

Abstract
A method based on the coupling of high resolution size-exclusion liquid chromatography using a polymer stationary phase with inductively coupled plasma mass spectrometry was developed to study the interactions of two metallodrugs – cisplatin and RAPTA-T – with the serum proteins albumin and transferrin. In contrast to previous approaches, the technique allowed the total recovery of the metals from the column and was able to discriminate between the different species of the metallodrugs and their complexes with the proteins at femtomolar detection levels. Metal binding was found to be dependent on the protein concentration and on the incubation time of the sample. Cisplatin was found to bind the serum proteins to the same extent, whereas RAPTA-T showed marked preference for transferrin. The affinity of the ruthenium complex for holo-transferrin was higher than for the apo-form suggesting a cooperative iron-mediated metal binding mechanism. RAPTA-T binding to holo-transferrin was further investigated by electrospray mass spectrometry using both the intact protein and a model peptide mimicking the iron-binding pocket.
Introduction
Dating back to the Renaissance and to Paracelsus’s alchemical studies, metals and minerals have profoundly fascinated physicians and have played a major role in the history of medicine.1 A large number of different metal salts and compounds are still used to treat a wide range of diseases. Notably, platinum coordination complexes are extensively used to treat cancer. Indeed, following the discovery of the anticancer properties of cisplatin in 1965 by Rosenberg,2 considerable efforts have been made to unravel the mechanism by which it exerts its anticancer effect and to develop alternative metal-based drugs.3–5 In recent years a vast number of non-platinum compounds have been prepared and evaluated as experimental anticancer drugs.6,7 Among them, ruthenium(II)-arene complexes bearing 1,3,5-triaza-7-phosphaadamantane (PTA) ligands – named RAPTA – have been found to exhibit promising antimetastatic properties in vivo.8 Their mechanism of action is still largely unknown, however, there is evidence to suggest that RAPTA compounds work on molecular targets other than DNA implying a biochemical mode of action profoundly different to classical platinum anticancer drugs.9,10 Indeed, ruthenium compounds have been shown to directly interfere with specific proteins involved in signal transduction pathways and/or to alter cell adhesion and migration processes.11
An understanding of metallodrug-protein interactions is crucial since the pharmacological activity of a drug, even for drugs where DNA is the ultimate target, is usually modified upon protein binding. Metal drug-protein interactions are not only important with regard to apoptosis, but also with respect to unwanted side-effects (due to indiscriminate protein binding), drug resistance (due to binding/deactivation by metal-lothioneins,12 glutathione transferases,13,14 etc.) and even possibly drug delivery and storage.15 Upon intravenous application, serum proteins including albumin, transferrin and globulins are thought to play a crucial role in the transport, delivery, and storage of anticancer metallodrugs. Human serum albumin (HSA) is the most abundant protein (about 52% of total serum protein) with a concentration of 40–45 g L−1 in healthy humans (ca. 600 μM; Mw 66–67 kDa). It comprises a single chain of 585 amino acids organized in three similar domains, each composed of two subdomains.16 HSA is known to bind a remarkably wide range of drugs, thereby restricting their free, active concentrations.17 Reducing this binding affinity represent a major challenge in drug development. However, HSA can also be exploited for targeted delivery strategies, as in the case of organometallic ruthenium(II)-arene anticancer compounds, to develop compounds that can selectively accumulate in tumors cells.18
Human serum transferrin (Tf) is a single-chain glycoprotein containing 679 amino acids with a molecular mass of about 80 kDa and is found in blood at a concentration of about 2.5 g L−1 (35 μM). Tf acts as an iron transporter and is capable of binding two iron(III) ions (Fe3+ is bound selectively over Fe2+).19 Tf is normally only 30% saturated with iron in the body,19 and at least 30 other metal ions can also bind to Tf.20 Therefore, it is possible to use Tf as a metal transporter within the body and the cellular uptake mechanism via the Tf-transferrin receptor transport system has the potential to be exploited for site-specific delivery of various therapeutic metal ions, drugs, proteins and genes.21 In the case of metallodrugs the resulting conjugates significantly improve the cytotoxicity and selectivity of the drugs itself. For example, anticancer Ru(III) complexes were reported not only to bind to Tf,22,23 but it has been shown that injection of Ru(III)-Tf conjugates resulted in high tumor uptake of the metal,24 which suggests that Tf uptake might be the more important mode of transport of Ru(III) anticancer complexes to the tumor. In some cases, the Ru-Tf adducts exhibit a significantly higher antitumor activity against human cancer cells than the Ru(III) complex itself, attributable to the Tf-mediated uptake mechanism, which may also lower drug toxicity.25
The development of analytical methods capable of providing a better understanding of how metallodrugs interact with proteins remains challenging.26,27 The direct monitoring of real-time protein-mediated metabolism of anticancer agents using hyphenated (coupled, tandem) techniques, that combine chromatographic separation with sensitive and element-specific detection techniques such as inductively coupled plasma-mass spectrometry (ICP-MS) have been effectively used.28–30 In terms of separation techniques, high performance liquid chromatography (HPLC)31,32 and capillary zone electrophoresis (CZE)29,33–35 were proposed for the study of platinum and ruthenium based drugs, to assess kinetic constants of hydrolysis and subsequent reactions with a variety of physiological targets. This concept has also been transferred to study the distribution of gallium-based pharmaceuticals in human serum.36
Within this frame, we propose size-exclusion liquid chromatography (SEC) coupled to ICP-MS to study metal complexes-proteins interactions. SEC has certain advantages compared to other separation techniques to characterize metallodrug binding to biomolecules in that it can be used together with other hyphenated systems to obtain complementary information. In comparison to ion exchange chromatography (IEC), the main advantage of the SEC approach lies in the more “gentle” conditions, as IEC usually requires buffers with high ionic strength and extreme pH values.37 The importance of working under more physiological-type conditions is crucial when studying metallodrugs to avoid interferences which might alter the metal-ligand bonds and the metal-protein adducts. With respect to capillary electrophoresis (CE) the major advantage of the SEC separation lies in the ease and robustness of the coupling to ICP-MS, which is considered one of the main problems of CE-ICP-MS, and usually leads to higher detection limits compared to LC-ICP-MS.35,38,39
Up to now, SEC coupled to ICP-MS has been used to probe protein interactions with ruthenium drugs, but suffers from poor recoveries because of the reactivity of the compounds with the column stationary phase.40 This problem was alleviated by using a short size-exclusion column at the expense of the chromatographic resolution, or it was simply used as a first separation technique to roughly discriminate between bound and unbound species prior to ion-exchange separation.40 Therefore, the primary objective of our research was to evaluate a polymeric stationary phase with regard to the unspecific retention, reactivity and carryover of an organometallic drug RAPTA-T (Ru(η6-C6H5Me)(PTA)Cl2) (Chart 1), and its adducts, with the serum proteins albumin and transferrin, in comparison to cisplatin. The binding of RAPTA-T towards intact Tf and the possible binding sites of the metallodrugs on a model peptide mimicking one of the iron-binding pockets of Tf were also investigated using electrospray ionization-mass spectrometry (ESI-MS).
