced by these ligands was independent of TRIF. However, when bone marrow from TRIF−/− mice was cultured with LPS, a reduction in the number of BMDCs was observed, although this was less pronounced than that observed in TRIF+/+ bone marrow cultures under similar conditions, suggesting that the effects of LPS were partially dependent on TRIF. By contrast, when TRIF-deficient bone marrow cultures were treated with Poly I or Poly I:C, CD11c+/MHCII+ BMDC production was at a level consistent with that observed in unstimulated cultures.
CpG ODN is a ligand for TLR9 and therefore was not expected to signal through TRIF because TLR9 signals exclusively via the MyD88-dependent pathway. As discussed above, TLR4, for which LPS is a ligand, utilizes either MyD88 or TRIF as adaptor molecules. In this case it appeared that the observed effects were diminished by deletion of either MyD88 or TRIF, but that the effect of MyD88 deletion was more marked. From this data it was concluded that signalling through both the MyD88-dependent and the MyD88-independent/TRIF-dependent pathways could initiate changes in lineage commitment in developing haematopoietic cells in vitro, which was dependent on the inducing ligand. The data from experiments involving influenza viruses demonstrated that, although they have been shown to activate MyD88-dependent signalling in B lymphocytes, in this instance their effects were mediated by a mechanism that was not dependent on either MyD88 or TRIF.
LPS requires TLR4 to induce changes in haematopoiesis
As the above evidence demonstrated that the effects of LPS on BMDC generation were dependent, in part, on both MyD88 and TRIF, and LPS has been shown to be a ligand for TLR4,16 which interacts with both adaptors, it was important to directly confirm the role of TLR4 in modulating the effects of LPS on BMDC production in vitro. To assess this, bone marrow cells from C57Bl/6 (TLR4+/+) and TLR4−/− mice were cultured in the presence of GM-CSF, with or without Poly I, Poly I:C, LPS or CpG, for 6 days. The production of BMDCs was assessed by monitoring the surface expression of CD11c and MHCII.
The results (Fig. 4) confirm a requirement for TLR4 in signalling initiated by LPS. Stimulation of TLR4+/+ bone marrow cultures with Poly I, Poly I:C, LPS or CpG resulted in a striking reduction in the production of CD11c+/MHCII+ BMDC, similar to that observed in BALB/c bone marrow cultures. A similar reduction in BMDC production was observed in cultures of TLR4-deficient bone marrow containing Poly I, Poly I:C or CpG. By contrast, TLR4-deficient bone marrow cultures containing LPS displayed a level of BMDC production comparable to that observed in unstimulated cultures.
Figure 4.
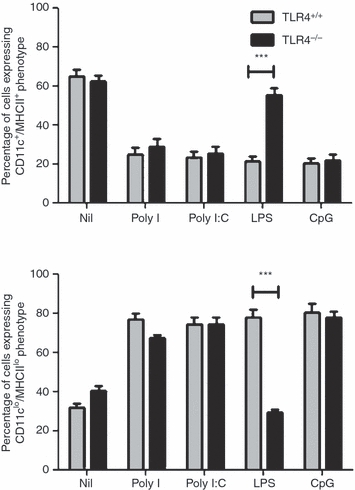
Lipopolysaccharide (LPS) requires toll-like receptor 4 (TLR4) to modulate the production of bone marrow-derived dendritic cells (BMDC). TLR4+/+ and TLR4−/− bone marrow cells (5 × 105 cells) were cultured for 6 days with granulocyte–macrophage colony-stimulating factor (GM-CSF) in the presence or absence of Poly I, Poly I:C, LPS or cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN). Surface markers were analysed by flow cytometry. The results are based on data for 10 000 gated events. The data shown are representative of two similar experiments. ***P < 0·0001, determined using the Student's t-test.
This evidence supports the previous findings that MyD88 and TRIF are involved in signalling downstream from LPS and confirms a role for TLR4 in regulating changes in BMDC production.
