

Abstract
CD8 T cells that recognize cytomegalovirus (CMV) -encoded peptides can be readily detected by staining with human leucocyte antigen (HLA) –peptide tetramers. These cells are invariably highly differentiated effector memory cells with high avidity T-cell receptors (TCR). In this report we demonstrate an HLA-A*0201 restricted CMV-specific CD8 T-cell response (designated YVL) that represents several percent of the CD8 T-cell subset, yet fails to bind tetrameric major histocompatibility complex (MHC) ligands. However, these tetramer-negative cells are both phenotypically and functionally similar to other CMV-specific CD8 T cells. YVL peptide-specific CD8 T-cell clones were generated and found to be of high avidity in both cytotoxicity and interferon-γ (IFN-γ) assays, and comparable with other CMV peptide-specific CD8 T-cell clones. However, under conditions of CD8 blockade, the response was almost nullified even at very high ligand concentrations. This was also the case in IFN-γ experiments using peripheral blood mononuclear cells stimulated with peptide ex vivo. In contrast, all other CMV specificities (tetramer-positive) displayed minimal or only partial CD8 dependence. This suggests that YVL-specific responses depict a low-affinity TCR–MHC–peptide interaction, that is compensated by substantial CD8 involvement for functional purposes, yet cannot engage multivalent soluble ligands for ex vivo analysis. It is interesting that such a phenomenon is apparent in the face of a persistent virus infection such as CMV, where the responding cells represent an immunodominant response in that individual and may present a highly differentiated effector phenotype.
Keywords: affinity, cytomegalovirus, T cells
Introduction
Cell-mediated immunity is essential for protection against a number of viral pathogens. CD8 T cells recognize peptide antigens, derived from intracellular processing of viral proteins, which are presented by major histocompatibility complex (MHC) class I molecules at the surface of infected cells. Such recognition occurs via the T-cell receptor (TCR),1 followed by T-cell activation, which leads to effector functions that can result in direct killing of infected cells or inhibition of viral replication by the secretion of anti-viral cytokines such as interferon-γ (IFN-γ). Following reduction of the viral burden, CD8 T cells persist as long-lived memory cells that are able to respond rapidly upon secondary challenge.
Traditional methods of studying virus-specific immunity have relied on long-term culture in vitro to expand T cells to large numbers. These methods prove labour and time intensive and are now known to vastly underestimate the size of the immune response. The advent of MHC–peptide tetramers has enabled researchers to rapidly analyse complex T-cell populations ex vivo for rare T-cell specificities when combined with flow cytometry.2 This has permitted a detailed insight into the biology of immune responses against a number of human pathogens, such as herpesviruses, hepatitis viruses, human immunodeficiency virus 1 and Mycobacterium tuberculosis,3–9 as well as tumour-associated antigens10,11 and auto-antigens.12 Human cytomegalovirus (CMV) is a widespread genetically stable herpesvirus that rarely poses clinical problems in the immunocompetent host. Evidence from murine CMV infection and immunocompromised humans suggests that cellular immunity is critical for protection from CMV-associated disease.13,14 The CD8 subset makes a significant investment in recognition of CMV-derived peptides in humans.15–18 These have been visualized using human leucocyte antigen (HLA)–peptide tetramers and can number several (1–5%) per cent of the CD8 subset, approaching 50% of all CD8 T cells in some elderly virus carriers.19
The MHC tetramers serve as TCR ligands because of their multivalent nature, allowing the simultaneous engagement of several TCR molecules. This overcomes the problem of low-affinity TCR–MHC–peptide interactions.20 Our work has also shown that not all CMV-specific responses can be studied using these tetrameric reagents and this can be demonstrated in healthy virus carriers displaying no signs of clinical infection.16 We have observed this phenomenon in two CMV seropositive donors, with strong responses to the immediate early 1 protein (IE-1) -derived peptide YVLEETSVM (hereafter referred to as the YVL response) in an IFN-γ enzyme-linked immunospot (ELISPOT) assay. This tetramer-negative phenotype may not be unexpected if very rare T cells, such as naive T cells or T cells specific for self antigens, or under-differentiated T cells are in question.21,22 However, CD8 T cells specific for persistent viruses usually represent large memory cell populations with highly differentiated membrane phenotypes8 and the YVL response was of very high frequency.
We proceeded to dissect the functional requirements of this T-cell population. Analyses were performed using IFN-γ production ex vivo and cytotoxicity assays with peptide-specific T-cell clones, generated in vitro. The CD8 dependence of T-cell responses to peptide antigen was also investigated to determine the relative avidity/affinity. Finally we used a number of peptide variants in an attempt to improve the immunogenicity of the native peptide. Our data reveal this response to be of high functional avidity but dependent almost completely on the CD8 receptor for T-cell function.
Materials and methods
Donors
All donors were healthy adult volunteers from whom informed consent was obtained before sample donation. Ethical approval was in place for this work. Blood samples were collected by venepuncture using heparinized vacutainers, and processed immediately for isolation of peripheral blood mononuclear cells (PBMC) by standard density gradient centrifugation.
Peptides, tetramer synthesis and staining
Peptides encoded by CMV proteins IE-1 (QIKVRVDMV: residues 88–96, YVLEETSVM: residues 315–323, VLEETSVML: residues 316–324, YVLEETSVML, ELKRKMIYM: residues 199–207 and all YVL peptide variants), pp50 (VTEHDTLLY: residues 245–253) and pp65 (YSEHPTFTSQY: residues 363–373, NLVPMVATV: residues 495–503, TPRVTGGGAM: residues 417–426), were purchased commercially (Invitrogen, Paisley, UK). The HLA-A*0201 peptide tetramers were synthesized as described previously.2 Briefly, class I heavy chains (with transmembrane and cytoplasmic domains substituted by a BirA target sequence) and β2-microglobulin proteins were expressed in Escherichia coli and purified as inclusion bodies solubilized in 8 m urea. Heavy chain and β2-microglobulin were refolded around the appropriate peptide for 48 hr at 4° and then biotinylated using the enzyme BirA. Refolded complexes were purified by fast protein liquid chromatography using gel filtration and ion exchange columns (Amersham Pharmacia, Bucks, UK). The presence of biotinylated and correctly refolded HLA-A2–YVL complexes was confirmed using a w6/32 capture enzyme-linked immunosorbent assay. Tetrameric complexes were made by addition of phycoerythrin (PE)-conjugated streptavidin (Invitrogen) in a molar ratio of 1 : 4 to the biotinylated monomer over 2–3 days. Tetramer staining of PBMC was performed at 37° for 15 min followed by washing with phosphate-buffered saline and then counter-staining with anti-CD8 monoclonal antibody (mAb; Caltag Labs, San Francisco, CA) for 20 min at 4°.