Chart 1.
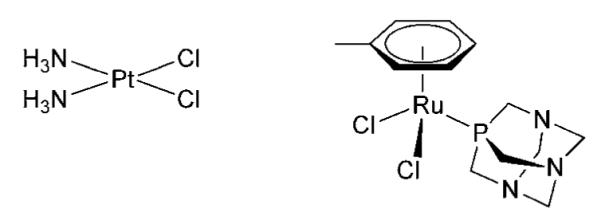
Structures of cisplatin (left) and RAPTA-T (right).
Experimental
Reagents
Cisplatin, human serum albumin (>99%, HSA), human holo transferrin (>98%, hTf) and human apo transferrin (>97%, apo-Tf) were obtained from Sigma-Aldrich. Sodium chloride, formic acid, Tris(hydroxymethyl)aminomethane hydrochloride, ammonium acetate and ammonium hydrogen carbonate were also purchased from Aldrich and were of analytical reagent grade. Buffers used for SEC-ICP-MS analysis were prepared as follows: ammonium acetate buffer (100 mM, pH 6.8) and Tris buffer saline (20 mM Tris-HCl, 100 mM NaCl, pH 7.2). Deionized water (18 MΩ/cm) (Millipore, Bedford, MA) was used throughout. RAPTA-T was synthesized as described elsewhere.8 The HPLC-purified model peptide (KDCHLAQVPSHTV) was purchased from PSL (Heidelberg, Germany).
ICP-MS instrumentation
An Agilent model 1200 binary pump, equipped with an auto-sampler, automatic injection system with a loop of 900 μL and a UV-VIS detector was used as the solvent delivery system and was controlled by Agilent ChemStation software. An Agilent 7500ce ICP-MS fitted with a Meinhardt nebulizer was used as the detector in SEC-ICP-MS experiments, employing the following conditions: plasma gas (Ar) 15 L min−1, forward power 1500 W, carrier gas 1.05 L min−1. The working conditions were optimised using a 1 μg L−1 solution of 7Li+, 89Y+ and 205Tl+ in 2% HNO3, with a dwell time of 100 ms for each isotope. For iron isotope monitoring, a collision cell was used to remove polyatomic interferences with H2 (3–3.5 ml min−1) and He (1.0 ml min−1) as auxiliary gases. Double charged ions and oxide levels were minimized by using 140Ce+ and were typically < 2%. The isotopes monitored (in time resolved analysis mode with a dwell time of 100 ms each) were 54Fe, 56Fe, 195Pt, 196Pt, 197Pt, 198Pt, 100Ru, 101Ru and 102Ru. The 54Fe, 56Fe, 102Ru, 195Pt and isotopes were used for quantification of binding and uptake.
Size-exclusion chromatography
Samples were analyzed on a Superdex™ 200 HR 10/30 (10 mm × 300 mm × 13 mm) (GE Healthcare) exclusion column with approximate bed volume of 24 mL and a linear separation range of 10–600 kDa for globular proteins. The column was calibrated as specified by the manufacturer with a mixture of thyroglobulin (670 kDa), hTf (81 kDa), bovine albumin (66 kDa), chicken egg albumin (44 kDa), Mn superoxide dismutase (39.5 kDa), Cu/Zn superoxide dismutase (32.5 kDa), carbonate dehydratase (29 kDa) and myoglobin (16.7 kDa) using UV/VIS detection at 280 nm with baseline evaluation at 800 nm. Retention times (in min) plotted versus the logarithm of molecular mass (in kDa) showed linear correlation (y=− 0.134x + 4.385; r2 0.992). Elution was carried out isocratically at 0.7 mL min−1 using 100 mM ammonium acetate (pH 6.8) as the mobile phase. Solvent flow from the chromatographic system was introduced into the ICP-MS nebulizer by means of PEEK tubing.
Sample preparation for SEC-ICP-MS
Cisplatin and RAPTA-T stock solutions were prepared by dissolving the compounds in ammonium acetate buffer or in Tris buffer. Proteins were dissolved in ammonium acetate at a concentration 0.5 mM. To investigate the interactions between serum proteins and the metal complexes, samples were initially prepared with various protein concentrations (1, 50 and 250 μM) and adding the appropriate amounts of metal complex to achieve different protein : metal stoichiometric ratios (1 : 1, 1 : 2 or 1 : 5) at each protein concentration. In the case of 250 μM protein samples only a maximum two-fold molar excess of the metal complex was applied due to protein precipitation phenomena. Samples were then incubated for 1 h at 37 °C and then diluted in ammonium acetate buffer to yield a final protein concentration of 1 μM prior to injection into the SEC column. Alternatively, for the evaluation of maximum metal binding, the protein concentration was fixed at 50 μM, while each metal complex was added in stoichiometric amount or in 2, 5, 10 and 18-fold excesses. Metal complex solutions were added freshly prepared (sample Set A) or 24 h after preparation (sample Set B; solution stored at 4 °C) in ammonium acetate buffer. Samples were then incubated for 1 h at 37 °C and diluted to 1 μM protein before injection. Quantification of the metal content in each chromatographic fraction was calculated directly from the integrated signal from the SEC-ICP-MS chromatograms using the formula cb = Mb × cinj × D, where cb represents the initial concentration of metal in the original sample, Mb is the fraction of metal bound to protein in the chromatogram (calculated as a percentage of bound metal signal with respect to the total sum of signals), cinj is the concentration of metal injected for analysis and D is the dilution factor applied to the samples prior to injection.
ESI-MS analysis
For ESI-MS experiments with the model peptide, samples were prepared with a metallodrug : peptide ratio of 1 : 1 (25 μM) in 20 mM carbonate buffer (pH 7.4) and incubated for 24 h at 37 °C prior to analysis. Samples were then mixed with acetonitrile and formic acid (final concentration 30% and 0.1%, respectively) and placed into a 96-well plate in an Advion TriVersa robot (Advion Biosciences, Ithaca, NY) equipped with a 5.5 mm-nozzle chip. The ESI robot was controlled with ChipSoft v7.2.0 software employing the following parameters: gas pressure 0.45 psi; voltage 1.7 kV. The samples were analyzed in positive ion mode using a LTQ XL mass spectrometer (ThermoFisher Scientific, Bremen, Germany). The Xcalibur software bundle was utilized for data acquisition and data analysis.