Influenza viruses activate a type 1 IFN response to induce changes in BMDC production
Type A influenza viruses have been shown to be strong inducers of type 1 IFNs,17 which are a major component of the antiviral response, inducing an antiviral state in uninfected cells18. It therefore seemed possible that the effects seen in our experiments in response to influenza A viruses could be mediated by type 1 IFNs. To investigate this we first examined the effects of influenza viruses on the generation of BMDCs in cultures of bone marrow cells from IFNAR−/− and IFNAR+/+ mice in the presence of GM-CSF. Cultures containing IFNAR+/+ bone marrow cells displayed reduced CD11c+/MHCII+ BMDC production in response to the addition of Jap, X31 or PR8 virus (Fig. 5a), as observed previously in BALB/c bone marrow cultures. By contrast, when IFNAR−/− bone marrow cells were cultured with influenza viruses, the proportion of CD11c+/MHCII+ BMDCs generated was similar to that observed in untreated cultures, suggesting that the IFNAR was required to mediate these effects.
Figure 5.
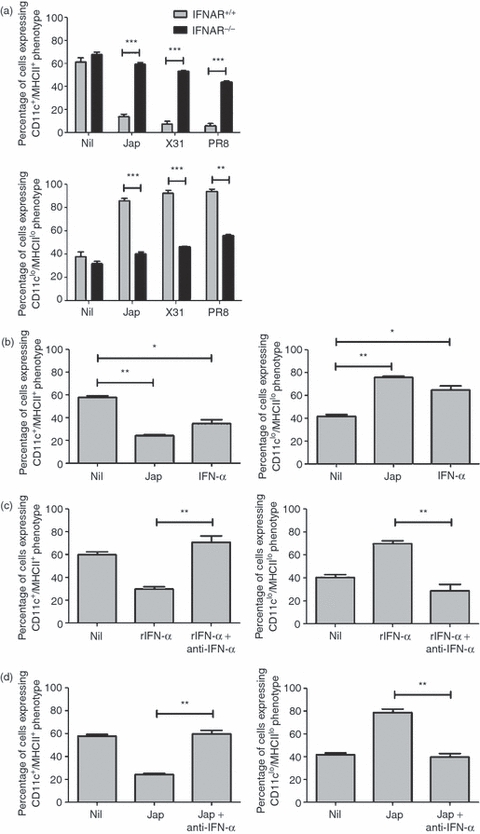
Influenza viruses require type 1 interferons (IFNs) to modulate the production of bone marrow-derived dendritic cells (BMDCs). (a) Type 1 IFN receptor (IFNAR)+/+ and IFNAR−/− bone marrow cells (5 × 105 cells) were cultured for 6 days with granulocyte–macrophage colony-stimulating factor (GM-CSF) in the presence or absence of Jap, X31 or PR8. **P < 0·001 and ***P < 0·0001, determined using the Student's t-test. (b) BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with GM-CSF in the presence of Jap or recombinant IFN-α. *P < 0·05 and **P < 0·001, determined using the Student's t-test with respect to Nil. (c) BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with GM-CSF in the presence of 100 pg/ml of recombinant IFN-α (rIFN-α) or in the presence of rIFN-α plus 200 pg/ml of anti-IFN-α. **P < 0·001, determined using the Student's t-test with respect to rIFN-α. (d) BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with GM-CSF in the presence of Jap or in the presence of Jap plus 200 pg/ml of anti-IFN-α. Surface markers were analysed by flow cytometry. The results are based on data for 10 000 gated events. Data shown are representative of two similar experiments. **P < 0·001, determined using the Student's t-test with respect to Jap.
To further investigate the role of type 1 IFN, BALB/c bone marrow was cultured in the presence of GM-CSF, with or without Jap or recombinant IFN-α. The data (Fig. 5b) demonstrated that cultures treated with IFN-α showed a reduction in BMDC production similar to that observed in cultures stimulated with Jap virus. We next examined the effects of neutralizing IFN. Cultures were treated with IFN-α in the presence or absence of neutralizing antibody to IFN-α. The results (Fig. 5c) showed that in the presence of neutralizing antibody the effects of IFN-α were negated and CD11c+/MHCII+ BMDC production was restored to levels corresponding to those observed in unstimulated cultures. To investigate whether the effects of influenza virus were mediated by IFN-α, cultures were treated with the Jap virus in the presence or absence of neutralizing anti-IFN-α (Fig. 5d). The addition of antibody clearly reversed the effects induced by the virus.
Taken together, this evidence clearly demonstrates a role for type 1 IFN, signalling through the IFNAR, in mediating responses to influenza viruses that lead to the observed changes in BMDC generation.
