

Abstract
It is clear that CD4+ CD25+ Foxp3+ regulatory T (Treg) cells inhibit chronic inflammatory responses as well as adaptive immune responses. Among the CD4+ T-cell population in the skin, at least one-fifth express Foxp3. As the skin is constantly exposed to antigenic challenge and is a common site of vaccination, understanding the role of these skin-resident Treg cells is important. Although the suppressive effect of Treg cells on T cells is well documented, less is known about the types of innate immune cells influenced by Treg cells and whether the Treg cells suppress acute innate immune responses in vivo. To address this we used a mouse melanoma cell line expressing Fas ligand (B16FasL), which induces an inflammatory response following subcutaneous injection of mice. We demonstrate that Treg cells limit this response by inhibiting neutrophil accumulation and survival within hours of tumour cell inoculation. This effect, which was associated with decreased expression of the neutrophil chemoattractants CXCL1 and CXCL2, promoted survival of the inoculated tumour cells. Overall, these data imply that Treg cells in the skin are rapidly mobilized and that this activity serves to limit the amplification of inflammatory responses at this site.
Keywords: neutrophils, regulatory T cells, skin, tumour
Introduction
CD4+ CD25+ Foxp3+ regulatory T (Treg) cells can suppress both antigen-specific and inflammatory responses.1 Indeed, studies of mice lacking Foxp3 have revealed that the cells play a key role in controlling autoimmunity and inflammatory disease, and in maintaining normal immune homeostasis.2–4 In addition, immune responses to pathogens are modulated by the activity of Treg cells, probably in an attempt to limit pathogen-induced immune-mediated damage to the host.5 Although the physiological role of Treg cells is to prevent immunopathology, studies in animal models and in humans indicate that Treg cells can be manipulated for the purpose of augmenting immunogenicity.6 This may prove useful, particularly for the treatment of diseases such as cancer, generally characterized by a paucity of effective immune responses. In fact, many laboratories including our own have shown that immune responses to tumour antigens can be enhanced in the absence of Treg cells.7 Detailed knowledge of the types of cells suppressed by Treg cells and how Treg cells alter the immune environment should inform the design of more successful immunotherapeutic strategies.
The suppressive effects of Treg cells have been studied mainly in the context of their ability to limit T-cell responses. However, studies have shown that Treg cells can suppress the activity of natural killer cells and neutrophils in vitro, and that they also limit chronic inflammatory responses in vivo. In this study we specifically sought to determine whether Treg cells impact on acute innate immune responses in vivo. For this purpose, we used a mouse model of melanoma. Mouse melanoma cells (B16F10), and particularly those engineered to express Fas ligand (B16FasL), induce an innate immune response following their subcutaneous inoculation into C57BL/6 (B6) mice.8 This innate immune response is important because it clearly contributes to tumour rejection. We have previously reported that an in vivo reduction in Treg-cell numbers promotes rejection of both B16 and B16FasL and that this is at least partly the result of enhanced inflammatory responses in these animals compared with those with an intact Treg-cell population.9 As B16FasL induces a more readily detectable and measurable inflammatory response compared with B16, this model provided an opportunity to address whether Treg cells limit acute innate immune responses in the skin, a site where at least one-fifth of skin-resident CD4+ T cells are Treg cells.
Materials and methods
Mice
The C57BL/6 (B6) mice were bred and maintained at Biomedical Services (Cardiff, UK). All experiments were performed in compliance with UK Home Office regulations.
Antibodies
Hybridomas secreting CD25 (PC61, rat IgG1), Escherichia coliβ-galactosidase- (GL113, rat IgG1, non-depleting isotype control antibody), Gr-1- (RB6-8C5, rat IgG2b) specific monoclonal antibodies (mAbs) have been described previously.8,10,11 Briefly, 0·5 mg PC61 or GL113 was administered intraperitoneally (i.p.) 1 and 3 days before tumour inoculation. As we have previously shown, PC61 administration in this way efficiently depletes the majority of CD25+ cells.9,11,12 However, it is also clear that many Foxp3+ Treg cells (20–50%) do not express CD25 and therefore escape the depleting effect of PC61 administration.13 Administration of PC61 therefore results in a reduction rather than a complete loss of Treg-cell activity. Neutrophils were depleted by administration of 0·3 mg of RB6-8C5 every second day from 1 day before tumour inoculation. The efficiency with which RB6-8C5 depletes neutrophils has been described elsewhere.9
Tumour cell lines
B16F10 (B16) and B16F10 transfected with the Fas ligand B16FasL were generated as previously described 14 and were maintained in R10, which consists of RPMI-1640 medium (Gibco – Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum (Gibco – Invitrogen), penicillin–streptomycin, l-glutamine, non-essential amino-acids (Life Technologies – Invitrogen, Carlsbad, CA) and 50 μm 2β-mercaptoethanol (Sigma-Aldrich, St Louis, MO). In the case of B16FasL, G418 was added to the media at a final concentration of 1·5 mg/ml to maintain expression of FasL. Tumour cells were either injected subcutaneously (s.c.) (105 in 100 μl phosphate-buffered saline) or i.p. (2 × 106 in 100 μl PBS).