ESI-MS measurements of apo transferrin-metallodrug interactions were carried out on an Ultima II q-TOF mass spectrometer (Waters, Manchester, UK) operated in positive mode. Data acquisition and analysis were carried out using the MassLynx software bundle (Waters). The instrument was calibrated daily using a 0.01% phosphoric acid solution in 50% acetonitrile. Samples were prepared with a metallodrug : protein ratio of 5 : 1 in 20 mM carbonate buffer (pH 7.4) and incubated for up to 24 h at 37 °C. Prior to analysis, samples were ultracentrifuged using 30 kDa cut-off filters (VWR, Switzerland) to remove unbound metallodrugs and injected into the ESI-MS system after dilution with acetonitrile and formic acid (final concentrations 25% and 0.05%, respectively).
Results and discussion
Size-exclusion chromatography of cisplatin and RAPTA-T
A SEC-ICP-MS system was initially used to evaluate solutions of the two metal drugs, cisplatin and RAPTA-T (Chart 1), in the absence of any protein in order to establish the evolution of metal species. Based on the molecular weight of the complexes (300–400 Da) an expected single peak with a total bed column volume (≈24 mL), corresponding to 35 min at flow rate of 0.7 mL min−1, might be expected to elute from the column. Two different buffers, namely Tris buffer saline (TBS) and ammonium acetate (pH ~ 7), were evaluated as incubation media and eluent for cisplatin and RAPTA-T hydrolysis. For method development, 50 μL of 1 μM cisplatin (195 ng mL−1, 50 pmol of Pt) or RAPTA-T (102 ng mL−1, 50 pmol of Ru) in the respective buffers were injected into the SEC column either freshly prepared or pre-incubated for 12 h at 37 °C.
Elution profiles for the two drugs in TBS are shown in Fig. 1. In the case of cisplatin (Fig. 1A), freshly prepared solution gave signals at 32 and 42 min, while for the sample obtained after 12 h incubation an additional peak appeared at 28 min. In both cases the peak at 42 min (intact cisplatin) was the most intense. Peaks at 32 and 28 min corresponded to cisplatin hydrolysis products (mono- and bis-aqua species, respectively) as confirmed by ESI-MS analysis. In ammonium acetate buffer, these compounds were detected at the same elution times, but more extensive hydrolysis of cisplatin was observed due to the absence of chlorido ions in the buffer. It is worth mentioning that the separation of different forms of metallodrugs arise from aspecific interactions with the stationary phase, since no differentiation is possible based on their low molecular weights. Elution times were found to be constants after multiple injection of metallodrug. Only for peaks eluting at 40 min, well after the total column volume, minor shifts of ±2 min in the elution time were observed.
Fig. 1.

SEC-ICP-MS elution profiles of the investigated metallodrugs and Tf. A: The ion chromatogram (195Pt+) compares a freshly prepared cisplatin solution (red bold line) to a cisplatin solution after 12 h incubation at 37 °C (red dotted line). Peak 3 at 28 min: di-aqua complex; peak 2 at 31 min: mono-aqua complex; peak 1 at 40 min: intact cisplatin. B: Chromatogram shows the single elution peak (peak 4, 102Ru+) of RAPTA-T at 28 min. C: Overlay of ion chromatograms for both hTf (grey trace) and apo-Tf (black trace) eluting at 19 min (peak 5: Tf; 56Fe+).
When RAPTA-T was injected, either freshly prepared or after 12 h of incubation, only a single symmetric peak at 28 min of the chromatographic run was observed regardless of the buffer employed (Fig. 1B). This peak probably corresponds to the intact complex or to the mono-aqua [Ru(η6-C6H5Me)(PTA)ClH2O] species as suggested by ESI-MS analysis and UV-visible spectrophotometry (data not shown).
Recovery experiments, based on the comparison of the metal content in the injected sample and the eluate, showed that the metals were not retained on the stationary phase. Indeed, recovery values for cisplatin and RAPTA-T were 100 ± 2% and 93 ± 3% respectively, in both buffers. The baseline remained stable during the analyses and a post-analysis injection of HSA gave a blank chromatogram.
Interaction of metallodrugs with serum proteins
The binding of cisplatin and RAPTA-T to transferrin – both holo- (hTf) and apo-transferrin (apo-Tf) – and human serum albumin was investigated by SEC-ICP-MS. Since no differences in the chromatographic separation of the complexes were observed as a function of the buffer, ammonium acetate was selected as it exhibited sufficient ionic strength to limit the interactions of the proteins with the stationary phase and is better tolerated in ICP-MS.41 The two proteins investigated in this study have a molecular mass which is not expected to be separated by this SEC column. In fact, calculated elution times for transferrin and albumin from column calibration curve are 18.5 and 18.9 min when molecular weights of 80 and 70 kDa are taken into account. Holo- and apo-transferrin were analyzed first in order to establish their elution time; note that hTf carries iron which can be used for ICP-MS analysis. Fig. 1C shows the chromatograms (56Fe+) of 1 μM hTf and apo-Tf samples in comparison to the ion chromatograms of the metallodrugs (195Pt+, 102Ru+) (Fig. 1A and 1B) to demonstrate the baseline separation of the iron-containing protein peak from the other metal species present in solution. The results show that the apo-Tf is not completely iron-free. To establish the amount of iron present in holo- and apo-transferrin, the proteins were digested and analyzed by ICP-MS. The results reveal a stoichiometric metal : protein ratio of 1.22 ± 0.12 for hTf and 0.20 ± 0.05 for apo-Tf. Notably, there is no change in retention time for both Tf and HSA upon adduct formation with the metallodrugs, since the resulting increase in the molecular mass is negligible. Moreover, in the case of the incubation mixtures between Tf and HSA with both the drugs, recovery experiments give values similar to those calculated for the free compounds.
Due to the high resolution separation of the metal-containing species and their excellent recovery rates, quantitative data on the amount of drug binding could be extracted directly from the chromatograms. With the optimized method two types of experiments were set up. These experiments aimed at, first, observing the effect of different protein concentrations and relative protein : metal ratios on the adduct formation, and second, quantitating the extent of adduct formation at increasing metallodrug concentrations.
Effect of protein concentration on adduct formation
Different concentrations (1, 50 and 250 μM) of single serum proteins were incubated with each metal complex at different protein : metal ratios (1 : 1, 1 : 2 and 1 : 5) for 1 h at 37 °C. Representative SEC-ICP-MS chromatograms for samples of HSA incubated with RAPTA-T are given in Fig. 2, the whole dataset for interactions of the serum proteins with RAPTA-T and cisplatin is summarized in Table 1.