Surface and cytoplasmic staining
For intracellular IFN-γ detection, PBMC were stimulated for either 6 hr or overnight with 5 μg/ml peptide at 37° in 5% CO2. After 2 hr of incubation brefeldin A (Sigma, Poole, UK) was added to a final concentration of 10 μg/ml. At the end of the incubation, cells were harvested and washed twice before surface staining with PE-conjugated anti-CD3 (Beckman-Coulter, High Wycombe, UK) and tricolour conjugated anti-CD8 mAb at 4°. For phenotyping studies, PBMC were stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD45RA, anti-CCR7, anti-CD27 or anti-CD28 mAb (all BD Biosciences, Oxford, UK). After another wash the cells were fixed and permeabilized according to the manufacturer's instructions using the Intraprep kit (Beckman-Coulter). Cells were then stained with a FITC-conjugated antibody against human IFN-γ or FITC-conjugated immunoglobulin G2a isotype control (BD Biosciences) and washed once more followed by analysis on a Coulter XL flow cytometer. For analysis of perforin expression, staining with FITC-conjugated anti-perforin mAb (BD Biosciences) was performed in combination with PE-conjugated IFN-γ mAb (BD Biosciences). Later analysis was performed using WinMDI version 2·8 software (downloaded from http://facs.scripps.edu/software.html).
Single cell sorting
The PBMC were stimulated overnight with peptide and then subjected to IFN-γ capture according to the manufacturer's protocol (Miltenyi Biotech, Bisley, UK). Then, IFN-γ-positive CD8-positive T cells were sorted into 96-well plates by seeding at 1 cell/well. Each well contained 1 × 105 autologous peptide-pulsed EBV transformed lymphoblastoid cell lines (LCL) and 1 × 106 allogeneic feeder cells in RPMI-1640 medium supplemented with 10% fetal calf serum, 1% human serum, 50 U/ml interleukin-2 (IL-2; Chiron, Emeryville, CA), and 5 ng/ml IL-7 (Peprotech, London, UK). After 14 days growing micro-cultures were expanded to 2-ml cultures and fed twice weekly with fresh medium. Re-stimulations were carried out after another 14 days with peptide-pulsed autologous LCL at a responder : stimulator ratio of 10 : 1. Clones were tested for specificity in either standard 5-hr chromium-release assays or by cytoplasmic IFN-γ staining after short 3-hr stimulation with cognate peptide/irrelevant peptide.
Cytotoxicity assays
Autologous LCL or fibroblasts were used as target cells. Targets were labelled with 51Cr for 1 hr before pulsing with 1 μg/ml peptide for an additional hour. For virus infections, fibroblasts were infected with CMV strains AD169 (G. Wilkinson, Cardiff, UK) overnight or RV798 (T. Jones, Wyeth Research Institute, Chazy, NY) for 36 hr, both at multiplicity of infection (MOI) of 5 : 1 before radioactive labelling. For determination of antigen specificity, cells were infected overnight with recombinant modified virus ankara (MVA) expressing either IE-1 or pp65, at an MOI of 5 : 1. Targets were washed twice, counted and then plated out at 2500 cells/well. T cells were added at various effector : target ratios in triplicate. After the 5-hr incubation at 37° in 5% CO2, supernatants were harvested and lysis was determined using a top-counter. Lysis values were calculated by subtracting spontaneous release from test release and dividing by (maximal release – spontaneous release). For CD8 blocking studies, T-cell clones were incubated with the anti-CD8 blocking mAb B9.11 (Beckman-Coulter) for 15 min before adding to the target cells.
Results
Detection of peptide-specific CD8 T cells by cytoplasmic IFN-γ staining
Our previous report16 showed that significant numbers of PBMC could make IFN-γ in response to two novel CMV IE-1 peptides. Interestingly we identified an unusual HLA-A*0201 restricted epitope (YVLEETSVM) characterized by a strong peptide-specific response but an inability to bind to our HLA-A*0201 YVL peptide tetramer. This was the case with multiple batches of tetramer, made from monomers refolded with YVL peptide commercially synthesized at high scales of purity. HLA-A*0201 YVL monomers were refolded successfully providing high yields with stability comparable to other monomers (see Supplementary material Fig. S1a,b). Figure 1(a) shows ELISPOT IFN-γ responses of PBMC from a CMV-seropositive donor after stimulation with different MHC class I restricted CMV-encoded peptides. Cells from this subject, Y26, made a number of responses of different magnitude which were mostly above the normal level of detection (250 spot-forming cells/well) unless a significantly lower peptide concentration (1 nm) was used. The ELISPOT assay is at best a semi-quantitative test and so a precise measurement of the frequency of responding cells was not derived, although we could conclude that the response to the YVL peptide was comparable to other immunodominant responses such as B8-QIK (IE-1 epitope), A1-VTE (pp50 epitope) and the A2-NLV (pp65 epitope). We then proceeded to measure the response by detection of cytoplasmic IFN-γ after short 6-hr stimulations. Figure 1(b) shows flow cytometric plots of responses against each peptide used in the ELISPOT assay with frequencies of responding CD8 T cells measured with more accuracy. Strikingly, this method revealed that over 5% of CD8 T cells were specific for the YVL peptide; this was the second highest response (the VTE response measured over 8%). The YVL peptide induced the largest IE-1 response and also exceeded the combined pp65 responses. Figure 1(c) shows that all of these responses could be visualized using specific MHC–peptide tetramers except for the A2-YVL response. As shown in Fig. 1(c), the frequency of tetramer binding cells identifies a larger number of cells than those producing IFN-γ, allowing us to speculate that the frequency of YVL-specific cells may also be well in excess of 5% of the CD8 subset. We have also detected this YVL-specific response in one other donor (O29) out of another 35 HLA-A2+ healthy virus carriers tested, albeit at lower frequencies (0·4% of CD8 T cells). This was comparable to that donor's measured frequency of NLV-specific CD8 T cells (0·5%), but once again the YVL tetramer did not bind PBMC of the donor (data not shown). Hence, YVL-specific CD8 T-cell responses are rare in the population, but when detected they are immunodominant yet fail to bind the appropriate MHC–peptide tetramer. In fact polyclonal cytotoxic T lymphocyte (CTL) lines, generated by peptide stimulation in vitro, did not stain our tetramer either. Such was the case using multiple batches of YVL tetramer, over a range of concentrations up to 2 mg/ml. This finding warranted further investigation.
Figure 1.