Histology
Tissue was taken from the area surrounding the inoculation site and fixed in zinc fixative as previously described.15 Tissue was then embedded in paraffin wax and 5-μm serial sections were taken every 300 μm. Sections were then either stained with haematoxylin & eosin (H&E) to estimate the tumour mass and infiltrate or subjected to immunohistochemistry to identify neutrophils and Treg cells. The length (l) and width (w) of tumour mass plus infiltrate on each section was measured on a calibrated microscope. An estimate was made of the total tumour volume based on the area of tumour mass and infiltrate (πlw) on adjacent sections and the distance between sections (h): i.e. hπ(√lw + √LW + (√lw* √LW))/3. It was assumed that the tumour mass and infiltrate terminated at the mid-point between the last section in which it was observed and the next. The sum of these volumes resulted in an estimation of the tumour mass and infiltrate.
Immunohistochemistry
For staining of neutrophils, sections were dehydrated then microwaved in 10 mm citrate buffer pH 6. Sections were equilibrated in PBS before blocking of peroxidase activity with 1% H2O2. Non-specific antibody binding was blocked by incubation with PBS supplemented with 1% bovine serum albumin and 2% rabbit serum. Neutrophils were detected using rabbit anti-mouse interleukin-8 receptor B (IL-8RB; K-19; Santa Cruz Biotechnology, Santa Cruz, CA) followed by incubation with biotinylated swine anti-rabbit abs (Dako, Glostrup, Denmark). Neutrophils were then visualized by incubation with horseradish peroxidase-conjugated Extravidin (Sigma-Aldrich) followed by development with diaminobenzidine (DAB) substrate kit (VectorLabs, Burlingame, CA) according to the manufacturer's instructions and counterstaining with haematoxylin.
For staining of Foxp3, sections were dehydrated and microwaved in 50 mm Tris–HCl, 2 mm EDTA, pH 9. Endogenous biotin was blocked by incubation in avidin followed by biotin (VectorLabs). Non-specific binding sites were subsequently blocked with horse serum. Foxp3 cells were stained using rat anti-Foxp3 antibodies (FJK-16; eBioscience, San Diego, CA, USA), then biotinylated anti-rat abs (BDBiosciences, San Jose, CA, USA) and stained cells were visualized by incubation with horseradish peroxidase-conjugated Extravidin and DAB as described above.
Mouse peritoneal lavage cells
The peritoneal lavage cells were collected by injecting 6 ml PBS with 2 mm EDTA and 0·5% bovine serum albumin into the peritoneum of killed mice with 6 ml fluid recovered in every case. Cytofunnels were assembled as described in the manufacturer's instructions. A 240-ml sample of lavage fluid was spun for 10 min at 112.9 g. Slides were then air dried and stained using a Wright–Giemsa stain, rinsed in deionized water and allowed to air dry.
Neutrophil cytotoxicity assay
Bone marrow (BM) was collected from naive mice and neutrophils were isolated by density centrifugation. Briefly, BM cells were layered on top of 72%, 64% and 52% Percoll solutions, with the cells at the lower interphase constituting mainly mature neutrophils after centrifugation. Purity of neutrophils was assessed by Giemsa staining of methanol-fixed cells and was > 90%. These cells were then incubated at a ratio of 40 : 1 with 51Cr-labelled B16 or B16FasL cells for 4 hr at 37°. For minimal and maximal lysis, cells were incubated with medium or 5% Triton-X-100, respectively. Lytic activity was measured by 51Cr release with the formula: % lysis = [(sample − min)/(max − min)] × 100.
Real-time PCR
B6 mice were injected i.p. with 0·5 mg of PC61 or GL113 antibodies 4 days and 1 day before s.c. injection of 0·5 × 106 B16-FasL cells. Twenty-four hours later, the skin area including the tumour cells was dissected, snap-frozen in liquid nitrogen and RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). Total RNA was reverse transcribed using Superscript III (Invitrogen), and subsequently cDNA was amplified in triplicate by real-time PCR using 1 × Platinum SYBR Green qPCR SuperMix (Invitrogen) with primers for glyceraldehyde 3-phosphate dehydrogenase (GAPDH), CXCL1/KC or CXCL2/MIP-2. Messenger mRNA levels were normalized relative to GAPDH mRNA expression. The average C(t) values were taken from three mice per group and data are presented as gene expression in PC61-treated mice relative to control GL113-treated mice. Primer pairs were as follows: GAPDH, 5′-TGACCTTGCCCACAGCCTTG-3′ (sense) and 5′-GAACGGGAAGCTTGTCATCA-3′ (anti-sense): CXCL1/KC, 5′-CTCAAGAATGGTCGCGAGGCT-3′ (sense) and 5′-GCACAGTGGTTGACACTTAGTGGTCTC-3′ (anti-sense); CXCL2/MIP-2 5′-CCACTCTCAAGGGCGGTCAAA-3′ (sense) and 5′-TACGATCCAGGCTTC-CCGGGT-3′ (anti-sense).