Fig. 2.
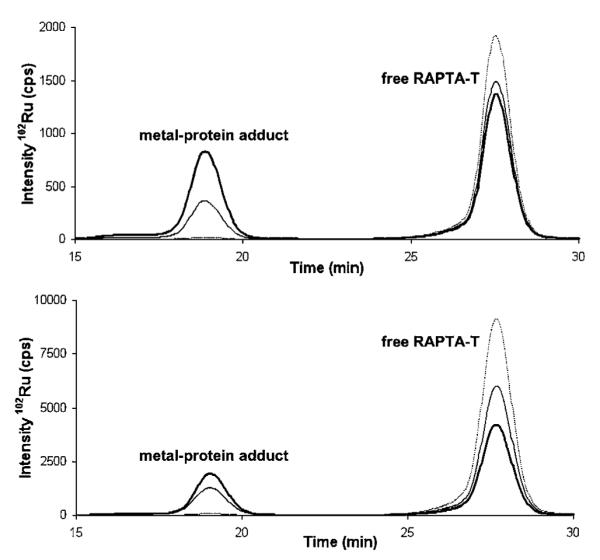
SEC-ICP-MS chromatogram of HSA incubated with RAPTA-T at different protein : metal ratios for 1 h at 37 °C. 1 : 1 (top) and 1 : 5 (bottom). Protein concentrations are 1 μM (dotted line), 50 μM (thin line) and 250 μM (bold line). All samples were diluted to 1 μM (protein) prior to injection into the SEC-ICP-MS system. The peak at 19 min corresponds to the HSA-RAPTA-T adduct, the peak at 28 min to free RAPTA-T.
Table 1.
Evaluation of metal binding by SEC-ICP-MS for samples with different metallodrug : protein ratios, incubated for 1 h at 37 °C. Results are the average of three independent experiments with S.D. < 2%
| Protein (μM) | Drug (μM) | Molar ratio metal : protein |
Metal bound (%) |
|
|---|---|---|---|---|
| cisplatin | RAPTA-T | |||
| hTf | ||||
| 250 | 250 | 1 | 17.3 | 38.4 |
| 250 | 500 | 2 | 16.0 | 30.4 |
| 50 | 50 | 1 | 5.9 | 19.0 |
| 50 | 250 | 5 | 5.7 | 16.9 |
| 1 | 1 | 1 | 0.2 | 1.1 |
| 1 | 5 | 5 | 0.3 | 0.8 |
| apo-Tf | ||||
| 250 | 250 | 1 | 14.5 | 37.2 |
| 250 | 500 | 2 | 14.8 | 36.6 |
| 50 | 50 | 1 | 2.9 | 10.1 |
| 50 | 250 | 5 | 3.2 | 9.7 |
| 1 | 1 | 1 | 0.2 | 0.7 |
| 1 | 5 | 5 | 0.2 | 0.7 |
| HSA | ||||
| 250 | 250 | 1 | 21.1 | 18.7 |
| 250 | 500 | 2 | 20.8 | 18.1 |
| 50 | 50 | 1 | 6.6 | 7.5 |
| 50 | 250 | 5 | 5.3 | 6.8 |
| 1 | 1 | 1 | 0.5 | 0.3 |
| 1 | 5 | 5 | 0.6 | 0.4 |
The data indicate that RAPTA-T can bind equally to both hTf and apo-Tf but to a significantly greater extent compared to cisplatin, regardless of the applied concentrations and metal complex : protein ratios. In contrast, similar metal binding of both metallodrugs with HSA was observed. In particular, since the short incubation time does not allow reaching the thermodynamic equilibrium, a kinetic preference of RAPTA-T for binding to Tf was detected with respect to HSA. However, it must be considered that HSA is about 20-times more concentrated than Tf in blood and therefore favoured for metal binding, as shown for the ruthenium(III) anticancer compound KP1019.42 Interestingly, the incubation concentrations affect the formation of protein-metal adducts, metal binding being favoured at higher concentrations of both the metallodrugs and the proteins.
Influence of hydrolysis on protein binding
Further experiments on protein binding and relative binding affinities of the metallodrugs were performed using a fixed concentration of proteins, 50 μM, (which represents a concentration similar to that of Tf in blood) incubated with different amounts of the metallodrugs. Quantitative metal binding data were calculated based on the fraction eluted at 19 min, related to the complexed forms of protein with metallodrugs, and the evaluation of all the other forms of free metallodrugs in the SEC-ICP-MS profiles by the equation stated in the Experimental Part.
It should be noted that the same results are obtained if the equation developed by Timerbaev et al.33 is applied to the integrated values for the eluted species in our SEC-ICP-MS chromatogram. In Table 2, results from each protein sample treated with RAPTA-T or cisplatin and incubated for 1 h at 37 °C are shown. Dataset A represents data for samples with freshly prepared solutions of the metal complexes and Dataset B for samples with metal complex solutions pre-incubated for 24 h prior to addition to the protein to study the effect of metallodrug hydrolysis on protein binding.
Table 2.