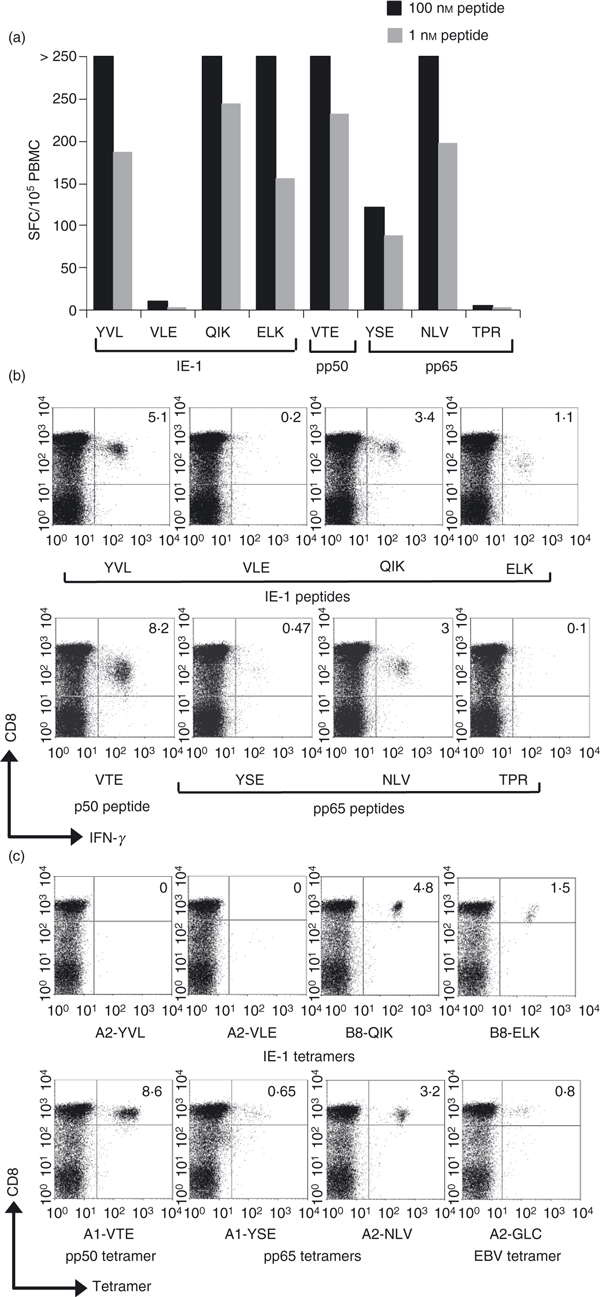
(a) ELISPOT interferon-γ (IFN-γ) responses against cytomegalovirus (CMV) -encoded peptides by donor Y26. Responses are expressed as spot-forming cells (SFC) per 100 000 peripheral blood mononuclear cells (PBMC) after subtracting the background responses against dumethyl sulphoxide (< 20 SFC/105 PBMC). HLA-B7-restricted TPR peptide was used as a further control. These responses were also visualized by flow cytometry (b) after staining for cytoplasmic IFN-γ induced by a 6-hr incubation with each peptide. The PBMC from donor Y04 were also incubated with major histocompatibility complex–peptide tetramers (c), representing CMV immediate early 1 protein (IE-1) epitopes (YVL, QIK, ELK), CMV early pp50 (VTE) and late pp65 epitopes (YSE and NLV) and an Epstein–Barr virus epitope (GLC). Cells were counter-stained with anti-CD8. Frequencies (%) of CD8 T cells binding the given tetramer are indicated in the upper right quadrant.
YVL-specific CD8 T cells display the classical effector memory phenotypic like other CMV epitope-specific CD8 T cells
It was unclear whether this ‘tetramer-negative’ (tetr–) response could be linked with other phenotypic differences between these and other, namely ‘tetramer-positive’ (tetr+) CD8 T cells. A possibility was that YVL-specific cells may have a deficiency in surface TCR expression, which would explain why the tetramer does not bind with sufficient avidity. This lack of TCR expression may be the result of recent activation or some unknown defect in the T cells. Testing for TCR expression on responding cells was problematic because TCR molecules are down-regulated after antigenic stimulation. Furthermore, YVL peptide-induced IFN-γ+ CD8 T cells displayed low levels of activation marker expression (CD38 and HLA-DR) which was in common with other CMV-specific responses that were tetr+ (data not shown). We also compared the levels of CD27, CD28, CD45RA, CCR7 and perforin between the different responses and observed (see example in Fig. 2) that YVL-specific cells displayed a highly differentiated effector memory phenotype (CD27lo CD28lo CCR7lo perforinhi). This resembled the phenotype associated with CMV-specific CD8 T cells in general but surprisingly in both donors YVL-specific cells were more differentiated (by lower expression of CD27 and CD28) and contained higher levels of intracellular perforin than NLV-specific cells. Furthermore, YVL-specific cells were also predominantly CD45RAhi, which again is associated with highly differentiated effector memory cells.23 This subset is also known to represent high-avidity T-cell responses that can sense very low levels of antigen. Indeed we observed that the YVL peptide could induce IFN-γ production by CD8 T cells at very low concentrations; 50% maximal response was induced at concentrations as low as 0·01 nm of peptide. These levels of sensitivity were comparable to responses against the NLV peptide (Fig. 3).
Figure 2.
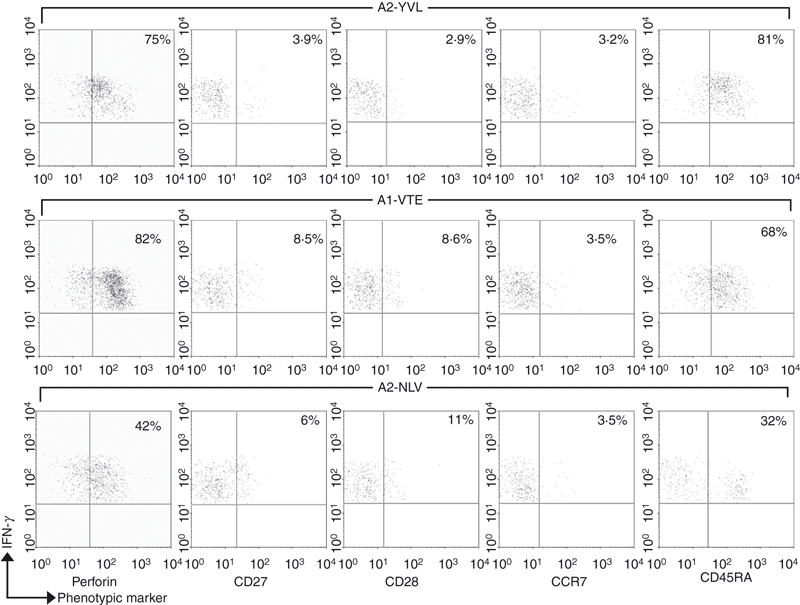
YVL-specific CD8 T cells display an effector memory phenotype like other cytomegalovirus (CMV) -specific CD8 T cells. Donor Y26 peripheral blood mononuclear cells were stimulated for 6 hr with synthetic peptides and surface stained for cell surface markers followed by cytoplasmic staining with anti-interferon-γ monoclonal antibody (IFN-γ mAb). Perforin expression was also determined by cytoplasmic staining. IFN-γ-positive CD8 T cells were gated and analysed for membrane phenotype. Values in the upper right quadrant indicate the frequency of IFN-γ-producing cells that express the given marker. Data are shown for three different responses: YVL (tetr– response) VTE (tetr+ response) and NLV (tetr+ response).
Figure 3.
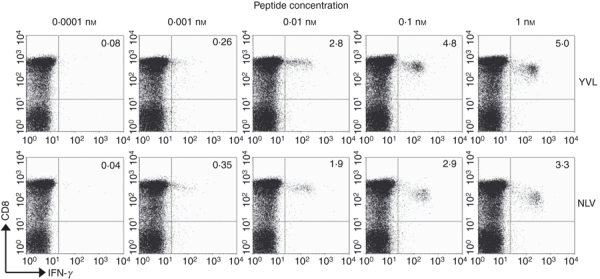
YVL-specific CD8 T cells represent a high avidity response. Peripheral blood mononuclear cells were stimulated with serial dilutions of synthetic peptides (YVL or NLV) ranging from 1 nm to 0·0001 nm for 6 hr at 37°. After surface staining and cytoplasmic staining, responses were visualized by flow cytometry. Values shown in upper right quadrants indicate the frequency of CD8 T cells that produce interferon-γ in response to specific peptide. Data shown are from donor Y26.