Signalling to induce changes in BMDC production in response to LPS and CpG ODN is partially dependent on TNF-α
As described above, ligands for TLRs 3, 4 and 9 were shown to initiate changes in haematopoiesis, inducing a marked reduction in BMDC production. In many cells the cytokine TNF-α is produced in response to MyD88-dependent TLR signalling and this cytokine has also been shown to inhibit haematopoiesis19. To examine a possible role for TNF in mediating the observed effects, recombinant TNF-α was added to bone marrow cultures containing GM-CSF. The results (Fig. 6a) show that the addition of TNF-α led to a reduction in the production of CD11c+/MHCII+ BMDC similar to that observed in cultures stimulated with influenza viruses or TLR ligands. The addition of a neutralizing antibody, anti-TNF-α (Fig. 6b), restored the production of CD11c+/MHCII+ BMDCs, confirming that TNF-α was responsible and that the antibody could abolish its effects. To assess whether TNF-α was mediating the effects of LPS and CpG ODN, bone marrow cells were cultured with GM-CSF and these stimuli in the presence or absence of the neutralizing antibody, anti-TNF-α. The resulting data (Fig. 6c) showed that anti-TNF-α had no effect on the modulation of BMDC production by LPS or CpG ODN. Data compiled from cell numbers (Fig. 6d) revealed that although there was little change in the proportion of cells displaying a CD11c+/MHCII+ phenotype, anti-TNF-α did appear to suppress the increase in cell number usually observed to occur in response to LPS and CpG ODN. To address the possibility that TNF-α production was sufficient to neutralize the effects of antibody added at the onset of cultures, bone marrow cells were cultured with GM-CSF in the presence or absence of LPS or CpG, and anti-TNF-α was added daily throughout the culture period. The data obtained (Fig. S1) were essentially identical to those shown in Fig. 6c when anti-TNF-α was added on day 0 only. Therefore, although TNF-α was capable of modulating BMDC production, it did not appear to be directly involved in the changes induced by ligands for TLR4 or TLR9, suggesting that other molecules were likely to be responsible.
Figure 6.
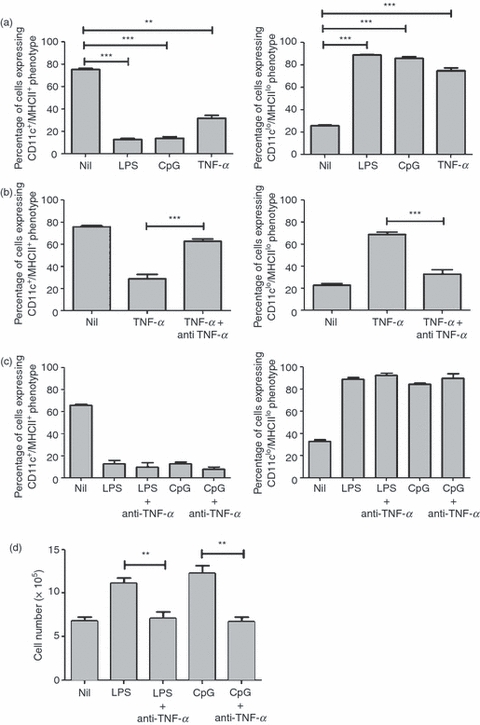
Signalling to modulate changes in the production of bone marrow-derived dendritic cells (BMDCs) in response to lipopolysaccharide (LPS) or cytosine–phosphate–guanosine (CpG) oligodeoxynucleotide (ODN) is partially dependent on tumour necrosis factor-α (TNF-α). BALB/c bone marrow cells (5 × 105 cells) were cultured for 6 days with granulocyte–macrophage colony-stimulating factor (GM-CSF) in the presence or absence of (a) LPS, CpG ODN or recombinant TNF-α. **P < 0·001 and ***P < 0·0001, determined using the Student's t-test with respect to Nil. (b) Recombinant TNF-α in the presence or absence of anti-TNF-α. ***P < 0·0001, determined using the Student's t-test with respect to TNF-α. (c) LPS or CpG ODN in the presence or absence of anti-TNF-α for 6 days. Surface markers were analysed using flow cytometry. The results are based on data for 10 000 gated events. Data shown are representative of two similar experiments. (d) Cell numbers were determined by counting on a Neubauer haemocytometer. The counts represent the mean values of triplicate wells for three similar experiments. **P < 0·001, determined using the Student's t-test with respect to LPS or CpG ODN, as appropriate.