Quantification of adduct formation between cisplatin and RAPTA-T with the serum proteins hTf, apo-Tf and HSA. Protein concentration was fixed at 50 μM, the metal : protein ratio varied between 1 : 1 and 18 : 1. Samples were incubated for 1 h at 37 °C. (Set A: freshly prepared metal complex solution added. Set B: 24 h pre-incubated metal complex solution added.) Results are the average of three independent experiments with S.D. < 2%
| Compound | Molar ratio metal : protein (theoret.) |
Set A (μM) |
Molar ratio metal : protein (experimental) |
Set B (μM) |
Molar ratio metal : protein (experimental) |
|---|---|---|---|---|---|
|
hTf Cisplatin |
1 | 1.4 | 0.03 | 1.7 | 0.03 |
| 2 | 2.7 | 0.05 | 3.3 | 0.07 | |
| 5 | 6.4 | 0.13 | 7.5 | 0.15 | |
| 10 | 8.1 | 0.16 | 14.4 | 0.29 | |
| 18 | 11.4 | 0.23 | 16.9 | 0.34 | |
| RAPTA-T | 1 | 8.5 | 0.17 | 8.2 | 0.16 |
| 2 | 14.7 | 0.29 | 15.3 | 0.31 | |
| 5 | 33.1 | 0.66 | 32.3 | 0.65 | |
| 10 | 56.8 | 1.14 | 57.0 | 1.14 | |
| 18 | 70.9 | 1.42 | 69.4 | 1.39 | |
|
apo-Tf Cisplatin |
1 | 1.0 | 0.02 | 1.7 | 0.03 |
| 2 | 2.0 | 0.04 | 3.1 | 0.06 | |
| 5 | 4.4 | 0.09 | 7.0 | 0.14 | |
| 10 | 7.6 | 0.15 | 14.0 | 0.28 | |
| 18 | 12.2 | 0.24 | 19.2 | 0.38 | |
| RAPTA-T | 1 | 3.2 | 0.06 | 4.6 | 0.09 |
| 2 | 7.8 | 0.16 | 8.8 | 0.18 | |
| 5 | 16.0 | 0.32 | 18.5 | 0.37 | |
| 10 | 26.1 | 0.52 | 34.7 | 0.69 | |
| 18 | 33.1 | 0.66 | 52.9 | 1.06 | |
|
HSA Cisplatin |
1 | 2.1 | 0.04 | 3.9 | 0.08 |
| 2 | 4.3 | 0.09 | 6.8 | 0.14 | |
| 5 | 7.3 | 0.15 | 11.8 | 0.24 | |
| 10 | 10.0 | 0.20 | 16.7 | 0.33 | |
| 18 | 11.2 | 0.16 | 17.4 | 0.35 | |
| RAPTA-T | 1 | 3.3 | 0.07 | 3.8 | 0.08 |
| 2 | 6.7 | 0.13 | 7.1 | 0.14 | |
| 5 | 13.3 | 0.27 | 15.9 | 0.32 | |
| 10 | 20.3 | 0.41 | 30.4 | 0.61 | |
| 18 | 24.2 | 0.48 | 32.0 | 0.64 |
The most notable difference between the two preparations corresponds to the availability of the different forms of the metallodrugs. For instance, in Dataset B higher contents of hydrolyzed products are present for cisplatin with respect to Set A. Since cisplatin hydrolysis products carry a positive charge, their interaction with negatively charged amino acid residues on the protein chains (possible under these conditions considering calculated pI values are 5.92 and 6.81 for HSA and Tf, respectively)43 potentially increases interactions compared to the neutral starting compound. Moreover, the hydrolyzed product is more reactive as the aqua ligands are more labile than chlorido ligands. In Set B, platinum adducts are formed to a greater extent with respect to Set A at any concentration. In general, for both datasets, cisplatin shows a slightly increased affinity towards HSA at lower molar excess of drug and no apparent preference for the two forms of transferrin. Upon increasing the drug excess, the reaction proceeds more rapidly and, as expressed by an increased number of metal ions bound per protein molecule, the proteins are metallated to a higher degree.
For RAPTA-T, a higher affinity is found for hTf with respect to apo-Tf and HSA, and the amount of ruthenium bound to hTf is similar in Set A and Set B. For apo-Tf and HSA, increased binding is observed for the Set B samples, especially at high metal : protein ratios. Based on the ratio of the metal concentration of the bound fraction, for cisplatin maximum binding was in the range from 0.2 to 0.35 for all three proteins. For RAPTA-T the highest metal binding was observed for hTf, with an average of ca. 1.4 ruthenium ions per protein. For apo-Tf and HSA an average 0.5 to 1.0 ruthenium ions bind per protein, which is higher than observed for cisplatin. In comparison to previously reported data using CZE-ICP-MS, which shows extensive binding of cisplatin toward the protein (up to 4 Pt ions per molecule of HSA for a sample containing a 10-fold excess of metal complex after 24 h incubation in PBS buffer), the values obtained for cisplatin-HSA adduct formation in our experiments were unexpected.33 Therefore, protein samples incubated for 24 h with a 10-fold excess of cisplatin or RAPTA-T were studied. For all the three proteins increased metal binding for both metallodrugs was detected (Table 3), although not to the same levels observed by CZE-ICP-MS.
Table 3.
Influence of different incubation times (1 and 24 h) on the extent of adduct formation between metallodrugs and the serum proteins. Protein concentration was fixed at 50 μM with a metal complex : protein ratio = 10 : 1. Results are the average of three independent experiments with S.D. < 2%
| Incubation time |
Cisplatin |
RAPTA-T |
|||
|---|---|---|---|---|---|
| metal bound (μM) |
metal : protein ratio |
metal bound (μM) |
metal : protein ratio |
||
| 1 h | hTf | 8.1 | 0.16 | 56.8 | 1.14 |
| apo-Tf | 7.6 | 0.15 | 26.1 | 0.52 | |
| HSA | 10.0 | 0.20 | 20.3 | 0.41 | |
| 24 h | hTf | 31.6 | 0.6 | 93.5 | 1.9 |
| apo-Tf | 25.6 | 0.5 | 50.8 | 1.0 | |
| HSA | 36.6 | 0.7 | 46.3 | 0.9 | |
Overall, the low detection limits of the applied method allowed analysis of samples containing 400 fmol of metals bound to the proteins, with as little as 2 pmol of protein injected into SEC column. In the case of RAPTA-T, the higher amount of ruthenium bound to hTf with respect to apo-Tf suggests that the presence of iron bound to protein might influence the formation of metal complex adducts, most likely maintaining a protein folding which favours metallodrug binding.
Metallodrug binding versus iron loss in holo-transferrin
Despite the higher affinity of RAPTA-T than cisplatin for hTf, no differences in terms of the released iron were observed. Fig. 3 shows the binding behaviour of both metal complexes for hTf, together with the decrease of the iron signal recorded by ICP-MS, for each concentration of metal complex. The graph shows that the iron is not released in a linear fashion. In fact, the iron signal decreases only slightly until a 10-fold molar excess (500 μM) of the metallodrug and then immediately drops to the half of its original value. As the employed hTf was only ca. 60% loaded with iron, these data suggest that initially metallodrug binding occurs at the iron-free protein active sites and therefore does not affect the iron release. Afterwards, in the presence of excess of metallodrug, significant iron release may result from the partial denaturation or conformational changes of Tf upon addition of excess of metal, in agreement with previous observation on cisplatin binding to hTf by Hoshino et al.33
Fig. 3.
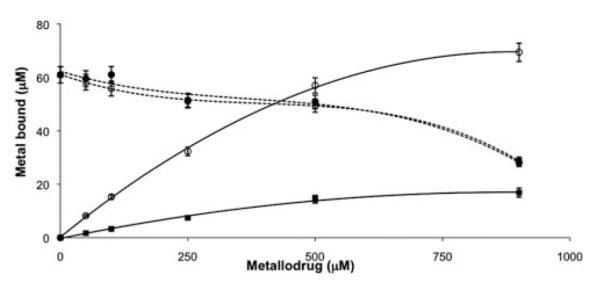
Concentration dependent binding of cisplatin and RAPTA-T towards holo-transferrin. Pt = ■; Ru=○. Dotted traces represent the corresponding iron signal.