Cloning of peptide-specific CD8 T cells by IFN-γ capture
For more detailed analysis of these non-tetramer binding cells we sought to expand them in vitro and generate peptide-specific CTL clones. As the YVL tetramer did not bind cognate CD8 T cells, we used the IFN-γ capture method to sort for YVL-specific CD8 T cells. Hence, IFN-γ-secreting cells were sorted into 96-well plates and expanded as single-cell clones. After outgrowth, clones were tested for specificity by a chromium-release assay and the majority recognized YVL peptide-pulsed autologous LCL targets and HLA-A2-matched allogeneic LCL targets to confirm HLA-A2 restriction. The YVL-specific T-cell clones did not recognize the two internal octomeric peptides, which are potentially also HLA-A2-binding epitopes (see Supplementary material Fig. S2). Typical of other IE-1-specific CD8 clones, YVL-specific clones displayed poor recognition of CMV-infected (AD169 strain) fibroblasts but strong recognition of targets infected with recombinant CMV (RV798 strain lacking US2-US11 region) (Fig. 4a). In addition we confirmed recognition of processed antigen using MVA-IE-1-infected fibroblast targets (Fig. 4a). These YVL-specific clones were also incubated with YVL tetramer to verify the lack of tetramer staining, and results confirmed all 14 clones to be tetr–. This was the case for clones from both donors and suggested that the interaction between the TCR and our tetramer might be very weak because of an intrinsic low-affinity of the TCR binding to this HLA-A*0201 YVL complex. This was also the case using an HLA-A2 tetramer incorporating a natural variant YILEETSVM,24 suggesting that the lack of binding was not the result of an incorrect peptide sequence. It was possible that these T-cell clones expressed low levels of surface TCR, which fell below the threshold of molecules required for ligand engagement. However, high levels of staining with a pan-TCR mAb (Fig. 4a–c) ruled out this possibility.
Figure 4.
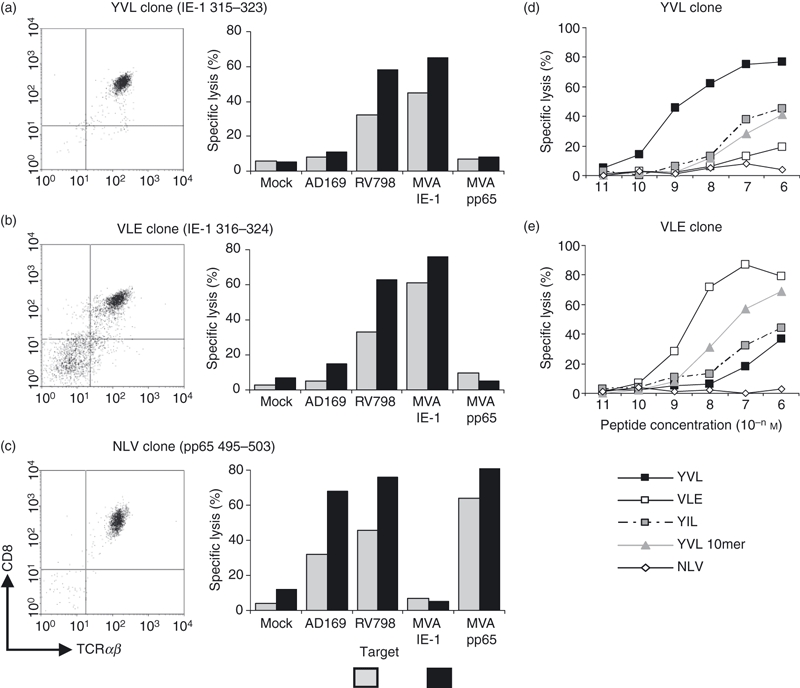
Recognition of processed antigen exquisite specificity of YVL-specific CD8 T cells. T-cell clones specific for different cytomegalovirus (CMV) peptides were confirmed as CD8+ with comparable levels of surface T-cell receptor (TCR) -αβ, for both tetr– clones (YVL) and tetr+ clones (VLE and NLV). YVL-specific CD8 T-cell clones were tested for lysis of virus-infected autologous fibroblasts (a) using AD169 and RV798 (US2-US11-deficient) strains of CMV. Antigen specificity was confirmed by co-incubation of T-cell clones with autologous targets infected with recombinant MVA expressing immediate early 1 protein (IE-1) or pp65. VLE-specific CD8 T-cell clones from donor Y14 (b) and NLV-specific CD8 T-cell clones from donor Y26 (c) were tested as human leucocyte antigen (HLA)-A2-restricted CMV-specific clones that stained positively with the appropriate major histocompatibility complex–peptide tetramer. YVL-specific clones were also tested in peptide titration assays for cross-recognition (d) of the VLE peptide (offset from YVL by one amino acid) or the variant peptide YIL (V→I variant) and this test was also performed with VLE-specific clones (e) The NLV peptide was used as an HLA-A2-restricted control peptide for both sets of clones. Effector : target ratios were 4 : 1 in all assays shown. Results show the mean of assays performed in triplicate (the standard deviation in the assay was < 5% of maximal lysis).
Interestingly, YVL-specific clones recognized the peptide VLEETSVML (designated VLE), only at very high concentrations (Fig. 4d). This is interesting because the VLE peptide is only offset from the YVL epitope by a single amino acid and is also an HLA-A*0201-binding peptide that is strongly immunogenic.16 Use of the decameric peptide that encompasses both nonameric peptides also showed recognition, but again only at high peptide concentrations. This inability to recognize an almost identical peptide implied that there were significant differences in the conformation of peptide recognition between YVL- and VLE-specific CTL.
As controls, we also generated HLA-A*0201-restricted NLV-specific and HLA-B*0801-restricted ELK-specific clones from the same donor and HLA-A*0201-restricted NLV- and VLE-specific clones from another donor using IFN-γ as the method for selection. These clones all displayed specific recognition of peptide-loaded target cells and stained tetramer at high levels of mean fluorescence, confirming the YVL response to be different from all other detected CMV epitope-specific responses.
Functional avidity of peptide-specific CD8 T cells
To indirectly estimate the relative affinity of these tetr– cells, YVL-peptide-specific CTL clones were tested for their ability to lyse target cells loaded with varying concentrations of peptide. Figure 5 shows that YVL-specific CTL clones do in fact lyse target cells quite well at low peptide concentrations compared with clones from the same donor that recognize another CMV peptide (NLVPMVATV) and stain with appropriate tetramer. Although the NLV-specific clones (tetr+) did lyse at lower peptide concentrations than YVL-specific clones (tetr–) this represented a < 10-fold difference in peptide concentration (respectively). Furthermore, the concentration at which YVL clones showed minimal killing was as strong as some tetr+ NLV-specific clones from other donors. This suggested that the observed difference was not significant and was not an adequate explanation for the lack of tetramer binding.
Figure 5.