Discussion
The aim of the present study was to investigate whether bacterial and viral products are able to affect the generation of DCs from BM in vitro. Our data suggested that inactivated influenza A viruses and the TLR3 ligands Poly I and Poly I:C reduce cellular proliferation in the cultures and cause a diminution in BMDC production. These data complement and extend those of previous studies, which suggest that Poly I:C inhibits granulocyte colony formation by bone marrow cells in vivo.20.
Viral infections result in the secretion of type 1 IFNs (IFN-αβ), which are crucial mediators of the antiviral response, and there is evidence to suggest that IFN-αβ inhibits the in vitro differentiation of DC from CD14+ precursors.21 Experiments with IFNAR-deficient bone marrow cells have shown that the IFNAR is required to modulate the changes in BMDC production induced by culture with influenza viruses. This role was confirmed by observations showing that recombinant IFN-α was able to replicate the effects, and neutralizing antibody to IFN-α was able to block them. These data are supported by other studies demonstrating an inhibitory effect of IFN-αβ on DC differentiation from monocyte-derived precursors,21 and by evidence which suggests that type 1 IFNs are cytotoxic for granulocytic progenitor cells in vitro.22 More recently, transient suppression of haematopoiesis in vivo has been shown to be caused by high levels of IFN-αβ.23 Taken together, this evidence suggests that IFN-αβ inhibits the differentiation of haematopoietic progenitors in a way that leads to reduced BMDC production.
In vivo infection with influenza virus induces a transient, but significant, loss of bone marrow B-lineage cells.24 A similar reduction in bone marrow B-lineage cells was observed during acute infection with lymphocytic choriomeningitis virus (LCMV) in mice.4 This bone marrow B-cell depletion accompanying acute influenza infection was found to be mediated by a mechanism involving TNF-α and LT-α. Interestingly, bone marrow B-cell depletion following infection with LCMV or influenza virus does not appear to be mediated by IFN-αβ.4 This contrasts with our data which show that in vitro BMDC depletion in response to influenza virus is IFN-αβ dependent, suggesting that there are differences in the signalling pathways activated in BMDC and bone marrow B-precursor cells following the recognition of influenza virus.
Although it is well documented that LPS and CpG ODN are potent activators of DCs in vitro and in vivo, their ability to modulate the development of DCs from immature precursors is relatively unknown. Studies have demonstrated that developing haematopoietic cells express TLRs7,25 and therefore would be expected to be sensitive to stimulation with their ligands. Our experiments indicate that the presence of TLR4 or TLR9 ligands (LPS and CpG ODN, respectively) during the generation of BMDCs in the presence of GM-CSF inhibits the differentiation of cells with the phenotype of BMDCs. This is in agreement with other studies which show that LPS or CpG ODN inhibit in vitro differentiation of DCs.26–28 Bartz et al.28 demonstrated that the generation of myeloid DCs from murine bone marrow was impaired by stimulation with LPS or CpG ODN. The cells generated exhibited reduced expression of CD11c and MHCII and a reduced ability to activate T lymphocytes. In humans, LPS stimulation has been shown to influence both early and late monocyte differentiation by blocking their ability to differentiate into DCs in vitro.25 The addition of LPS to cultures of monocytes containing GM-CSF and IL-4 reduced the cell yields, altered the morphology and phenotype of the cells generated, and compromised their capacity to present antigen.27,28 We did not explore the antigen-presentation function of the cells generated, but their phenotype, CD11clo/MHCIIlo, suggests a reduced antigen-presentation capacity because of the crucial role of MHCII in this process. Taken together, these findings confirm the inhibitory effects of LPS and CpG ODN stimulation during DC generation.
