

Abstract
The human cytomegalovirus virion is composed of a DNA genome packaged in an icosahedral capsid, surrounded by a tegument of protein and RNA, all enclosed within a glycoprotein-studded envelope. Achieving this intricate virion architecture requires a coordinated process of assembly and egress. We show here that pUL71, a component of the virion tegument with a previously uncharacterized function, is required for the virus-induced reorganization of host cell membranes, which is necessary for efficient viral assembly and egress. A mutant that did not express pUL71 was able to efficiently accumulate viral genomes and proteins that were tested but was defective for the production and release of infectious virions. The protein localized to vesicular structures at the periphery of the viral assembly compartment, and during infection with a pUL71-deficient virus, these structures were grossly enlarged and aberrantly contained a cellular marker of late endosomes/lysosomes. Mutant virus preparations exhibited less infectivity per unit genome than wild-type virus preparations, due to aggregation of virus particles and their association with membrane fragments. Finally, mutant virus particles accumulated within the cytoplasm of infected cells and were localized to the periphery of large structures with properties of lysosomes, whose formation was kinetically favored in mutant-virus-infected cells. Together, these observations point to a role for pUL71 in the establishment and/or maintenance of a functional viral assembly compartment that is required for normal virion trafficking and egress from infected cells.
IMPORTANCE
In addition to causing disease in immunocompromised individuals, human cytomegalovirus is the leading known infectious cause of birth defects. To induce these pathologies, the virus must spread from its site of introduction to various organs and tissues in the body. The processes of viral assembly and egress, which underlie the spread of infection, are incompletely understood. We elucidate a role for a virus-coded protein, pUL71, in these processes and demonstrate the importance of maintaining an intricate, virus-induced reorganization of host cell membranes for efficient virus spread.
INTRODUCTION
Human cytomegalovirus (HCMV) is a betaherpesvirus that displays the signature virion architecture of all herpesviruses (1). HCMV virions contain about 70 viral proteins (2), representing about a third of its ~200 viral protein-coding open reading frames (ORFs) (3), including capsid constituents, tegument species, and envelope glycoproteins (1). To achieve its intricate virion architecture, as well as to ensure high-fidelity packaging of virion proteins and efficient release of infectious progeny, HCMV employs a highly coordinated, but incompletely understood, process of assembly and egress (4). Assembly begins in the nucleus where capsids are formed and loaded with viral genomes. These nucleocapsids likely associate with a subset of tegument proteins that accumulate in the nucleus. Next, the phosphorylation of nuclear lamins is altered, allowing the nucleocapsids and associated tegument proteins to translocate into the cytoplasm by a proposed envelopment/de-envelopment process across the inner and outer nuclear envelopes. The nucleocapsids associate with additional tegument proteins and acquire their final envelope in the cytoplasm within a region termed the viral assembly compartment (vAC) (5–9). The vAC is a juxtanuclear collection of membranes, virion proteins, and cellular proteins that include markers of the exocytic and endocytic networks. During secondary envelopment, tegumented nucleocapsids bud into vesicles that are believed to be derived from the trans-Golgi network (TGN)/endosome to acquire their final envelopes and traffic out of cells (10–13). The reorganization of membranes into the vAC is believed to be essential for the sorting of virion glycoproteins and for coordination of cellular trafficking required for egress of virions (6, 7, 14).
HCMV pUL71 is a tegument protein (2, 15) whose function has been unknown. It is a positional homologue of herpes simplex virus type 1 (HSV-1) UL51, which has been shown to be a tegument-associated, late protein (16) that is palmitylated (17). A UL51-deficient derivative of HSV-1 has been shown to exhibit a 100-fold growth defect and to be defective for nuclear egress (18). While HCMV UL71 and HSV-1 UL51 are positional homologues, they do not share significant homology at the nucleotide or amino acid level. HCMV pUL71 is, however, a member of the herpesvirus U44 superfamily, containing a conserved, but functionally unclassified, U44 domain (19).
The growth of UL71-null viruses has been examined in two global mutagenesis studies, where mutants exhibited a >104-fold growth defect relative to wild-type virus (20) or did not grow at all (21). While the studies reached somewhat different conclusions regarding the requirement for pUL71, both illustrated its importance during replication. Sera from HCMV-positive individuals contain pUL71-specific antibodies (22), illustrating that the protein is a target of the humoral response to infection.