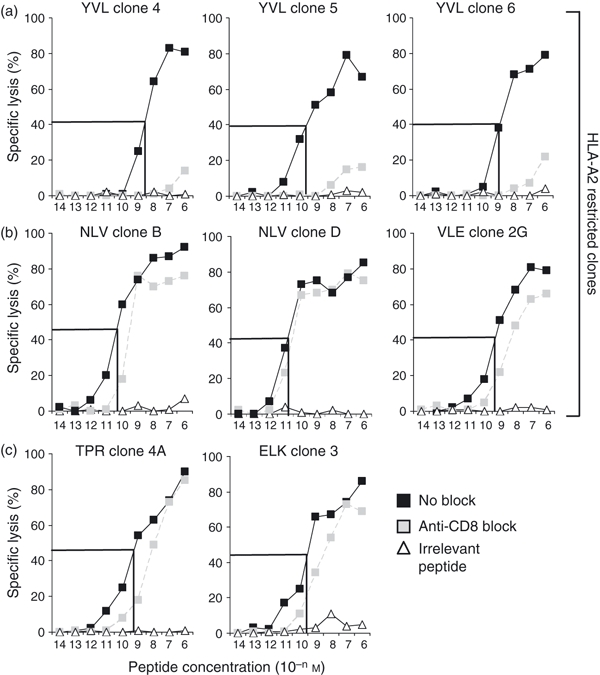
Extreme levels of CD8 dependence by YVL-specific clones. CD8 dependence of T-cell clones specific for (a) YVL, (b) NLV and VLE and (c) TPR and ELK peptide epitopes was determined in chromium-release assays. YVL clones 4 and 5, NLV clone B and ELK clone 3 were all derived from donor Y26. YVL clone 6 and NLV clone D was derived from donor O29. VLE clones were donor Y14 derived and TPR clones were donor Y05 derived. Targets were autologous lymphoblastoid cell lines pulsed with serial dilutions of cognate peptide for each set of clones shown (black symbols). For CD8 blocking, T-cell clones were coated with B911 anti-CD8 monoclonal antibody before incubation with peptide-loaded targets (grey symbols). Irrelevant peptides restricted by the same HLA allele were used as controls (white symbols); NLV was used as control peptide for YVL and VLE clones, the respiratory syncytial virus-encoded NPK epitope was used as control for HLA-B7-restricted TPR clones the Epstein–Barr virus-encoded RAK peptide was used as control peptide for HLA-B8-restricted ELK clones. Effector : target ratios were 2 : 1. The overall functional avidity is highlighted by marking the concentration of peptide that mediates 50% of optimal response. Mean values from triplicate wells are shown (the standard deviation was < 5% of maximal lysis).
In parallel with these experiments, we also performed CD8 blocking studies, using an anti-CD8 blocking mAb. This would ascertain the degree of CD8 dependence involved for each clone to respond. Figure 5 shows that YVL-specific clones from donors Y26 and O29 were highly CD8 dependent. Only at the highest peptide concentration was there significant lysis when clones were incubated with anti-CD8. Below this there was an almost complete loss of sensitivity to the target cells. This property was common to all six YVL-specific clones tested for CD8 dependence (four from Y26 and two from O29). Conversely, with all the other clones, such as the NLV-specific CTL from the same donors, co-incubation with the blocking mAb had little effect at even very low peptide concentrations. The general pattern was that tetr+ clones exhibited far less CD8 dependence.
Immunogenicity of peptide variants
We then sought to evaluate whether specific alterations to the peptide sequence would increase the strength of the TCR–MHC–peptide interaction. This may be achieved by improving binding to HLA-A*0201 by changing the peptide anchors at positions 2 and/or 9. Alternatively, alterations at positions 3, 4, 5 and 7, (TCR contact residues) could improve the epitope as shown elsewhere for tumour antigens.25 Initial experiments were aimed at improving the peptide anchors as the YVL epitope lacked the usual common anchors at position 2 (leucine) and 9 (valine). However, substitution at either or both anchor positions caused a dramatic reduction in the ability to sensitize YVL-specific T-cell clones (Fig. 6a,b). This loss of activity was also observed in reduced IFN-γ staining of PBMC after stimulation with variant peptides (data not shown). This suggested that this TCR–MHC–peptide interaction was very sensitive, even to changes not directly affecting TCR contact points.
Figure 6.
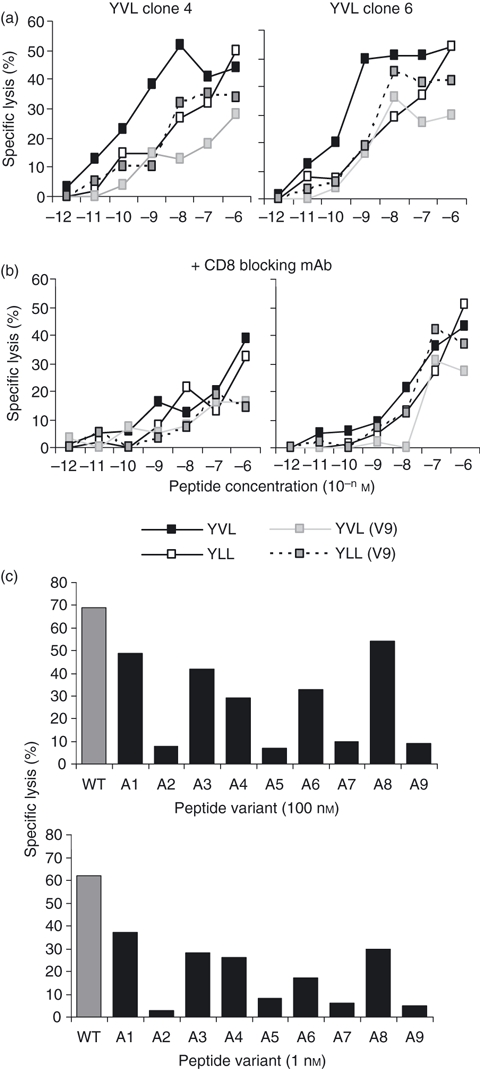
CD8 T-cell clones specific for YVL peptide were tested for the ability to recognize altered peptide ligands. These included YVL peptide analogues with changed human leucocyte antigen (HLA) -A2-binding anchor residues at position 2 (V→L, designated YLL) and/or position 9 (M→V, designated V9). Assays were performed either in the absence (a) or presence (b) of CD8 blocking monoclonal antibodies. Representative data from two clones are shown. Effector : target ratios used were 2 : 1 in these assays. (c) Second, YVL-specific clones were screened against different concentrations of YVL peptide variants with single alanine substitutions at positions 1–9 (A1–A9). The unaltered wild-type (WT) YVL peptide is indicated as grey bars. The data shown are derived from YVL clone 4 with similar results observed using other YVL clones. Mean values from triplicate wells are shown (the standard deviation was < 5% of maximal lysis).
The fine specificity of the YVL-specific response was further analysed by scanning T-cell clones against targets pulsed with single alanine substitutions from positions 1 to 9. As expected, alanine replacement at the HLA binding anchor residues, positions 2 and 9, abolished the immunogenicity of the peptide. A reduction in the cytotoxicity response was also observed using alanine variants at other positions, although this drop was particularly sharp when positions 5 and 7 were replaced (Fig. 6c). This suggested that the position 5 glutamic acid and the position 7 serine have considerable importance in T-cell contact.
Discussion
This report demonstrates that low-affinity CMV-specific CD8 T cells can persist in asymptomatic infection at very high frequencies albeit with the assistance of CD8 co-receptor binding in the event of antigen recognition. This represents a novel finding which contrasts with data showing that CMV, through providing a persistent antigenic stimulus, drives high-frequency responses that evolve high-avidity TCR with low dependence on CD8 for stable interactions with MHC–peptide ligands.26,27
One of the assumptions made during this study was that our HLA-A*0201 YVL tetramer was indeed a bona fide multimeric reagent that has not degraded at a faster rate than our other tetramers. Checks of monomer and tetramer integrity using gel filtration and sodium dodecylsulphate–polyacrylamide gel electrophoresis indicate that both YVL monomers and tetramers remain intact as a complex (and not as separate heavy chain and β2-microglobulin) and that heavy chains are not degraded to a lower molecular weight form by protease activity (indicating the loss of the biotinylation target sequence, which usually accounts for degraded monomers). As a further confirmation of both monomer and tetramer stability, we have succeeded in swapping the peptide from YVL monomers and tetramers for another HLA-A2 restricted peptide (NLV) to produce tetramers that bind T cells specific for the newly introduced peptide (M. Cobbold, unpublished data).