Our experiments indicate that TLR stimulation during the development of BMDCs in vitro inhibited the differentiation of CD11c+/MHCII+ cells while simultaneously enhancing the production of CD11clo/MHCIIlo cells. Experiments with knockout mice revealed that TLR4 (data not shown) and MyD88 were required to generate both of these effects. TLR4 and MyD88 have been shown to be expressed by developing haematopoietic cells,5 and this study demonstrated that MyD88-dependent signalling promoted myeloid lineage differentiation from HSC-enriched cultures stimulated with LPS in serum-free, stromal cell-free conditions. The differentiation potential of lymphoid progenitors has also been shown to be influenced by TLR9 ligation in a MyD88-dependent manner,29 and CpG ODN-induced inhibition of BMDC production is known to require TLR9.28
Although signalling via TLRs on granulocyte and macrophage progenitors has been shown to obviate the need for growth and differentiation factors to direct the differentiation of haematopoietic cells in vitro7 it was likely that the effects we observed were mediated by cytokines produced in response to TLR stimulation. This suggestion is supported by several reports which indicate that cytokines provide differentiation cues for developing haematopoietic cells.30–33 Tumour necrosis factors have been shown to inhibit haematopoiesis in vitro.18,34–36 We therefore investigated the role of TNF-α in modulating BMDC production. The data presented here show that although recombinant TNF-α was able to replicate the effects observed in response to LPS or CpG ODN, antibody to TNF-α was unable to reverse the effect of these ligands. However, anti-TNF-α did appear to suppress the proliferation of CD11clo/MHCIIlo cells that was observed in response to LPS or CpG ODN. TNF-α has previously been shown to reduce colony formation in bone marrow cultures containing stem cell factor and GM-CSF,36 and the suppressive effects of TNF-α on colony formation do not appear to be mediated by monocytes or T lymphocytes, both of which have been implicated in the regulation of granulopoiesis.37 However, TNF-α has also been demonstrated to provide positive cues for haematopoiesis in vivo.38,39 Recombinant TNF-α stimulates the production of G-CSF and GM-CSF by fibroblasts,38 and TNF-α enhances the proliferative effects of IL-3 and GM-CSF on CD34+ haematopoietic progenitor cells.39 This proliferative effect was revealed to be short term; after initial proliferation, TNF-α inhibited the in vivo differentiation of granulocytic cells while driving the development of maturing monocytic cells.40 More recently, Welner et al.28 demonstrated that reduced in vivo B-cell production from lymphoid precursors in response to TLR9 ligation was suppressed by TNF-α, while DC production observed under the same conditions was independent of TNF-α. Taken together, this evidence suggests that although TNF-α can affect the generation of BMDCs, other growth and differentiation factors may be required to generate all the effects observed in this study.
A major finding of the current study was the generation of CD11clo/MHCIIlo/B220+/Gr1+ cells in bone marrow cultures containing GM-CSF and stimulated with LPS or CpG ODN. These cells displayed a lymphoid morphology and also expressed PDCA, a marker thought only to be expressed on pDCs.41 This is in contrast to the results of a previous study,28 which showed that cells generated in response to LPS or CpG ODN in the presence of GM-CF in vivo displayed increased phagocytic capacity. However, another study29 demonstrated that lymphoid precursors generated pDCs and cDCs in response to in vivo stimulation with CpG ODN, suggesting that CpG ODN can provide differentiation cues that enhance the production of pDCs, in agreement with our findings.
Several cytokines have been shown to differentially promote the growth and differentiation of DC subsets. GM-CSF supports the differentiation of myeloid DCs from early haematopoietic progenitors and monocytes, whereas the FMS-like tyrosine kinase 3 ligand (Flt3L) is an essential factor for promoting the development of both human and murine cDCs and pDCs. Mice treated with murine Flt3L display a bias towards in vivo generation of pDCs and CD8+ cDCs,42,43 whereas the treatment of mice with GM-CSF enhances the in vivo production of CD8− cDCs.43 Although both GM-CSF and Flt3L play a crucial role in the development of cDCs, only Flt3 is required for the development of pDCs, and GM-CSF promotes the development of cDCs at the expense of Flt3L-mediated pDC development in vivo and in vitro.44,45 GM-CSF requires signal transducer and activator of transcription 5 (STAT5) to suppress Flt-3-driven pDC development.46 STAT5 activation by GM-CSF promptly reduces the expression of essential pDC-related genes in lin−Flt3+ haematopoietic progenitor cell cultures in the presence of Flt3L.46 By contrast, STAT3 has been shown to be essential for the proliferation of bone marrow progenitors in response to Flt3L,46 and pDC and cDC numbers were shown to be reduced in STAT3-deficient mice. However, STAT3 was not shown to be required for the commitment or development of pDCs, because STAT3-deficient pDCs responded to CpG ODN by producing IFN-α, a characteristic of differentiated pDCs. Taken together, these data reveal a suppressive role for STAT5 and a proliferative role for STAT3 in regulating the production of pDCs. Further to this, studies have demonstrated that TLR9 ligation by CpG ODN diminished STAT5 activation by IL-7,29 and LPS stimulation led to increased STAT3 activity in human immature monocyte-derived DCs.27 We therefore suggest that the mechanism driving pDC generation at the expense of BMDCs in response to stimulation with LPS or CpG ODN involves reduced GM-CSF-mediated signalling as a result of decreased STAT5 activity. As Flt3L has been shown to be produced by human bone marrow stromal cells,47 we also suggest that Flt3L is secreted in response to the stimuli and that the signal provided by Flt3L is boosted by increased STAT3 activity. This hypothesis could be tested by culturing bone marrow cells with GM-CSF in the presence or absence of LPS or CpG ODN and assessing the Flt3L-dependent production and phosphorylation of STAT3 and STAT5, and these experiments are under way.