In this study, we characterize a mutant virus that does not express pUL71. The mutant exhibited a severe growth defect in fibroblasts that occurs after late gene expression. The protein was required for the reorganization of the host cell endocytic/exocytic network that normally generates the proper architecture of the vAC. HCMV virions produced in the absence of pUL71 remained cell associated; mutant-virus-infected cells accumulated large intracytoplasmic inclusions, exhibiting lysosomal characteristics, with virus particles at their periphery.
RESULTS
pUL71 is required for the efficient accumulation of extracellular progeny.
To study the function of pUL71, we constructed a mutant virus, BADinUL71STOP, using bacterial artificial chromosome (BAC) recombineering technology (23). To design this mutant, we mapped the 5′ end of the UL71 transcript (see Fig. S1A in the supplemental material), testing the possibility that there might be additional ORFs upstream of the predicted UL71 coding region (24). We determined that the transcript begins 29 nucleotides upstream of an AUG that marks the start of the previously specified UL71 ORF and 14 nucleotides downstream of the start codon for the UL70 ORF, which resides on the opposite strand from UL71 (Fig. S1B and C). The mutant virus contains an 11-nucleotide stop codon frameshift cassette inserted after nucleotide 36 of the UL71 ORF (Fig. 1A).
FIG 1 .

BADinUL71STOP generates fewer infectious progeny than BADwt. (A) Schematic of the BADinUL71STOP genome. An 11-base-pair translation stop cassette was inserted after nucleotide 36 of the UL71 ORF. (B) BADinUL71STOP produces >100-fold-fewer infectious progeny than BADwt. Infected fibroblasts and supernatants were harvested at the indicated times after infection at a multiplicity of 2 PFU/cell, and virus titers were determined by 50% tissue culture infective dose (TCID50) assay. Inp, input virus. (C) Accumulation of RNA from neighboring genes is unaffected by the UL71STOP mutation. Fibroblasts were infected at a multiplicity of 2 PFU/cell with BADwt or BADinUL71STOP and harvested 72 h later. Total RNA was isolated. UL70 and UL72 message levels were quantified by reverse transcription-quantitative PCR (RT-qPCR) and normalized to actin RNA. Error bars represent standard errors of means calculated from technical repeats.
We observed a >100-fold reduction in total infectious virus accumulation after infection at a relatively high multiplicity (2 PFU/cell) with the mutant virus BADinUL71STOP compared to its parent, BADwt (Fig. 1B). A similar defect was evident in a second, independently derived pUL71-deficient virus (data not shown), arguing that the phenotype was not influenced by a spurious off-target mutation. Further, the observed defect was not due to disruption of the expression of neighboring genes, as RNA levels for the neighboring ORFs UL70 and UL72 were not significantly different in BADinUL71STOP and BADwt (Fig. 1C), and the translational starts for the surrounding ORFs are located far from the insertion in UL71. This conclusion is corroborated by the fact that UL70, the gene whose expression was more likely to be affected due to its position relative to UL71, is essential for viral DNA replication (25), and BADinUL71STOP accumulated viral DNA to wild-type levels (Fig. 2C).
FIG 2 .

BADinUL71STOP produces a higher proportion of cell-associated progeny than BADwt but accumulates normal levels of viral DNA and proteins. Infections were performed at a multiplicity of 2 PFU/cell. (A) BADinUL71STOP produces >100-fold-fewer extracellular infectious viral progeny and ~10-fold-fewer cell-associated infectious viral progeny than BADwt. Cultures were harvested on the indicated days postinfection (dpi), and viral titers were determined by TCID50 assay. Inp, input virus. Results are representative of an experiment conducted in duplicate with two separate isolates of BADinUL71STOP mutant virus. Error bars estimate standard deviations calculated from technical repeats. (B) Ratio of cell-associated to cell-free virus for mutant and wild-type viruses. The primary data displayed in panel A are plotted as ratios. Error bars represent standard errors of means calculated from technical repeats. (C) BADinUL71STOP-infected cells accumulate viral DNA normally. Fibroblasts were harvested at the indicated hour postinfection (hpi) with mutant or wild-type virus, and total cellular DNA was isolated. Viral genomes were quantified by qPCR with UL123-specific primers and normalized to cellular actin. Results are representative of an experiment conducted in duplicate with two separate isolates of BADinUL71STOP. Error bars represent standard errors of means calculated from technical repeats. (D) BADinUL71STOP-infected cells accumulate representative viral proteins from all kinetic classes normally. Cells were harvested at the indicated hpi, and whole-cell lysates were prepared. Proteins were analyzed by Western blotting using antibodies for IE1, pUL44, and pUL99. Tubulin was assayed as a loading control. Results are representative of an experiment conducted in duplicate with two separate isolates of BADinUL71STOP.