ESI-MS studies with transferrin and an iron binding site-model peptide
As the interactions of cisplatin with intact transferrin studied by ESI-MS have already been reported earlier,44 only the binding of RAPTA-T to transferrin was further investigated by ESI-MS in this study. Fig. 4 shows the spectrum of apo-Tf treated with a 5-fold excess of RAPTA-T and incubated for 1 h at 37 °C, in comparison to the pure protein. The spectra of the RAPTA-T treated samples show additional signals corresponding to adduct formation with the ruthenium compound. Based on the mass difference, these signals are attributable to RAPTA-T after loss of both chlorido and PTA ligands and subsequent addition of a carbonate anion. The fact that only the arene ligand stays attached to the metal centre is in good agreement with data obtained for interactions of RAPTA compounds with cytochrome c,45 and the coordination of carbonate is also not surprising under the conditions employed. It is worth noting that a synergistic carbonate anion interaction is also crucial for stabilizing the binding site to ensure efficient binding of iron to transferrin.
Fig. 4.

ESI-MS spectrum of pure apo-transferrin (left) and incubated with RAPTA-T for 1 h at 37 °C at a metallodrug : protein ratio of 5 : 1 (right). Stars indicate the additional peaks caused by the adduct formation.
Furthermore, the exact binding mode of cisplatin and RAPTA-T towards Tf and the nature of the species that bind to the protein as well as the identification of the amino acids involved were probed by ESI-MS utilizing a model peptide consisting of the active site residues 239KDCHLAQVPSHTV251 of Tf. This peptide contains the His249 residue, which is involved in the iron binding in Tf, but also threonine and cysteine, residues which are possible binding partners.44,46,47 The model peptide was incubated with the metallodrugs under simulated physiological conditions (pH 7.4, 37 °C) for 24 h and then analysed. The bound species were identified as RAPTA-T and cisplatin after loss of the chlorido ligands, indicating a bidentate binding to the peptide. Although loss of a chlorido ligand could also be induced during ionization, a multidentate binding mode seems more probably due to hydrolysis of the compounds under the used incubation conditions. Different amino acids depending on the metal complex were identified as binding partners upon collision induced dissociation (CID) of the modified peptide, as can be seen from Fig. 5 and 6. The insets show signals of metallated ions, exhibiting characteristic isotopic patterns induced by ruthenium and platinum, which facilitate the identification of modified residues. In general, the remaining ligands of the metallodrugs (amino for cisplatin, toluene and PTA for RAPTA-T) were dissociated more easily than the peptide bonds under fragmentation conditions, indicating the formation of strong bonds between the amino acids and the metals.
Fig. 5.
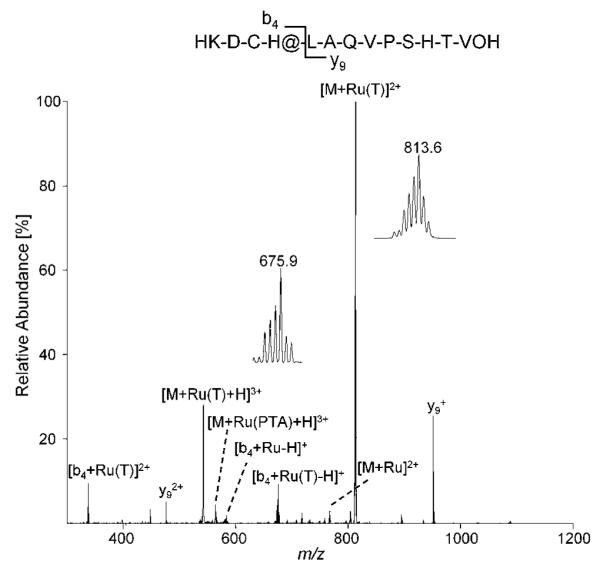
MS/MS spectra of the transferrin iron-binding site model peptide after incubation with RAPTA-T revealing histidine as the binding partner. The insets show the [M + Ru(T)]2+ (T = toluene) ion at 813.6 m/z as well as the [b4 + Ru(T)-H]+ ion at 675.9 m/z with an isotopic distribution characteristic for ruthenium. The fragmented parent ion corresponds to [M + Ru(PTA)(T)+H]3+.
Fig. 6.
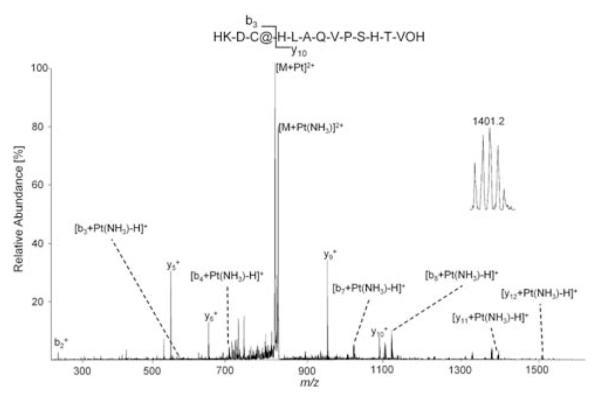
MS/MS spectra of the transferrin iron-binding site model peptide after incubation with cisplatin revealing cysteine as the binding partner. The insets shows the [y11 + Pt(NH3)-H]+ ion at 1401.2 m/z with an isotopic distribution characteristic of platinum. The fragmented parent ion corresponds to [M + Pt(NH3)2 + H]3+.
For RAPTA-T (Fig. 5), the first histidine residue appears to be the major binding partner, as evidenced by the presence of the modified b4 ion in contrast to the unmodified y9 ion following CID. This is not surprising since nitrogen donor ligands are known to form strong complexes with ruthenium. Interestingly, no binding towards the histidine residue in position 11 of the peptide (iron binding His249 in transferrin) could be detected for the ruthenium compound, indicating that the cysteine residue might also be involved in the binding of RAPTA-T. It is also worth mentioning that cleavage of the peptide occurs predominately next to the metal binding site, resulting in low relative abundances of other b and y fragment ions.