Disclosures
The authors report no conflict of interest.
Supporting information
Additional Supporting information may be found in the online version of this article:
Figure S1. Daily addition of TNF-α does not reverse the effects of LPS or CpG on BMDC production. BALB/c bone marrow cells (5 × 105) were cultured for 6 days with GM–CSF in the presence or absence of LPS or CpG ODN in the presence or absence of daily additions of 20 mg/ml anti-TNFα for 6 days. Surface markers were analysed by flow cytometry. Results are based on data for 10 000 gated events. Data shown are representative of two similar experiments..
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–41. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Ueda Y, Yang KY, Foster SJ, Kondo M, Kelsoe G. Inflammation controls B lymphopoiesis by regulating chemokine CXC12 expression. J Exp Med. 2004;199:47–57. doi: 10.1084/jem.20031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ueda Y, Kondo M, Kelsoe G. Inflammation and the reciprocal production of granulocytes and lymphocytes in bone marrow. J Exp Med. 2005;199:47–57. doi: 10.1084/jem.20041419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borrow P, Hou S, Gloster S, Ashton M, Hyland L. Virus infection-associated bone marrow B cell depletion and impairment of humoral immunity to heterologous infection mediated by TNF-alpha/LT-alpha. Eur J Immunol. 2005;35:524–32. doi: 10.1002/eji.200425597. [DOI] [PubMed] [Google Scholar]
- 5.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signalling pathways. Mol Cell. 1998;2:253–8. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 6.Yamamoto M, Sato S, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signalling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 7.Nagai Y, Garrett KP, Ohta S, et al. Toll-like receptors on haematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24:801–12. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McGettrick AF, O'Neill LAJ. Toll-like receptors: key activators of leukocytes and regulator of haematopoiesis. Br J Haematol. 2007;139:185–93. doi: 10.1111/j.1365-2141.2007.06802.x. [DOI] [PubMed] [Google Scholar]
- 9.Caux C, Dezutter-Dambuyant C, Schmitt D, Banchereau J. GM-CSF and TNF-alpha co-operate in the generation of dendritic langerhans cells. Nature. 1992;360:258–61. doi: 10.1038/360258a0. [DOI] [PubMed] [Google Scholar]
- 10.Mitani H, Katayama N, Araki H, et al. Activity of interleukin 6 in the differentiation of monocytes to macrophages and dendritic cells. Br J Haematol. 2000;109:288–95. doi: 10.1046/j.1365-2141.2000.02020.x. [DOI] [PubMed] [Google Scholar]
- 11.Brasel KA, Maliszewski CR. Effects of IL-6 on Flt3 ligand-mediated murine DC development. Blood. 2000;96:33A–33A. [Google Scholar]
- 12.Basham T, Smith W, Lanier L, Morhenn V, Merigan T. Regulation of expression of class-II major histocompatibility antigens on human peripheral-blood monocytes and langerhans cells by interferon. Hum Immunol. 1984;10:83–93. doi: 10.1016/0198-8859(84)90075-2. [DOI] [PubMed] [Google Scholar]
- 13.Radvanyi LG, Banerjee A, Weir M, Messner H. Low levels of interferon-alpha induce CD86 (B7.2) expression and accelerates dendritic cell maturation from human peripheral blood mononuclear cells. Scand J Immunol. 1999;50:499–509. doi: 10.1046/j.1365-3083.1999.00625.x. [DOI] [PubMed] [Google Scholar]
- 14.Santini SM, Lapenta C, Logozzi M, et al. Type 1 interferon as a powerful adjuvant for monocyte-derived dendritic cell development and activity in vitro and Hu-PBL-SCID mice. J Exp Med. 2000;191:1777–88. doi: 10.1084/jem.191.10.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asselin-Paturel C, Boonstra A, Dalod M, et al. Mouse type 1 IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immun. 2001;2:1144–50. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- 16.Janeway CA, Medzhitov R. Innate immunity: Lipoproteins take their Toll on the host. Curr Biol. 1999;9:879–882. doi: 10.1016/s0960-9822(00)80073-1. [DOI] [PubMed] [Google Scholar]