We conducted further analyses that revealed defects in the accumulation of both extracellular and intracellular infectious virus after infection with the mutant at a multiplicity of 2 PFU/cell (Fig. 2A). Interestingly, the ratio of cell-associated virus to cell-free virus is nearly 100-fold greater for BADinUL71STOP than for BADwt (Fig. 2B), indicating that most progeny virus fail to egress from the infected cell. Despite this reduction in infectious virus, BADinUL71STOP-infected cells accumulated viral DNA to the same extent as BADwt-infected cells (Fig. 2C). Additionally, representative immediate-early (IE1), early (pUL44), and late (pUL99) viral protein accumulated to wild-type levels with proper kinetics during BADinUL71STOP infection (Fig. 2D). To determine whether virus released from mutant-virus-infected cells contributed to spread and whether the intracellular infectious virus observed in mutant-infected cells was capable of spreading through a monolayer, cells were infected at a multiplicity of 0.01 PFU/cell and treated with neutralizing antibody to prevent spread due to release of extracellular infectious virus. Neutralizing antibody markedly reduced the spread of wild-type virus (Fig. 3A) but did not affect the spread of mutant virus (Fig. 3B), indicating that the contribution of released extracellular virus to BADinUL71STOP spread was minimal. The size of infectious centers increased during mutant infection (Fig. 3B), indicating that the mutant is competent for cell-to-cell spread.
FIG 3 .
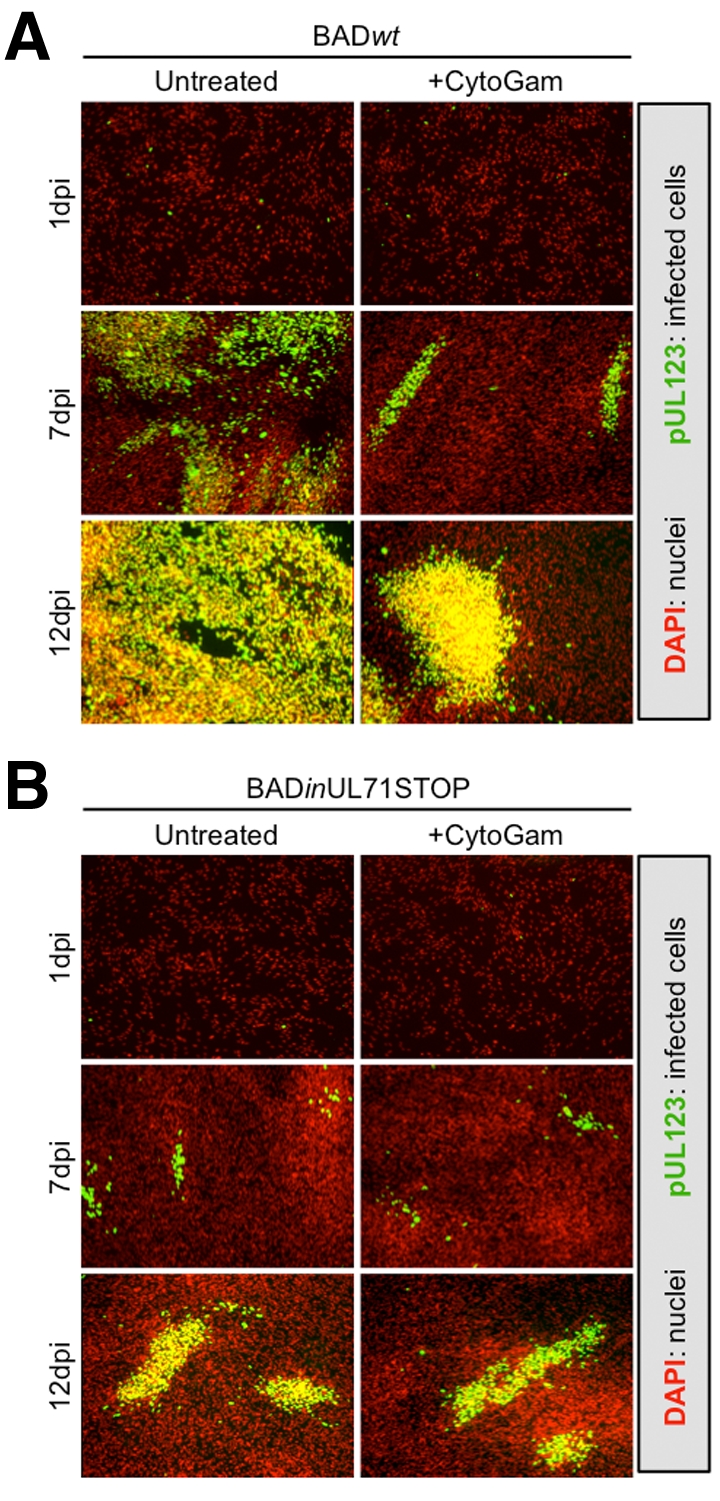
BADinUL71STOP virus spreads in a direct cell-to-cell manner. Infections with BADwt (A) or BADinUL71STOP (B) were performed at a multiplicity of 0.01 PFU/cell in the presence of nonsupplemented growth medium (untreated) or growth medium supplemented with 3% (vol/vol) CytoGam (+CytoGam). Nuclei were stained with DAPI (red), and infected cells were detected by probing for IE1 expression (green).