Results
Treg cells limit the inflammatory response induced by FasL-expressing tumour cells
We previously found that B16FasL cells are rejected more efficiently by C57BL/6 (B6) mice when Treg cells are partially depleted by in vivo administration of CD25-specific mAbs.9 Furthermore, this effect is attributable to the ability of Treg cells to suppress innate immune responses.9 To characterize the nature of the innate response inhibited by Treg cells, we injected mice partially depleted of Treg cells and control mice with B16FasL cells and assessed the response to this whole cell challenge at early time-points thereafter. We first performed histological analyses to study the cellular infiltrate at the non-palpable B16FasL inoculation site. B6 mice treated with depleting CD25-specific mAbs (PC61) or non-depleting control mAbs (GL113) were injected s.c. with 105 live B16FasL, then 4, 24 and 96 hr after tumour injection mice were killed and the injected skin was removed for histology. Tissue was embedded in paraffin and 5-μm sections were cut at 300-μm intervals throughout the skin. Sections were stained with H&E to locate the midsection of the tumour inoculation site (Fig. 1a–d). A large amount of cell death was observed at each inoculation site, as indicated by the lack of cellular cohesion and the presence of fragmented nuclei (Fig. 1b,d). Analyses at these early time-points revealed the presence of an inflammatory infiltrate evident within 24 hr of tumour cell inoculation and which was significantly larger in the PC61-treated group (Fig. 1c,d) compared with the GL113-treated group (Fig. 1a,b). During these analyses, it was noticed that there were two forms of cellular mass displaying different histological characteristics (Fig. 2). In one type, cells were confined to a single layer of the skin, surrounded by normal tissue (Fig. 2a,b); however, in the other type, inflammatory cells were found spread throughout the layers of the skin (Fig. 2c,d). Upon assessment of sections for these characteristics, none of the sections from PC61-treated mice, and around half of the GL113-treated mice, displayed the ‘confined’ phenotype (Fig. 2e). This is noteworthy when compared with the percentage of mice that reject these tumours; approximately 50% in GL113-treated mice and 100% in PC61-treated mice.9
Figure 1.
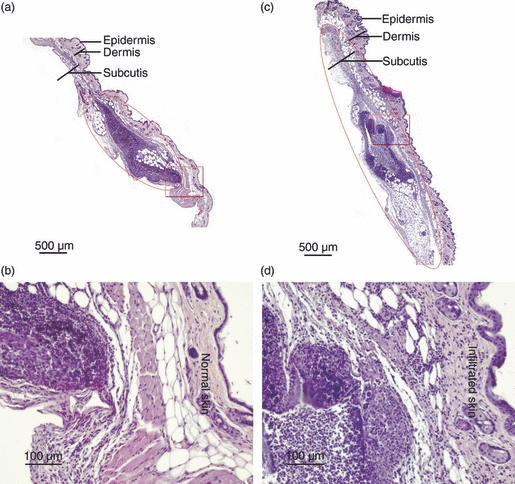
Histological examination of the site of B16Fas ligand cell inoculation in regulatory T cell-depleted and control mice. Mice treated either with isotype control monoclonal antibodies (GL113; a, b) or CD25-specific monoclonal antibodies (PC61; c, d) were injected subcutaneously 1 day later with 105 B16FasL. At 24 hr post-injection mice were killed and the skin surrounding the injection site was collected for histology. Haematoxylin & eosin-stained 5-μm paraffin-mounted sections were generated throughout the skin. Sections (a) to (d) are representative mid-sections of cellular masses from at least four mice per group.
Figure 2.
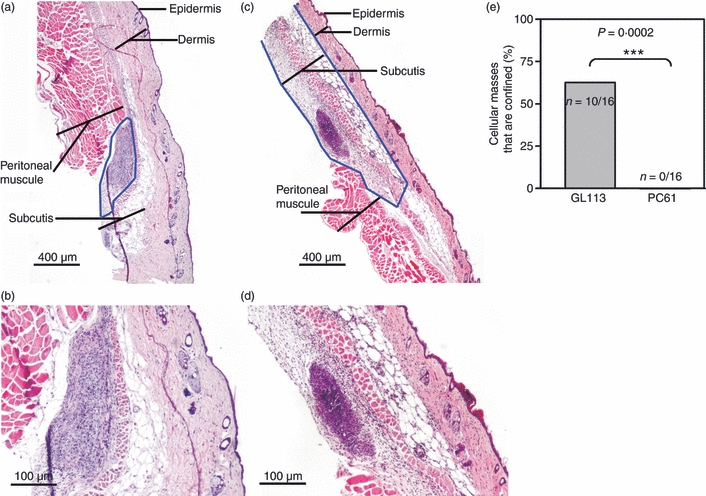
Distinct patterns in cell mass are observed at the site of B16Fas ligand cell inoculation in reulatory T cell-depleted versus control mice. Haematoxylin & eosin-stained sections were generated as described for Fig. 1 at 24 hr post-injection. Sections shown are representative of confined (a, b) and non-confined (c, d) cellular masses. The blue lines enclose the area of skin which histologically deviates from normal or PBS-injected skin. (e) The percentage of mice displaying the confined phenotype of cellular mass is given for each group. Statistical significance was evaluated by Fisher's exact test (***P < 0·001).
To perform a more quantitative assessment of the differences between cellular masses termed ‘confined’ versus those termed ‘non-confined’, the total volume of each cellular mass within the GL113-treated and PC61-treated groups (> 4 per group), 4 and 24 hr after tumour cell inoculation, was calculated. These data, shown in Fig. 3(a), corroborated our previous observation in that at 24 hr larger masses were observed in the PC61 group compared with those treated with GL113. At later time-points (96 hr), larger cellular masses were measured in the latter, control group of mice, coinciding with detection of live tumour cells in this group. Live tumour cells were identified by histological examination of H&E-stained sections in GL113-treated mice but not in PC61-treated mice. In the former group, within the tumour cell mass, amid cell debris, there are areas of homogeneous healthy cells, forming foci of organized tissue, similar to that seen in large, established tumours (Fig. 3b,c). These data are consistent with the observation that around 50% of mice inoculated with B16FasL develop palpable tumours whereas tumours are rarely seen in B16FasL-inoculated mice pre-treated with PC61.9 Overall, these data indicate that an inflammatory infiltrate into the tumour creates a disorganized, non-confined mass that is associated with tumour cell death and tumour rejection, favoured by depletion of Treg cells by PC61 mAbs.