In the case of cisplatin (Fig. 6), the sulfur containing cysteine residue seems to be responsible for the binding. CID resulted in a more extensive fragmentation of the peptide as compared to RAPTA-T, and unmodified b2 and y10 in contrast to modified b3 and y11 ions allow unambiguous identification of the binding site. Although these data are in agreement with previous studies which show that cisplatin readily reacts with sulfur containing biomolecules such as glutathione or metallothionein within the cell,48 caution should be applied to the validity of the model peptide in terms of comparability to intact proteins. As it has been shown in earlier proteomic studies of transferrin-cisplatin interaction44 as well as on samples extracted from E. coli upon incubation with a ruthenium-arene compound and cisplatin,49,50 oxygen atoms in the carboxy- and hydroxyl-functions of certain residues (e.g. aspartate, serin, threonine) may also serve as binding partners in biological systems. Consequently, protein conformation has a strong influence on the site and type of adduct formed as exposed residues might be more easily accessible and therefore more reactive than those buried in the folded protein. Although such structural features cannot be mimicked by a short, linear model peptide, the general reactivity of a compound in the presence of several possible binding amino acids can be assessed.
Conclusions
High resolution SEC-ICP-MS based on a polymer stationary phase was demonstrated, for the first time, to be a powerful tool for the analysis of metal drugs with serum proteins. The approach provides information on the affinity of each metallodrug for the selected proteins, quantification of metal uptake and on the stoichiometry of the metal-protein complexes. The primary advantage of this approach over the previous SEC-ICP-MS methods is that all the metal species are quantitatively recovered under the separation conditions. Although the SEC system was not suitable for the baseline separation of samples containing both HSA and Tf, it proved to be an efficient tool to analyze the binding of metallodrugs to single proteins and in principle could be developed to investigate protein mixtures. Indeed, recently the use of SEC hyphenated to ICP atomic-emission spectrometry has been successfully applied for metalloproteome studies of human plasma.51 The reported results highlight the different reactivity of cisplatin and the antimetastatic ruthenium-based drug RAPTA-T. The latter compound showed a preferential binding for Tf with respect to HSA, while cisplatin was less selective, being able to bind both proteins to a similar extent. Differences in the selectivity of cisplatin and RAPTA compounds were already identified using an ESI-MS based approach to study protein mixtures.52,53 In particular, cisplatin was found to be moderately reactive towards the protein without any discrimination/selectivity, whereas the RAPTA compound was considerably more reactive and could also bind selectively. Such selectivity has important implications for the mode of action of the metallodrugs in the cell, leading to the efficacy of the compound towards specific tumor types, and presumably also for their toxic side effects.
Acknowledgements
A.C gratefully acknowledges the Swiss National Science Foundation (AMBIZIONE project n° PZ00P2_121933), the Swiss Confederation (Action COST D39 - Accord de recherche - SER project n° C09.0027) and COST D39 for financial support. M.G. thanks the Austrian Science Foundation for financial support (Schrödinger Fellowship J2882-N19).
Footnotes
This article is part of a themed issue devoted to highlighting the work of outstanding young analytical scientists (YAS) working in the area of analytical atomic spectrometry. This 3rd YAS issue has been guest edited by Professor Spiros Pergantis.
References
- 1.Koehler CSW. Today’s Chemists at Work. 2001;10:61–65. [Google Scholar]
- 2.Rosenberg B, Vancamp L, Krigas T. Nature. 1965;205:698. doi: 10.1038/205698a0. [DOI] [PubMed] [Google Scholar]
- 3.Clarke MJ, Zhu FC, Frasca DR. Chem. Rev. 1999;99:2511–2533. doi: 10.1021/cr9804238. [DOI] [PubMed] [Google Scholar]
- 4.Reedijk J. Proc. Natl. Acad. Sci. U. S. A. 2003;100:3611–3616. doi: 10.1073/pnas.0737293100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casini A, Hartinger C, Gabbiani C, Mini E, Dyson PJ, Keppler BK, Messori L. J. Inorg. Biochem. 2008;102:564–575. doi: 10.1016/j.jinorgbio.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Nobili S, Mini E, Landini I, Gabbiani C, Casini A, Messori L. Med. Res. Rev. 2009 doi: 10.1002/med.20168. DOI: 10.1002/med.20168. [DOI] [PubMed] [Google Scholar]
- 7.Gianferrara T, Bratsos I, Alessio E. Dalton Trans. 2009;(37):7588–7598. doi: 10.1039/b905798f. [DOI] [PubMed] [Google Scholar]
- 8.Scolaro C, Bergamo A, Brescacin L, Delfino R, Cocchietto M, Laurenczy G, Geldbach TJ, Sava G, Dyson PJ. J. Med. Chem. 2005;48:4161–4171. doi: 10.1021/jm050015d. [DOI] [PubMed] [Google Scholar]
- 9.Dyson PJ, Sava G. Dalton Trans. 2006:1929–1933. doi: 10.1039/b601840h. [DOI] [PubMed] [Google Scholar]
- 10.Bergamo A, Sava G. Dalton Trans. 2007:1267–1272. doi: 10.1039/b617769g. [DOI] [PubMed] [Google Scholar]
- 11.Bergamo A, Masi A, Dyson PJ, Sava G. Int J Oncol. 2008;33:1281–1289. [PubMed] [Google Scholar]
- 12.Knipp M. Curr. Med. Chem. 2009;16:522–537. doi: 10.2174/092986709787458452. [DOI] [PubMed] [Google Scholar]