pUL71-deficient virus generates an abnormal viral assembly compartment (vAC).
Since the effect on yield was considerably greater for extracellular virus than for intracellular virus, we suspected that pUL71 plays a role in viral assembly and/or egress. To pursue this hypothesis, we constructed BADinUL71GFP, which expresses a pUL71GFP fusion protein (GFP, green fluorescent protein) (see Fig. S2A in the supplemental material). The accumulation of extracellular virus was monitored after infection at a multiplicity of 0.1 PFU/cell, and there was no significant difference between wild-type and UL71GFP-tagged virus (see Fig. S2B in the supplemental material), confirming that the addition of the tag to pUL71 did not interfere with its function.
Using BADinUL71GFP, we examined the expression kinetics and subcellular localization of pUL71. It was detected at 24 h postinfection (hpi), the earliest time examined, and increased throughout infection (see Fig. S2C in the supplemental material). At 48 hpi, pUL71 colocalized with pUL99 (pp28) and pUL55 (gB) in the vAC (Fig. 4). At 96 hpi, it continued to colocalize with pUL99 and pUL55 in large vesicular structures at the periphery of the vAC, which are thought to arise from the fusion of smaller, discrete classes of vesicles (26). Although pUL99 and pUL55 remain broadly distributed throughout the vAC at this late time, pUL71 is substantially restricted to the peripheral vesicles. We next examined the large viral vesicular structures at 96 hpi, comparing pUL71 localization to a number of cellular markers of the exocytic/endocytic network. The markers were reorganized by infection (see Fig. S3 in the supplemental material), consistent with previous studies of HCMV assembly (5–10), but only ERGIC53 substantially colocalized with pUL71.
FIG 4 .