Figure 3.
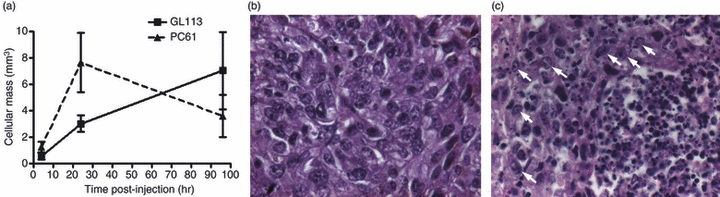
Live tumour cells observed in B16Fas ligand-injected mice. Haematoxylin & eosin-stained sections were generated as described for Fig. 1 at 4, 24 and 96 hr post-injection. (a) The total volume of cellular mass for each mouse was estimated based on the area of cellular mass on each section and the distance between them, as described in the Materials and methods. Data are shown ± SEM of at least four mice per group. (b) An example of live tumour cells in an established tumour. (c) Live cells within cellular masses of GL113-treated mice are identified as being histologically similar to those found within established tumours and are indicated by arrows.
We were struck by how rapidly Treg-cell depletion affected the accumulation of inflammatory cells at the site of the tumour cell inoculum. The ability of Treg cells to suppress an inflammatory response within hours of an antigenic challenge and at a peripheral site implies that skin-resident Treg cells are rapidly mobilized. To visualize Treg cells at the site of tumour cell challenge, skin sections were stained with Foxp3-specific mAbs. Foxp3+ cells were found in the skin and particularly at the site of tumour cell inoculation (Fig. 4). This is in agreement with other studies reporting Treg-cell identification in the skin of mice16 and humans.17 Stained cells were not observed in sections prepared from PC61-treated mice (data not shown).
Figure 4.
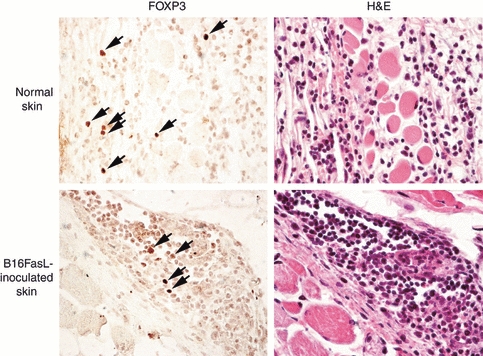
Foxp3+ cells in skin of B16Fas ligand-injected mice. Serial sections of normal and B16FasL-inoculated skin from GL113-treated mice were stained with either haematoxylin & eosin or Foxp3-specific monoclonal antibodies. The Foxp3+ cells are indicated by the arrows. No stained cells were observed in the PC61-treated mice (not shown).
Treg cells limit neutrophil infiltration at the site of B16FasL inoculation
Examination of the H&E-stained skin sections indicated that the cellular mass was composed largely of neutrophils. To confirm this, neutrophils were further identified as polymorphonuclear cells that express IL-8R (Fig. 5a–d). Furthermore, the results show an increased number of neutrophils in PC61-treated mice at 24 hr post-injection (Fig. 5d) reflecting the data on increased cellular mass in PC61-treated mice (Figs 1 and 3).
Figure 5.
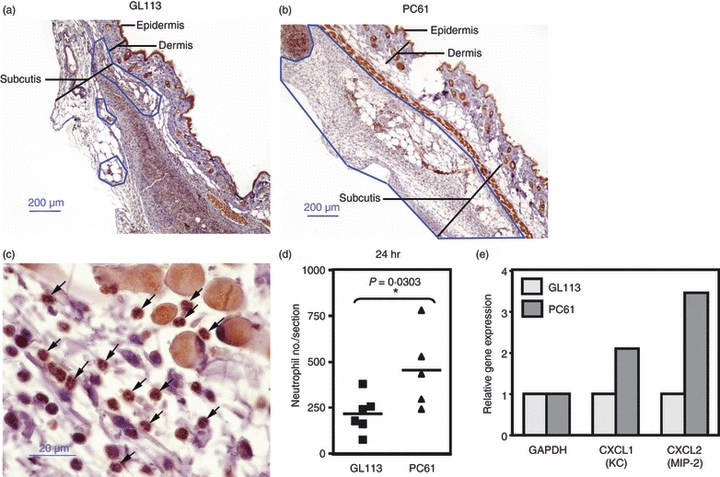
Depletion of CD25+ cells promotes neutrophil accumulation at the site of tumour inoculation. Mice were treated with either isotype control monoclonal antibodies (GL113; a) or CD25-specific monoclonal antibodies (PC61; b) and injected 1 day later with 105 B16Fas ligand. At 24 hr post-injection, mice were killed and the skin surrounding the injection site was collected for histology. Interleukin-8 receptor (IL-8R) -stained 5-μm paraffin-mounted sections were generated to identify neutrophils. Areas of neutrophil infiltration are enclosed by the blue lines. Sections are representative of at least five mice per group at 24 hr. (c) Examples of neutrophils (indicated by arrows) which stain dark brown with multi-lobed nuclei. (d) The numbers of neutrophils, assessed as polymorphonuclear cells with IL-8R expression, are given where each symbol represents the score for one mouse. Statistical significance was evaluated by Mann–Whitney U-test (*P < 0·05). (e) The relative expression of CXCL1 and CXCL2, assessed by real-time PCR, in the skin of GL113-treated and PC61-treated B16FasL-inoculated mice.