- 17.Miller JL, Anders EM. Virus-cell interactions in the induction of type 1 interferon by influenza virus in mouse spleen cells. J Gen Virol. 2003;84:193–202. doi: 10.1099/vir.0.18590-0. [DOI] [PubMed] [Google Scholar]
- 18.Isaacs A, Lindenmann J. Virus interference 1 The interferon. Proc R Soc B. 1957;147:258–67. [Google Scholar]
- 19.Broxmeyer HE, Williams DE, Lu L, Cooper S, Anderson SL, Beyer GS, Hoffman R. The suppressive influences of human-tumour necrosis factor on bone-marrow haematopoietic progenitor cells from normal donors and patients with leukemia – synergism of tumor-necrosis factor and interferon-gamma. J Immunol. 1986;136:4487–95. [PubMed] [Google Scholar]
- 20.McNeill TA. Effect of synthetic double-stranded polyribonucleotides on haematopoietic colony-forming cells in-vivo. Immunology. 1971;21:741–50. [PMC free article] [PubMed] [Google Scholar]
- 21.McRae BL, Nagai T, Semnani RT, van Seventer JM, van Seventer GA. Interferon-alpha and –beta inhibit the in vitro differentiation of immunocompetent human dendritic cells from CD14(+) precursors. Blood. 2000;96:210–7. [PubMed] [Google Scholar]
- 22.Greenberg PL, Mosny SA. Cytotoxic effects of interferon in vitro on granulocytic progenitor cells. Cancer Res. 1977;37:1794–9. [PubMed] [Google Scholar]
- 23.Binder D, Fehr J, Hengartner H, Zinkernagel RM. Virus-induced transient bone marrow aplasia: major role of interferon-alpha/beta during acute infection with the noncytopathic lymphocytic choriomeningitis virus. J Exp Med. 1997;185:517–30. doi: 10.1084/jem.185.3.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sedger LM, Hou S, Osvath SR, Glaccum MB, Peschon JJ, van Rooijen N, Hyland L. Bone marrow B cell apoptosis during in vivo influenza virus infection requires TNF-alpha and lymphotoxin-alpha. J Immunol. 2002;169:6193–201. doi: 10.4049/jimmunol.169.11.6193. [DOI] [PubMed] [Google Scholar]
- 25.Kadowaki N, Ho S, Antonenko S, de Waal Malefyt R, Kastelein RA, Bazan F, Liu Y. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med. 2001;194:863–9. doi: 10.1084/jem.194.6.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palucka KA, Taquet N, Sanchez-Chapuis F, Gluckman JC. Lipopolysaccharide can block the potential of monocytes to differentiate into dendritic cells. J Leukoc Biol. 1999;65:232–40. doi: 10.1002/jlb.65.2.232. [DOI] [PubMed] [Google Scholar]
- 27.Xie J, Qian JF, Wang S, Freeman ME, III, Epstein J, Yi Q. Novel and detrimental effects of lipopolysaccharide on in vitro generation of immature dendritic cells: involvement of mitogen-activated protein kinase p38. J Immunol. 2003;171:4792–800. doi: 10.4049/jimmunol.171.9.4792. [DOI] [PubMed] [Google Scholar]
- 28.Bartz H, Avolas NM, Baetz A, Heeg K, Dalpke AH. Involvement of suppressors of cytokine signalling in toll-like receptor-mediated block of dendritic cell differentiation. Blood. 2006;108:4102–8. doi: 10.1182/blood-2006-03-008946. [DOI] [PubMed] [Google Scholar]
- 29.Welner RS, Pelayo R, Nagai Y, et al. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during hepes infection. Blood. 2008;112:3753–61. doi: 10.1182/blood-2008-04-151506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu J, Emerson SG. Haematopoietic cytokines, transcription factors and lineage commitment. Oncogene. 2002;21:3295–313. doi: 10.1038/sj.onc.1205318. [DOI] [PubMed] [Google Scholar]