As this prompted us to question whether the actual lack of binding was a property attributed to the T cells rather than our reagent, we performed more detailed analyses of these tetr– T cells. One of the two donors studied has sizeable T-cell responses to at least six CMV-encoded peptides and the response against the YVL peptide is the second largest, when measured by IFN-γ staining (at 5·1% of CD8 T cells). These cells do not bind tetramers ex vivo, even as expanded effectors after short-term culture or as single-cell clones. Yet they maintain effector function in terms of both cytotoxicity and their ability to produce IFN-γ in response to antigen, which is a hallmark of CMV-specific CD8 T cells. In fact YVL-specific CD8 T cells appeared to be more differentiated and cytolytic than their NLV-specific counterparts within the same individual, ruling out functional incompetence of this response. The protective value of IE-1-specific CTL against CMV replication in both mice and humans is now well appreciated,28,29 suggesting that YVL-specific CD8 T cells will be biologically effective against virus-infected cells in vivo.
Peptide titration experiments showed little difference in functional avidity for antigen between both tetramer binding and non-binding T cells. However, CD8 blocking studies show that the overall avidity has considerable support from the CD8 interaction with class I MHC, possibly to stabilize the complex.30,31 Virus-specific responses have been shown to be composed of T-cell clones with different affinities that are compensated by variable CD8 contribution.26,32 Our experiments show a similar phenomenon but in a more exaggerated fashion with the response in this case being almost entirely CD8 dependent. The effects of anti-CD8 mAbs on TCR–MHC–peptide tetramer binding have also been debated,33–35 with some showing that certain anti-CD8 mAbs enhancing the binding of MHC tetramers to T cells.36,37 However, we found that our tetramer could not stain either PBMC or peptide-specific T-cell clones in conjunction with a number of different CD8 antibodies, or even in the absence of CD8 antibodies (data not shown).
Other reports also describe antigen-specific T cells that do not bind tetramers. In one case this was attributed to incomplete differentiation of the T cells causing an inability to cluster sufficient TCR molecules in close proximity for ligand binding.21 The authors described a sub-dominant T-cell response with a tetr– phenotype, which was overcome by prolonged stimulation resulting in the eventual differentiation to a tetr+ phenotype. This is thought to involve changes in lipid raft integrity that influence the surface distribution of TCRs to favour tetramer binding.38 A second report describes hepatitis B-specific HLA-A2-restricted CD8 T cells which also shifted to a tetr+ phenotype after repetitive in vitro stimulation.39 The tetr– response in our study is already a highly differentiated effector population and, unsurprisingly, did not change to a tetr+ phenotype after prolonged stimulation or culture, indicating that this was an inherent property of these virus-specific T cells.
To gain further insights into the TCR–HLA-A*0201–YVL interaction, surface plasmon resonance experiments could be performed. This would require knowledge of and recombinant expression of the TCR-α and TCR-β chains used by YVL-specific T cells. Such studies would provide data on the actual strength of the interaction between TCR and ligand (YVL monomer) independent of co-receptors and confirm whether there is an extremely low affinity relative to other known TCR–ligand interactions. The strength of this monomeric interaction may well be critical for successful tetramer binding. It is argued that the mean duration of the first TCR–MHC class I interaction has to be long enough to allow the next MHC class I complex to bind a second TCR and so allow for avidity to have its effect.40 Lower affinity interactions would therefore be at a significant disadvantage. In addition, a definitive role for CD8 may also be revealed for very-low-affinity interactions, adding to the debate on CD8 involvement in TCR–MHC–peptide binding.31,41 Price and colleagues have recently demonstrated that improving the CD8 interaction by introducing a CD8-binding site mutation on the MHC class I molecule (Q115E) can enhance tetramer staining of T cells with low TCR–MHC class I affinities.42 It would be of interest to know whether this modification would also result in successful YVL tetramer staining.
The absence of IE-1 sequence information for donor viral isolates raises the possibility that our YVL tetramer presents the wrong peptide. Detection of CMV genomes in healthy carriers has proved notoriously difficult for many laboratories and our endeavours to reproducibly amplify and sequence viral antigens from healthy persons have been unsuccessful. The use of published variant sequences (such as YILEETSVM) in cytotoxicity assays did not enhance responses detected against the parental YVL epitope. Furthermore, changes in anchor residues to improve HLA-A2 binding that may improve peptide immunogenicity43,44 always abrogated T-cell recognition, probably by altering the conformation of TCR contact residues. Alanine scanning of the YVL peptide has revealed positions 5 and 7 to be important positions in T-cell recognition. Substitutions at these positions with other peptides may cause unexpected increases in potency as described in the literature.25
From our studies a clear hierarchy of immunodominance with regards to IE-1 epitope choice is evident. This may be because of differences in avidity for antigen at the level of the antigen-presenting cell45 but the relative efficiency of epitope generation may also be a major influence on immunodominance. In most HLA-A2+-B7+ donors (9/10), CD8 T-cell responses are almost exclusively directed against the HLA-B7-restricted CRVLCCYVL peptide,46 which overlaps at its C terminus with both HLA-A2-restricted YVL and VLE peptides. One can envisage that HLA-B7–CRV complexes are more efficiently generated than HLA-A2–YVL/VLE complexes because proteasomal cleavage at the C-terminal leucine residue for generating the CRV epitope would curtail the formation of either HLA-A2-binding peptide. Higher levels of IE-1 protein may be required to generate YVL/VLE epitopes suggesting that the small minority of HLA-A2+-B7+ donors with detectable HLA-A2 YVL/VLE T-cell responses reflect greater viral burden.
Intriguingly, CD8 T-cell responses for the two peptides display exquisite ligand specificity with limited cross-reactivity for the alternate peptides. Our results contrast with published data47 showing T-cell clones specific for the decameric YVLEETSVML peptide that recognize both of the internal nonamers (YVL and VLE). This may be explained by possible differences in the TCR usage, and consequent flexibility, between their T-cell clones and ours. Differences in viral load and infectious strains may have driven the selection of cross-reactive TCR clonotypes over time, whereas in our donors one can hypothesize that more rigid T-cell responses have matured. The TCR usage may also be affected by the HLA background of the donor if virus-specific responses are cross-reactive with other self MHC–peptide complexes.48
To conclude, it is shown that CMV-specific CD8 T cells circulate invariably at high frequency and have a high functional avidity for their ligands. However, occasionally some of these high-frequency responses can be low-affinity T cells that overcome this problem through enhanced co-receptor involvement. Future work should explore TCR usage and biophysical aspects of this model of a low-affinity TCR–MHC class I–peptide interaction.
Acknowledgments
We thank David Lloyd for flow assisted cell sorting.