As neutrophils were more abundant in the Treg-depleted animals, we examined relative levels of neutrophil chemoattractants, CXCL1 (KC) and CXCL2 (MIP-2), in the skin of Treg-reduced and control mice 24 hr post-inoculation with B16FasL cells. Elevated levels of both chemokines were observed in the skin of Treg-depleted animals suggesting that Treg cells inhibit local neutrophil chemoattractant production (Fig. 5e).
Treg cells limit neutrophil survival at the site of B16FasL inoculation
As detailed phenotypic characterization of neutrophils from tissue sections is difficult, cytospins were generated from the lavage fluid of mice receiving B16FasL cells i.p., enabling us to compare neutrophils isolated from PC61-treated and GL113-treated mice (Fig. 6). No differences were observed in expression of the neutrophil activation marker, CD11b or ROS (data not shown). An effect of Treg cells on neutrophil activation cannot be ruled out, however, because it is possible that only activated neutrophils would be recovered in the lavage fluid (and similarly the site of tumour cell inoculation) so any impact of Treg cells on neutrophil activation may be difficult to observe in vivo. However, differences were observed between neutrophils isolated from PC61-treated and GL113-treated mice (Fig. 6). Figure 6(a,b) shows examples of neutrophils isolated from GL113-treated and PC61-treated mice, respectively. Examples of segmented nuclei are given in Fig. 6(c), where segments are joined by thin strands of chromatin. Upon enumeration, it was evident that the proportion of neutrophils with a higher number of segments was increased in PC61-treated mice (Fig. 6d,e), which results in an increase in the average number of segments per neutrophil (Fig. 6d,e). Hypersegmentation of nuclei in neutrophils has long been associated with more mature neutrophils, and is an indicator of prolonged neutrophil survival.18 Collectively, these data support the premise that Treg cells affect neutrophil accumulation at the site of antigenic challenge not through inhibiting their activation but through influencing local chemokine production and by limiting their survival.
Figure 6.
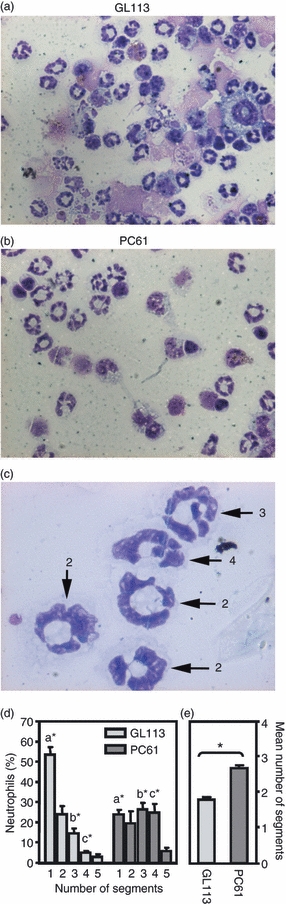
Increased number of nuclear segments in neutrophils recovered from mice depleted of CD25+ cells. Mice were treated with either isotype control monoclonal antibodies (GL113) or CD25-specific monoclonal antibodies (s (PC61) and injected intraperitoneally 1 day later with 2 × 106 B16Fas ligand. Eighteen hours later, mice were killed and the peritoneal cavity was lavaged. Cytospins generated of lavaged cells were stained and the number of segments in 100 neutrophils was assessed. Representative photographs of cytospins generated from GL113-treated (a) and PC61-treated (b) mice. (c) Examples of segmented neutrophils where the number of segments in each neutrophil is given. (d) The proportion of neutrophils with the indicated number of segments is shown. Mice were analysed individually and data shown are the mean ± SEM of four mice per group. Statistical significance between groups sharing the same letter was evaluated by Mann–Whitney U-test (*P> 0·05). (e) The average number or segments per neutrophil is given for each mouse. Statistical significance was evaluated by Mann–Whitney U-test (*P = 0·0286).
Suppression of neutrophil infiltration by Treg cells limits the immunogenicity of vaccination by B16FasL
To test the relevance of neutrophils in this model, we first determined, in an in vitro assay, whether neutrophils could impinge on tumour rejection through direct lysis of tumour cells. As shown in Fig. 7(a), neutrophils were capable of lysing both B16 and B16FasL cells. To test the hypothesis in vivo, mice were treated with both PC61 and RB6-8C5, to deplete CD25+ cells and neutrophils, respectively, followed by s.c. challenge with B16FasL (Fig. 7b). As expected, rejection of B16FasL tumour cells increased from around 50% in GL113-treated mice to 100% in mice depleted of Treg cells using PC61. When neutrophils were concurrently depleted this enhanced rejection was no longer observed. These data indicate that Treg cells can limit the extent of neutrophil activity in the skin at a very early time-point following antigenic challenge and highlight the connection between enhanced neutrophil accumulation observed in the skin of Treg-reduced mice and tumour rejection.
Figure 7.