- 31.Nagasawa T. Microenvironmental niches in the bone marrow in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6:107–16. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- 32.Acosta-Rodriguez EV, Merino MC, Montes CL, Motran CC, Gruppi A. Cytokines and chemokines shaping the B-cell compartment. Cytokine Growth Factor Rev. 2007;18:73–83. doi: 10.1016/j.cytogfr.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 33.Wu L, Liu YJ. Development of dendritic-cell lineages. Immunity. 2007;26:741–50. doi: 10.1016/j.immuni.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Peetre C, Gullberg U, Nilsson E, Olsson I. Effects of recombinant tumour-necrosis-factor on proliferation and differentiation of leukemic and normal haematopoietic-cells in vitro-relationship to cell surface receptor. J Clin Invest. 1986;78:1694–700. doi: 10.1172/JCI112764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murphy M, Perussia B, Trinchieri G. Effects of recombinant tumour necrosis factor, lymphotoxin, and immune interferon on proliferation and differentiation of enriched haematopoietic precursor cells. Exp Hematol. 1988;16:131–8. [PubMed] [Google Scholar]
- 36.Selleri C, Sato T, Anderson S, Young NS, Maciejewski P. Interferon-gamma and tumour-necrosis-factor-alpha suppress both early and late stages of haematopoiesis and induce programmed cell death. J Cell Physiol. 1995;165:538–46. doi: 10.1002/jcp.1041650312. [DOI] [PubMed] [Google Scholar]
- 37.Bagby GC, Rigas VD, Bennett RM, Vandenbark AA, Garewal HS. Interaction of lactoferrin, monocytes, and lymphocyte-T subsets in the regulation of steady-state granulopoiesis in vitro. J Clin Invest. 1981;68:56–63. doi: 10.1172/JCI110254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koeffler HP, Gasson J, Ranyard J, Souza L, Shepard M, Munker R. Recombinant human TNF-alpha stimulates production of granulocyte colony-stimulating factor. Blood. 1987;70:55–9. [PubMed] [Google Scholar]
- 39.Caux C, Saeland S, Favre C, Duvert V, Mannoni P, Banchereau J. Tumour necrosis factor-alpha strongly potentiates interleukin-3 and granulocyte-macrophage colony-stimulating factor-induced proliferation of human CD34+ haematopoietic progenitor cells. Blood. 1990;75:2292–8. [PubMed] [Google Scholar]
- 40.Caux C, Favre C, Saeland S, Duvert V, Durand I, Mannoni P, Manchereau J. Potentiation of early haematopoiesis by tumour-necrosis-factor-alpha is followed by inhibition of granulopoietic differentiation and proliferation. Blood. 1991;78:635–44. [PubMed] [Google Scholar]
- 41.Colonna M, Trinchieri G, Liu Y. Plasmacytoid dendritic cells in immunity. Nat Immun. 2004;5:1219–26. doi: 10.1038/ni1141. [DOI] [PubMed] [Google Scholar]
- 42.Bjorck P. Isolation and characterization of plasmacytoid dendritic cells from Flt3 ligand and granulocyte-macrophage colony-stimulating factor-treated mice. Blood. 2001;98:3520–6. doi: 10.1182/blood.v98.13.3520. [DOI] [PubMed] [Google Scholar]
- 43.O'Keeffe M, Hochrein H, Vremec D, Pooley J, Evans R, Woulfe S, Shortman K. Effects of administration of propenipoietin1, Flt3 ligand, granulocyte colony-stimulating factor, and pegylated granulocyte-macrophage colony-stimulating factor on dendritic cell subsets in mice. Blood. 2002;99:2122–30. doi: 10.1182/blood.v99.6.2122. [DOI] [PubMed] [Google Scholar]
- 44.Gilliet M, Boonstra A, Paturel C, et al. The development of murine plasmacytoid dendritic cell precursors is differentially regulated by FLT3-ligand and granulocyte/macrophage colony-stimulating factor. J Exp Med. 2002;195:953–8. doi: 10.1084/jem.20020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Naik SH, Corcoran LM, Wu L. Development of murine plasmacytoid dendritic cell subsets. Immunol Cell Biol. 2005;83:563–70. doi: 10.1111/j.1440-1711.2005.01390.x. [DOI] [PubMed] [Google Scholar]
- 46.Esashi E, Wang YH, Perng O, Qin X, Liu Y, Watowich SS. The signal transducer stat5 inhibits plasmacytoid dendritic cell development by suppressing transcription factor IRF8. Immunity. 2008;28:509–20. doi: 10.1016/j.immuni.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lisovsky M, Braun SE, Ge Y, et al. Flt3-ligand production by human bone marrow stromal cells. Leukemia. 1996;10:1012–8. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.