Disclosures
The authors have no conflicts of interests to declare.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Analysis of human leucocyte antigen A2 (HLA-A2) YVL monomer stability. YVL monomers were successfully prepared using standard in vitro methods of class I major histocompatibility complex (MHC) –peptide refolding and purification. (a) Purification of refolded HLA-A2 YVL monomers by gel filtration using a Superdex S75 column. (b) sodium dodecylsulphate–polyacrylamide gel electrophoresis determination of heavy chain stability and sample purity. A Coomassie-stained 12% gel is shown with approximately 5 μg protein loaded into each well. Lanes 1, 2 and 3 contain YVL monomers taken out of −80° storage (lane 1), YVL monomers stored at 4° for 7 days (lane 2) and YVL monomers stored at room temperature for 3 hr (lane 3). As a control, HLA-A2-NLV monomer stored at −80°, used to prepare ‘working’ NLV tetramer, is shown (lane 4). Each sample gave a positive biotinylation result by w6/32 enzyme-linked immunosorbent assay. Molecular weight standards are also shown to confirm heavy chain and β2-microglobulin size.
Figure S2. YVL-specific T-cell clones show minimal recognition of internal octomeric peptides. YVL-specific T cells were cloned in vitro and tested in 5-hr cytotoxicity assays for recognition of shorter octomeric peptides (YVLEETSV and VLEETSVM) that may also be human leucocyte antigen A2 (HLA-A2) -restricted T-cell epitopes. Variants containing isoleucine (YILEETSV and ILEETSVM) were also tested for both octomers. Target cells were HLA-A2-matched lymphoblastoid cell lines at effector : target ratio of 4 : 1.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Davis MM, Boniface JJ, Reich Z, Lyons D, Hampl J, Arden B, Chien Y. Ligand recognition by alpha beta T cell receptors. Annu Rev Immunol. 1998;16:523–44. doi: 10.1146/annurev.immunol.16.1.523. [DOI] [PubMed] [Google Scholar]
- 2.Altman JD, Moss PA, Goulder PJR, Barouch DH, McHeyzer-Williams MG, Bell JI, McMichael AJ, Davis MM. Phenotypic analysis of antigen-specific T lymphocytes. Science. 1996;274:94–6. [published erratum appears in Science 1998; 280: 1821] [PubMed] [Google Scholar]
- 3.Tan LC, Gudgeon N, Annel NE, et al. A re-evaluation of the frequency of CD8+ T cells specific for EBV in healthy virus carriers. J Immunol. 1999;162:1827–35. [PubMed] [Google Scholar]
- 4.Gillespie GM, Wills MR, Appay V, et al. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8+ T lymphocytes in healthy seropositive donors. J Virol. 2000;74:8140–50. doi: 10.1128/jvi.74.17.8140-8150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maini MK, Boni C, Lee CK, et al. The role of virus-specific CD8+ cells in liver damage and viral control during persistent hepatitis B virus infection. J Exp Med. 2000;191:1269–80. doi: 10.1084/jem.191.8.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lechner F, Wong DK, Dunbar PR, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wilson JD, Ogg GS, Allen RL, et al. Direct visualization of HIV-1-specific cytotoxic T lymphocytes during primary infection. AIDS. 2000;14:225–33. doi: 10.1097/00002030-200002180-00003. [DOI] [PubMed] [Google Scholar]
- 8.Appay V, Dunbar P, Callan M, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–85. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 9.Tully G, Kortsik C, Höhn H, et al. Highly focused T cell responses in latent human pulmonary Mycobacterium tuberculosis infection. J Immunol. 2005;174:2174–84. doi: 10.4049/jimmunol.174.4.2174. [DOI] [PubMed] [Google Scholar]
- 10.Pittet MJ, Valmori D, Dunbar PR, et al. High frequencies of naive Melan-A/MART-1-specific CD8+ T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med. 1999;190:705–15. doi: 10.1084/jem.190.5.705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, Davis MM. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6:1018–23. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- 12.Reijonen H, Kwok WW, Nepom GT. Detection of CD4+ autoreactive T cells in T1D using HLA class II tetramers. Ann N Y Acad Sci. 2003;1005:82–7. doi: 10.1196/annals.1288.009. [DOI] [PubMed] [Google Scholar]
- 13.Reddehase M. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol. 2002;2:831–44. doi: 10.1038/nri932. [DOI] [PubMed] [Google Scholar]
- 14.Gandhi MK, Khanna R. Human cytomegalovirus: clinical aspects, immune regulation, and emerging treatments. Lancet Infect Dis. 2004;4:725–8. doi: 10.1016/S1473-3099(04)01202-2. [DOI] [PubMed] [Google Scholar]
- 15.Khan N, Shariff N, Cobbold M, Bruton R, Ainsworth JA, Sinclair AJ, Nayak L, Moss PA. Cytomegalovirus seropositivity drives the CD8 T cell repertoire towards greater clonality in healthy elderly individuals. J Immunol. 2002;169:1984–92. doi: 10.4049/jimmunol.169.4.1984. [DOI] [PubMed] [Google Scholar]
- 16.Khan N, Cobbold M, Keenan R, Moss PA. Comparative analysis of CD8+ T cell responses against human cytomegalovirus proteins pp65 and IE-1 shows similarities in precursor frequency, oligoclonality and phenotype. J Infect Dis. 2002;185:1025–34. doi: 10.1086/339963. [DOI] [PubMed] [Google Scholar]
- 17.Elkington R, Walker S, Crough T, Menzies M, Tellam J, Bharadwaj M, Khanna R. Ex vivo profiling of CD8+-T-cell responses to human cytomegalovirus reveals broad and multi-specific reactivities in healthy virus carriers. J Virol. 2003;77:5226–40. doi: 10.1128/JVI.77.9.5226-5240.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sylwester A, Mitchell B, Edgar J, et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ T cells dominate the memory compartments of exposed subjects. J Exp Med. 2005;202:673–85. doi: 10.1084/jem.20050882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan N, Hislop AD, Gudgeon N, Cobbold M, Khanna R, Nayak L, Rickinson AB, Moss PA. Herpesvirus-specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a co-resident EBV infection. J Immunol. 2004;173:7481–9. doi: 10.4049/jimmunol.173.12.7481. [DOI] [PubMed] [Google Scholar]
- 20.Matsui K, Boniface JJ, Reay PA, Schild H, Fazekas de St Groth B, Davis MM. Low affinity interaction of peptide–MHC complexes with T cell receptors. Science. 1991;254:1788–91. doi: 10.1126/science.1763329. [DOI] [PubMed] [Google Scholar]
- 21.Spencer JV, Braciale TJ. Incomplete CD8+ T cell differentiation as a mechanism for sub-dominant lymphocyte responses to a viral antigen. J Exp Med. 2000;191:1687–98. doi: 10.1084/jem.191.10.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gebe JA, Falk BA, Rock KA, Kochik SA, Heninger AK, Reijonen H, Kwok WW, Nepom GT. Low-avidity recognition by CD4+ T cells directed to self-antigens. Eur J Immunol. 2003;33:1409–17. doi: 10.1002/eji.200323871. [DOI] [PubMed] [Google Scholar]