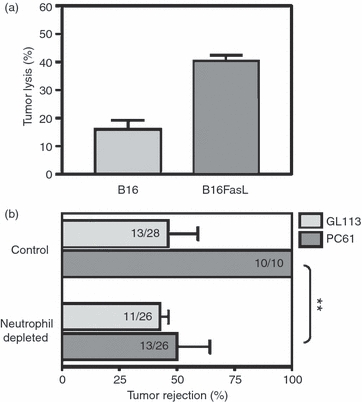
Neutrophils play a significant role in tumour rejection and in induction of adaptive immunity in regulatory T cell-depleted mice. (a) Neutrophils isolated from the bone marrow of mice were used directly as effectors against 51Cr-labelled B16 or B16Fas ligand cells at a ratio of 40 : 1. Bars indicate mean ± SD of duplicates and are representative of two independent experiments. (b) Mice were treated with either isotype control monoclonal antibodies (mAbs) (GL113) or CD25-specific mAbs (PC61), with or without Gr1-specific mAbs (RB6-8C5) for the depletion of neutrophils. Bars indicate the mean percentage rejection ± SEM in three and two experiments respectively, with the number of tumour-free mice/total number indicated. Statistical significance was evaluated by Fisher's exact test (**P < 0·01).
Discussion
Previous reports indicate that B16FasL is associated with the accumulation of neutrophils following subcutaneous injection of the cells into B6 mice.8 Our own previous work using B16FasL confirmed this finding but highlighted important roles for macrophages and natural killer cells for rejection of the tumour cells.9 This current report extends our understanding of the model by showing that neutrophils can also contribute to tumour rejection but that this ability is normally suppressed by Treg cells. In this study we used the FasL-expressing tumour cell line to study the effect of Treg cells on neutrophils. Collectively, our data indicate that skin-resident Treg cells act rapidly to limit the extent of neutrophil accumulation at the site of tumour cell challenge. This occurs partly through the influence of Treg cells on neutrophil survival, as evidenced by a significantly enhanced nuclear hypersegmentation in neutrophils recovered from mice with reduced Treg-cell numbers. Nuclear hypersegmentation is strongly associated with non-infectious inflammatory conditions 19–21 and is historically associated with older neutrophils and prolonged survival. More recently, hypersegmented neutrophils resulting from granulocyte colony-stimulating factor treatment,22 exhibited increased survival and increased phagocytic and cytolytic capacity.23,24 In addition, hypersegmentation was associated with prolonged chemotaxis towards C5a and IL-8 and sustained expression of chemokine receptors CXCR1 and CXCR2.25 Our in vivo data relating to the relationship between Treg cells and neutrophil survival is supported by previous in vitro studies indicating that lipopolysaccharide-activated human Treg cells promoted neutrophil apoptosis and death.26 A previous report by Engeman et al.27 indicated that the extent of the neutrophil response to a given antigenic challenge correlated with the number of CD8+ T cells recruited to the challenge site. Although not addressed in our study, these data collectively support the possibility that Treg cells can impact on adaptive immune responses indirectly, through limiting early neutrophil activity.
As migration of inflammatory cells is regulated by various chemoattractants and adhesion molecules produced/up-regulated in response to injury or infection, we surmised that manipulation of Treg cells might alter chemokine production in response to B16FasL challenge. Indeed, quantitative PCR performed at the site of tumour cell inoculation revealed an increase in levels of the neutrophil-attracting chemokines CXCL1 and CXCL2 in animals partially depleted of Treg cells compared with controls. We speculate that one highly effective mechanism through which Treg cells limit neutrophil responses is to reduce chemokine production perhaps by a variety of cells, including epithelial cells, macrophages (CXCL1 and CXCL2) and neutrophils (CXCL1). This finding expands upon previous observations showing that anergic regulatory T cells inhibit tissue invasion by T cells and granulocytes through chemokine metabolism.28 Similarly, Sarween et al.29 reported that CD4+ CD25+ Treg cells prevent tissue invasion by other T cells through effects on chemokine receptor and chemokine expression.
The impact of Treg cells on inflammatory responses is not confined to B16FasL. Enhanced rejection of B16 cells observed after partial depletion of Treg cells is dependent on innate immune responses and B16 tumours grow more rapidly in RAG−/− mice receiving CD4+ CD25+ cells compared with those receiving CD4+ CD25− cells. Full characterization of the early events following tumour cell inoculation in these mice is not possible because the inflammatory response, although biologically relevant, cannot be readily detected by immunohistochemistry. Hence, the B16FasL cell line serves a useful purpose as enhanced immunogenicity facilitates characterization of early events occurring after tumour cell inoculation. Other studies support a role for Treg cells in controlling neutrophil responses. Previous studies of Helicobacter hepaticus-driven inflammatory responses in the gut indicated that adoptive transfer of Treg cells reduced neutrophil numbers in the spleen and lamina propria of chronically infected RAG−/− mice.30
There are many mechanisms involved in controlling immune responses in the skin. The ability of Treg cells to control the activity of CD8+ T-cell responses in the skin has been previously demonstrated.31,32 Our results show that Treg cells also control innate responses in the skin. These Treg cells may be activated by tissue damage or stimuli from melanoma cells and thereafter act rapidly in an antigen non-specific fashion, to be able to control early innate immune activation. We found no detectable increase in the level of skin Treg cells after inoculation of tumour cells further supporting the premise that skin-resident Treg cells are rapidly mobilized, controlling innate immune activation without the need for expansion of recruitment of Treg cells into the skin. In line with the rapid manifestation of Treg-cell activity in the skin, previous reports indicate that the majority of skin Treg cells express CCR4 and high levels of CD103, a molecule implicated as a marker of effector memory Treg cells,33 suggesting that the Treg cells are ready to exert their effects early in an immune response. In addition, a recent report by Rubtsov et al.34 indicated that Treg cells, present at environmental surfaces like skin and gut, keep immune responses at these sites in check through the production of the immunosuppressive cytokine, IL-10. Interleukin-10 exerts potent and widespread immunosuppressive effects including the inhibition of chemokine production. (reviewed in ref. 35) It is therefore possible that IL-10, produced by a small number of skin-resident Treg cells, mediates potent anti-inflammatory effects by serving to limit the amplification of inflammatory networks. With this in mind it is therefore tempting to speculate that in our model, IL-10 produced by skin-resident Treg cells, acts to suppress the accumulation and survival of neutrophils at the site of antigenic challenge thereby reducing the overall immunogenicity of the antigen. These findings have implications for vaccine efficacy because they indicate that even partial removal of Treg cells will alter vaccine immunogenicity through limiting the influence of the cells on both innate and adaptive immune responses.