- 13.Ang WH, Parker LJ, De Luca A, Juillerat-Jeanneret L, Morton CJ, Lo Bello M, Parker MW, Dyson PJ. Angew. Chem., Int. Ed. 2009;48:3854–3857. doi: 10.1002/anie.200900185. [DOI] [PubMed] [Google Scholar]
- 14.Ang WH, De Luca A, Chapuis-Bernasconi C, Juillerat-Jeanneret L, Lo Bello M, Dyson PJ. ChemMedChem. 2007;2:1799–1806. doi: 10.1002/cmdc.200700209. [DOI] [PubMed] [Google Scholar]
- 15.Reedijk J. Chem. Commun. 1996:801–806. [Google Scholar]
- 16.He XM, Carter DC. Nature. 1992;358:209–215. doi: 10.1038/358209a0. [DOI] [PubMed] [Google Scholar]
- 17.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. J. Mol. Biol. 2005;353:38–52. doi: 10.1016/j.jmb.2005.07.075. [DOI] [PubMed] [Google Scholar]
- 18.Ang WH, Daldini E, Juillerat-Jeanneret L, Dyson PJ. Inorg. Chem. 2007;46:9048–9050. doi: 10.1021/ic701474m. [DOI] [PubMed] [Google Scholar]
- 19.Gomme PT, McCann KB. Drug Discovery Today. 2005;10:267–273. doi: 10.1016/S1359-6446(04)03333-1. [DOI] [PubMed] [Google Scholar]
- 20.Sun HZ, Li HY, Sadler PJ. Chem. Rev. 1999;99:2817–2842. doi: 10.1021/cr980430w. [DOI] [PubMed] [Google Scholar]
- 21.Li HY, Qian ZM. Med. Res. Rev. 2002;22:225–250. doi: 10.1002/med.10008. [DOI] [PubMed] [Google Scholar]
- 22.Smith CA, SutherlandSmith AJ, Keppler BK, Kratz F, Baker EN. JBIC, J. Biol. Inorg. Chem. 1996;1:424–431. [Google Scholar]
- 23.Kratz F, Hartmann M, Keppler B, Messori L. J. Biol. Chem. 1994;269:2581–2588. [PubMed] [Google Scholar]
- 24.Som P, Oster ZH, Matsui K, Guglielmi G, Persson BRR, Pellettieri ML, Srivastava SC, Richards P, Atkins HL, Brill AB. Eur. J. Nucl. Med. Mol. Imaging. 1983;8:491–494. doi: 10.1007/BF00598908. [DOI] [PubMed] [Google Scholar]
- 25.Kratz F, Keppler BK, Hartmann M, Messori L, Berger MR. Met.-Based Drugs. 1996;3(1):15–23. doi: 10.1155/MBD.1996.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Chem. Rev. 2006;106:2224–2248. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 27.Sun XS, Tsang CN, Sun HZ. Metallomics. 2009;1:25–31. [Google Scholar]
- 28.Becker JS, Jakubowski N. Chem. Soc. Rev. 2009;38:1969–1983. doi: 10.1039/b618635c. [DOI] [PubMed] [Google Scholar]
- 29.Brouwers EEM, Tibben M, Rosing H, Schellens JHM, Beijnen JH. Mass Spectrom. Rev. 2008;27:67–100. doi: 10.1002/mas.20159. [DOI] [PubMed] [Google Scholar]
- 30.Lobinski R, Schaumloffel D, Szpunar J. Mass Spectrom. Rev. 2006;25:255–289. doi: 10.1002/mas.20069. [DOI] [PubMed] [Google Scholar]
- 31.Hann S, Stefanka Z, Lenz K, Stingeder G. Anal. Bioanal. Chem. 2005;381:405–412. doi: 10.1007/s00216-004-2839-z. [DOI] [PubMed] [Google Scholar]
- 32.Esteban-Fernandez D, Montes-Bayon M, Gonzalez EB, Gomez MMG, Palacios MA, Sanz-Medel A. J. Anal. At. Spectrom. 2008;23:378–384. [Google Scholar]
- 33.Timerbaev AR, Aleksenko KS, Polec-Pawlak K, Ruzik R, Semenova O, Hartinger CG, Oszwaldowski S, Galanski M, Jarosz M, Keppler BK. Electrophoresis. 2004;25:1988–1995. doi: 10.1002/elps.200305984. [DOI] [PubMed] [Google Scholar]
- 34.Polec-Pawlak K, Abramski JK, Semenova O, Hartinger CG, Timerbaev AR, Keppler BK, Jarosz M. Electrophoresis. 2006;27:1128–1135. doi: 10.1002/elps.200500694. [DOI] [PubMed] [Google Scholar]
- 35.Groessl M, Hartinger CG, Polec-Pawlak K, Jarosz M, Keppler BK. Electrophoresis. 2008;29:2224–2232. doi: 10.1002/elps.200780790. [DOI] [PubMed] [Google Scholar]
- 36.Groessl M, Bytzek A, Hartinger CG. Electrophoresis. 2009;30:2720–2727. doi: 10.1002/elps.200800745. [DOI] [PubMed] [Google Scholar]
- 37.Michalke B. TrAC, Trends Anal. Chem. 2002;21:142–153. [Google Scholar]
- 38.Moini M. Anal. Bioanal. Chem. 2002;373:466–480. doi: 10.1007/s00216-002-1283-1. [DOI] [PubMed] [Google Scholar]
- 39.Yin XB, Li Y, Yan XP. TrAC, Trends Anal. Chem. 2008;27:554–565. [Google Scholar]
- 40.Szpunar J, Makarov A, Pieper T, Keppler BK, Lobinski R. Anal. Chim. Acta. 1999;387:135–144. [Google Scholar]
- 41.Makarov A, Szpunar J. Analusis. 1998;26:M44–M48. [Google Scholar]
- 42.Sulyok M, Hann S, Hartinger CG, Keppler BK, Stingeder G, Koellensperger G. J. Anal. At. Spectrom. 2005;20:856–863. [Google Scholar]
- 43. www.expasy.org.
- 44.Khalaila I, Allardyce CS, Verma CS, Dyson PJ. ChemBioChem. 2005;6:1788–1795. doi: 10.1002/cbic.200500067. [DOI] [PubMed] [Google Scholar]
- 45.Casini A, Mastrobuoni G, Ang WH, Gabbiani C, Pieraccini G, Moneti G, Dyson PJ, Messori L. ChemMedChem. 2007;2:631. doi: 10.1002/cmdc.200600258. [DOI] [PubMed] [Google Scholar]
- 46.Allardyce CS, Dyson PJ, Coffey J, Johnson N. Rapid Commun. Mass Spectrom. 2002;16:933–935. doi: 10.1002/rcm.662. [DOI] [PubMed] [Google Scholar]
- 47.Will J, Wolters DA, Sheldrick WS. ChemMedChem. 2008;3:1696–1707. doi: 10.1002/cmdc.200800151. [DOI] [PubMed] [Google Scholar]
- 48.Wang D, Lippard SJ. Nat. Rev. Drug Discovery. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 49.Will J, Sheldrick WS, Wolters D. JBIC, J. Biol. Inorg. Chem. 2008;13:421–434. doi: 10.1007/s00775-007-0333-8. [DOI] [PubMed] [Google Scholar]
- 50.Will J, Kyas A, Sheldrick WS, Wolters D. JBIC, J. Biol. Inorg. Chem. 2007;12:883–894. doi: 10.1007/s00775-007-0242-x. [DOI] [PubMed] [Google Scholar]
- 51.Manley SA, Gailer J. Expert Rev. Proteomics. 2009;6:251–265. doi: 10.1586/epr.09.44. [DOI] [PubMed] [Google Scholar]
- 52.Casini A, Gabbiani C, Michelucci E, Pieraccini G, Moneti G, Dyson PJ, Messori L. JBIC, J. Biol. Inorg. Chem. 2009;14:761–770. doi: 10.1007/s00775-009-0489-5. [DOI] [PubMed] [Google Scholar]
- 53.Casini A, Karotki A, Gabbiani C, Rugi F, Vasak M, Messori L, Dyson PJ. Metallomics. 2009;1:434–441. doi: 10.1039/b909185h. [DOI] [PubMed] [Google Scholar]