- 23.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 24.Retiere C, Prod'homme V, Imbert-Marcille BM, Bonneville M, Vie H, Hallet MM. Generation of cytomegalovirus-specific human T-lymphocyte clones by using autologous B-lymphoblastoid cells with stable expression of pp65 or IE1 proteins: a tool to study the fine specificity of the antiviral response. J Virol. 2000;74:3948–52. doi: 10.1128/jvi.74.9.3948-3952.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tangri S, Ishioka GY, Huang X, Sidney J, Southwood S, Fikes J, Sette A. Structural features of peptide analogs of human histocompatibility leukocyte antigen class I epitopes that are more potent and immunogenic than wild-type peptide. J Exp Med. 2001;194:833–46. doi: 10.1084/jem.194.6.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price DA, Brenchley JM, Ruff LE, et al. Avidity for antigen shapes the clonal dominance of CD8 T cell populations specific for persistent DNA viruses. J Exp Med. 2005;202:1349–61. doi: 10.1084/jem.20051357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trautmann L, Rimbert M, Echasserieau K, Saulquin X, Neveu B, Dechanet J, Cerundolo V, Bonneville M. Selection of T cell clones expressing high-affinity public TCRs within human cytomegalovirus-specific CD8 T cell responses. J Immunol. 2005;175:6123–32. doi: 10.4049/jimmunol.175.9.6123. [DOI] [PubMed] [Google Scholar]
- 28.Del Val M, Schlicht HJ, Volkmer H, Messerle M, Reddehase MJ, Koszinowski UH. Protection against lethal cytomegalovirus infection by a recombinant vaccine containing a single nonameric T-cell epitope. J Virol. 1991;65:3641–6. doi: 10.1128/jvi.65.7.3641-3646.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bunde T, Kirchner A, Hoffmeister B, et al. Protection from cytomegalovirus after transplantation is correlated with immediate early 1-specific CD8 T cells. J Exp Med. 2005;201:1031–6. doi: 10.1084/jem.20042384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, Teyton L. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384:577–81. doi: 10.1038/384577a0. [erratum in Nature 1997; 387:634] [DOI] [PubMed] [Google Scholar]
- 31.Wooldridge L, van den Berg HA, Glick M, et al. Interaction between the CD8 coreceptor and major histocompatibility complex class I stabilizes T cell receptor antigen complexes at the cell surface. J Biol Chem. 2005;280:27491–501. doi: 10.1074/jbc.M500555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Couedel C, Bodinier M, Peyrat MA, Bonneville M, Davodeau F, Lang F. Selection and long-term persistence of reactive CTL clones during an EBV chronic response are determined by avidity, CD8 variable contribution compensating for differences in TCR affinities. J Immunol. 1999;162:6351–8. [PubMed] [Google Scholar]
- 33.Daniels MA, Jameson SC. Critical role for CD8 in T cell receptor binding and activation by peptide/major histocompatibility complex multimers. J Exp Med. 2000;191:335–46. doi: 10.1084/jem.191.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Denkberg G, Cohen CJ, Reiter Y. Critical role for CD8 in binding of MHC tetramers to TCR: CD8 antibodies block specific binding of human tumor-specific MHC–peptide tetramers to TCR. J Immunol. 2001;167:270–6. doi: 10.4049/jimmunol.167.1.270. [DOI] [PubMed] [Google Scholar]
- 35.Holman PO, Walsh ER, Jameson SC. Characterizing the impact of CD8 antibodies on class I MHC multimer binding. J Immunol. 2005;174:3986–91. doi: 10.4049/jimmunol.174.7.3986. [DOI] [PubMed] [Google Scholar]
- 36.Campanelli R, Palermo B, Garbelli S, Mantovani S, Lucchi P, Necker A, Lantelme E, Giachino C. Human CD8 co-receptor is strictly involved in MHC–peptide tetramer–TCR binding and T cell activation. Int Immunol. 2002;14:39–44. doi: 10.1093/intimm/14.1.39. [DOI] [PubMed] [Google Scholar]
- 37.Wooldridge L, Hutchinson SL, Choi EM, et al. Anti-CD8 antibodies can inhibit or enhance peptide–MHC class I (pMHCI) multimer binding: this is paralleled by their effects on CTL activation and occurs in the absence of an interaction between pMHCI and CD8 on the cell surface. J Immunol. 2003;171:6650–60. doi: 10.4049/jimmunol.171.12.6650. [DOI] [PubMed] [Google Scholar]
- 38.Drake DR, 3rd, Braciale TJ. Cutting edge: lipid raft integrity affects the efficiency of MHC class I tetramer binding and cell surface TCR arrangement on CD8+ T cells. J Immunol. 2001;166:7009–13. doi: 10.4049/jimmunol.166.12.7009. [DOI] [PubMed] [Google Scholar]
- 39.Reignat S, Webster D, Brown G, et al. Escaping high viral load exhaustion: CD8 T cells with altered tetramer binding in chronic hepatitis B infection. J Exp Med. 2002;195:1089–101. doi: 10.1084/jem.20011723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wooldridge L, Lissina A, Cole DK, van den Berg HA, Price DA, Sewell AK. Tricks with tetramers: how to get the most from multimeric peptide–MHC. Immunology. 2009;126:147–64. doi: 10.1111/j.1365-2567.2008.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wyer JR, Willcox BE, Gao GF, Gerth UC, Davis SJ, Bell JI, Van der Merwe PA, Jacobsen BK. T cell receptor and coreceptor CD8 αα bind peptide–MHC independently and with distinct kinetics. Immunity. 1999;10:219–25. doi: 10.1016/s1074-7613(00)80022-9. [DOI] [PubMed] [Google Scholar]
- 42.Melenhorst JJ, Scheinberg P, Chattopadhyay PK, et al. Detection of low avidity CD8+ T cell populations with coreceptor-enhanced peptide–major histocompatibility complex class I tetramers. J Immunol Methods. 2008;338:31–9. doi: 10.1016/j.jim.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valmori D, Fonteneau JF, Lizana CM, et al. Enhanced generation of specific tumor-reactive CTL in vitro by selected Melan-A/MART-1 immunodominant peptide analogues. J Immunol. 1998;160:1750–8. [PubMed] [Google Scholar]
- 44.Chen JL, Dunbar PR, Gileadi U, et al. Identification of NY-ESO-1 peptide analogues capable of improved stimulation of tumor-reactive CTL. J Immunol. 2000;165:948–55. doi: 10.4049/jimmunol.165.2.948. [DOI] [PubMed] [Google Scholar]
- 45.Kedl RM, Rees WA, Hilderman DA, Schaefer B, Mitchell T, Kappler J, Marrack P. T cells compete for access to antigen-bearing antigen-presenting cells. J Exp Med. 2000;192:1105–13. doi: 10.1084/jem.192.8.1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kern F, Surel IP, Faulhaber N, Frömmel C, Schneider-Mergener J, Schönemann C, Reinke P, Volk HD. Target structures of the CD8+-T-cell response to human cytomegalovirus: the 72-kilodalton major immediate-early protein revisited. J Virol. 1999;73:8179–84. doi: 10.1128/jvi.73.10.8179-8184.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prod'homme V, Retiere C, Imbert-Marcille BM, Bonneville M, Hallet MM. Modulation of HLA-A*0201-restricted T cell responses by natural polymorphism in the IE1(315-324) epitope of human cytomegalovirus. J Immunol. 2003;170:2030–6. doi: 10.4049/jimmunol.170.4.2030. [DOI] [PubMed] [Google Scholar]
- 48.Burrows SR, Silins SL, Moss DJ, Khanna R, Misko IS, Argaet VP. T cell receptor repertoire for a viral epitope in humans is diversified by tolerance to a background major histocompatibility complex antigen. J Exp Med. 1995;182:1703–15. doi: 10.1084/jem.182.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.