Acknowledgments
This work was supported by an MRC non-clinical senior fellowship (G117/488), an MRC collaboration grant (G0500617) and project grants from the AICR (05-028) and the Wellcome Trust (067046).
Disclosures
The authors declare that there are no conflicts of interest.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–87. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Brunkow ME, Jeffery EW, Hjerrild KA, et al. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet. 2001;27:68–73. doi: 10.1038/83784. [DOI] [PubMed] [Google Scholar]
- 3.Dudda JC, Perdue N, Bachtanian E, Campbell DJ. Foxp3+ regulatory T cells maintain immune homeostasis in the skin. J Exp Med. 2008;205:1559–65. doi: 10.1084/jem.20072594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Godfrey VL, Wilkinson JE, Russell LB. X-linked lymphoreticular disease in the scurfy (sf) mutant mouse. Am J Pathol. 1991;138:1379–87. [PMC free article] [PubMed] [Google Scholar]
- 5.Belkaid Y, Rouse BT. Natural regulatory T cells in infectious disease. Nat Immunol. 2005;6:353–60. doi: 10.1038/ni1181. [DOI] [PubMed] [Google Scholar]
- 6.Dannull J, Su Z, Rizzieri D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Invest. 2005;115:3623–33. doi: 10.1172/JCI25947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gallimore A, Godkin A. Regulatory T cells and tumour immunity – observations in mice and men. Immunology. 2008;123:157–63. doi: 10.1111/j.1365-2567.2007.02748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seino K, Kayagaki N, Okumura K, Yagita H. Antitumor effect of locally produced CD95 ligand. Nat Med. 1997;3:165–70. doi: 10.1038/nm0297-165. [DOI] [PubMed] [Google Scholar]
- 9.Simon AK, Jones E, Richards H, Wright K, Betts G, Godkin A, Screaton G, Gallimore A. Regulatory T cells inhibit Fas ligand-induced innate and adaptive tumour immunity. Eur J Immunol. 2007;37:758–67. doi: 10.1002/eji.200636593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lowenthal JW, Corthesy P, Tougne C, Lees R, MacDonald HR, Nabholz M. High and low affinity IL-2 receptors: analysis by IL-2 dissociation rate and reactivity with monoclonal anti-receptor antibody PC61. J Immunol. 1985;135:3988–94. [PubMed] [Google Scholar]
- 11.Jones E, Dahm-Vicker M, Simon AK, Green A, Powrie F, Cerundolo V, Gallimore A. Depletion of CD25+ regulatory cells results in suppression of melanoma growth and induction of autoreactivity in mice. Cancer Immun. 2002;2:1. [PubMed] [Google Scholar]
- 12.Betts G, Twohig J, Van den Broek M, Sierro S, Godkin A, Gallimore A. The impact of regulatory T cells on carcinogen-induced sarcogenesis. Br J Cancer. 2007;96:1849–54. doi: 10.1038/sj.bjc.6603824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Regulatory T cell lineage specification by the forkhead transcription factor foxp3. Immunity. 2005;22:329–41. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 14.Simon AK, Gallimore A, Jones E, Sawitzki B, Cerundolo V, Screaton GR. Fas ligand breaks tolerance to self-antigens and induces tumor immunity mediated by antibodies. Cancer Cell. 2002;2:315–22. doi: 10.1016/s1535-6108(02)00151-4. [DOI] [PubMed] [Google Scholar]
- 15.Beckstead JH. A simple technique for preservation of fixation-sensitive antigens in paraffin-embedded tissues. J Histochem Cytochem. 1994;42:1127–34. doi: 10.1177/42.8.8027531. [DOI] [PubMed] [Google Scholar]
- 16.Suffia I, Reckling SK, Salay G, Belkaid Y. A role for CD103 in the retention of CD4+ CD25+ Treg and control of Leishmania major infection. J Immunol. 2005;174:5444–55. doi: 10.4049/jimmunol.174.9.5444. [DOI] [PubMed] [Google Scholar]
- 17.Clark RA, Kupper TS. IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood. 2007;109:194–202. doi: 10.1182/blood-2006-02-002873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu MJ, Feldman BF, Zinkl JG, Jain NC. Using red blood cell creatine concentration to evaluate the equine erythropoietic response. Am J Vet Res. 1983;44:1427–32. [PubMed] [Google Scholar]
- 19.Anderson RS, Thomson SM, Gutshall LL., Jr Comparative effects of inhaled silica or synthetic graphite dusts on rat alveolar cells. Arch Environ Contam Toxicol. 1989;18:844–9. doi: 10.1007/BF01160299. [DOI] [PubMed] [Google Scholar]
- 20.Gagnon ZE, Newkirk C, Conetta JA, Sama MA, Sisselman S. Teratogenic effect of broad-band electromagnetic field on neonatal mice (Mus musculus) J Environ Sci Health A Tox Hazard Subst Environ Eng. 2003;38:2465–81. doi: 10.1081/ese-120024449. [DOI] [PubMed] [Google Scholar]
- 21.Tsuruta D, Mochida K, Hamada T, Ishii M, Wakasa K, Hashimoto S, Takekawa KE. Chemotherapy-induced acral erythema: report of a case and immunohistochemical findings. Clin Exp Dermatol. 2000;25:386–8. doi: 10.1046/j.1365-2230.2000.00670.x. [DOI] [PubMed] [Google Scholar]
- 22.Ulich TR, del Castillo J, Souza L. Kinetics and mechanisms of recombinant human granulocyte colony-stimulating factor-induced neutrophilia. Am J Pathol. 1988;133:630–8. [PMC free article] [PubMed] [Google Scholar]
- 23.Brach MA, deVos S, Gruss HJ, Herrmann F. Prolongation of survival of human polymorphonuclear neutrophils by granulocyte–macrophage colony-stimulating factor is caused by inhibition of programmed cell death. Blood. 1992;80:2920–4. [PubMed] [Google Scholar]
- 24.Spiekermann K, Roesler J, Emmendoerffer A, Elsner J, Welte K. Functional features of neutrophils induced by G-CSF and GM-CSF treatment: differential effects and clinical implications. Leukemia. 1997;11:466–78. doi: 10.1038/sj.leu.2400607. [DOI] [PubMed] [Google Scholar]
- 25.Wolach B, van der Laan LJ, Maianski NA, Tool AT, van Bruggen R, Roos D, Kuijpers TW. Growth factors G-CSF and GM-CSF differentially preserve chemotaxis of neutrophils aging in vitro. Exp Hematol. 2007;35:541–50. doi: 10.1016/j.exphem.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Lewkowicz P, Lewkowicz N, Sasiak A, Tchorzewski H. Lipopolysaccharide-activated CD4+ CD25+ T regulatory cells inhibit neutrophil function and promote their apoptosis and death. J Immunol. 2006;177:7155–63. doi: 10.4049/jimmunol.177.10.7155. [DOI] [PubMed] [Google Scholar]
- 27.Engeman T, Gorbachev AV, Kish DD, Fairchild RL. The intensity of neutrophil infiltration controls the number of antigen-primed CD8 T cells recruited into cutaneous antigen challenge sites. J Leukoc Biol. 2004;76:941–9. doi: 10.1189/jlb.0304193. [DOI] [PubMed] [Google Scholar]
- 28.James MJ, Belaramani L, Prodromidou K, et al. Anergic T cells exert antigen-independent inhibition of cell–cell interactions via chemokine metabolism. Blood. 2003;102:2173–9. doi: 10.1182/blood-2003-02-0637. [DOI] [PubMed] [Google Scholar]
- 29.Sarween N, Chodos A, Raykundalia C, Khan M, Abbas AK, Walker LS. CD4+ CD25+ cells controlling a pathogenic CD4 response inhibit cytokine differentiation, CXCR-3 expression, and tissue invasion. J Immunol. 2004;173:2942–51. doi: 10.4049/jimmunol.173.5.2942. [DOI] [PubMed] [Google Scholar]
- 30.Maloy KJ, Salaun L, Cahill R, Dougan G, Saunders NJ, Powrie F. CD4+ CD25+ T(R) cells suppress innate immune pathology through cytokine-dependent mechanisms. J Exp Med. 2003;197:111–9. doi: 10.1084/jem.20021345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorbachev AV, Fairchild RL. CD4+ T cells regulate CD8+ T cell-mediated cutaneous immune responses by restricting effector T cell development through a Fas ligand-dependent mechanism. J Immunol. 2004;172:2286–95. doi: 10.4049/jimmunol.172.4.2286. [DOI] [PubMed] [Google Scholar]
- 32.Kish DD, Gorbachev AV, Fairchild RL. CD8+ T cells produce IL-2, which is required for CD(4+) CD25+ cell regulation of effector CD8+ T cell development for contact hypersensitivity responses. J Leukoc Biol. 2005;78:725–35. doi: 10.1189/jlb.0205069. [DOI] [PubMed] [Google Scholar]
- 33.Huehn J, Siegmund K, Lehmann JC, et al. Developmental stage, phenotype, and migration distinguish naive- and effector/memory-like CD4+ regulatory T cells. J Exp Med. 2004;199:303–13. doi: 10.1084/jem.20031562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–58. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 35.de Vries JE. Immunsuppressive and anti-inflammatory properties of interleukin-10. Ann Med. 1995;27:537–41. doi: 10.3109/07853899509002465. [DOI] [PubMed] [Google Scholar]