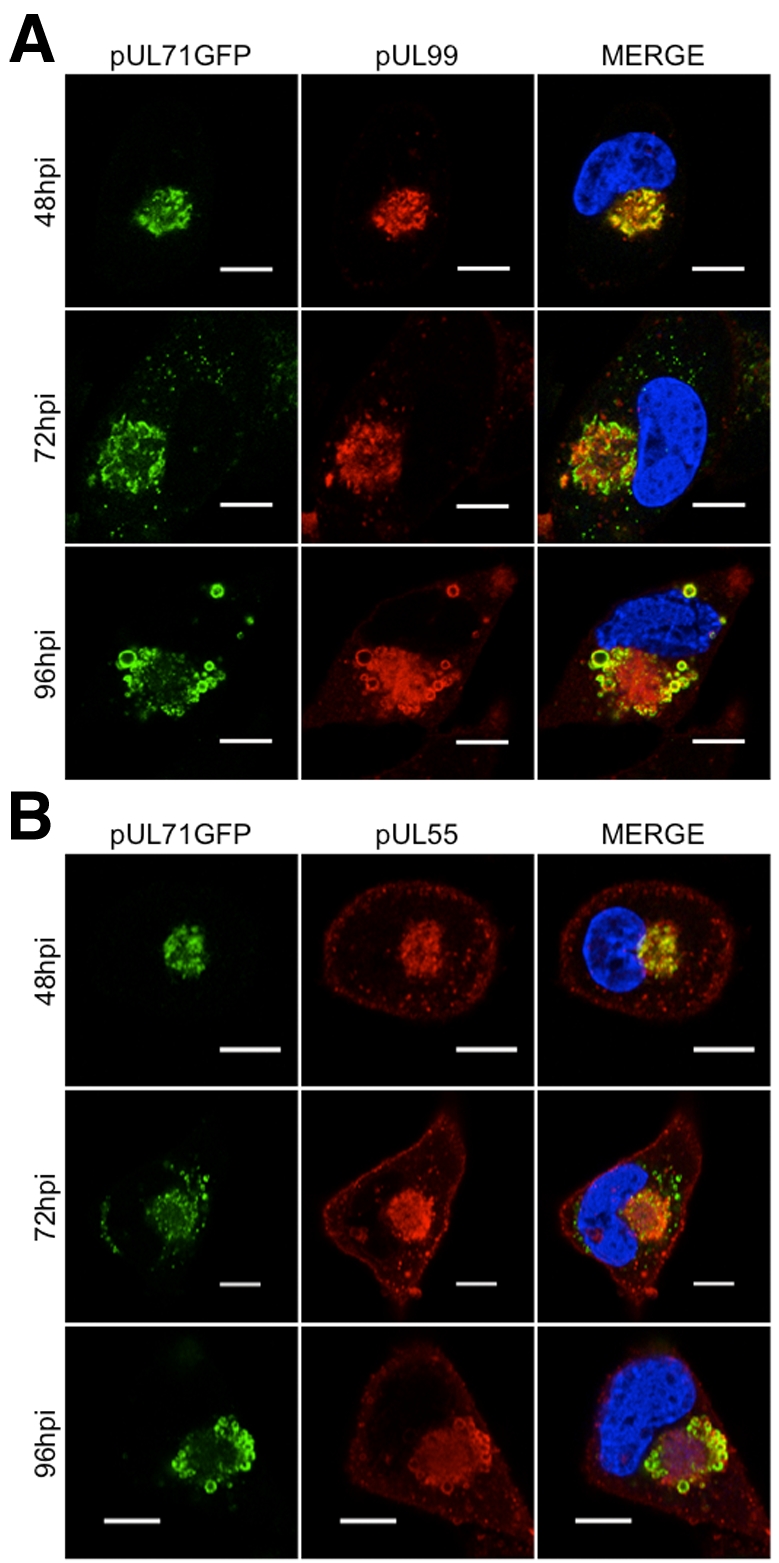
pUL71GFP partially colocalizes with pUL99 (pp28) and pUL55 (gB) in the viral assembly compartment (vAC). pUL71GFP (green) was tested for colocalization with pUL99 (red) (A) and pUL55 (red) (B). Fibroblasts were infected at a multiplicity of 1 PFU/cell and processed for immunofluorescence at the indicated time postinfection (hpi). DAPI-stained nuclei are blue in merged images. Results are representative of an experiment conducted with two separate isolates of BADinUL71GFP. Bars, 10 µm.
We also examined the cytoplasmic structures of BADinUL71STOP-infected cells for changes in morphology of the vAC and its surrounding vesicles. At 96 h after BADinUL71STOP infection, we observed pUL99-containing vesicular structures that were on average enlarged (~5 µm) compared to the vesicles present during BADwt infection (~1 to 2 µm) (Fig. 5A, top and middle panels). Vesicular structures containing pUL55 were also enlarged in mutant-virus-infected cells (Fig. 5A), which is consistent with previous observations that pUL99 and pUL55 merge into larger vesicles at late times postinfection (26). Interestingly, a subset of ≤10% of BADinUL71STOP-infected cells displayed extremely enlarged pUL99- and pUL55-containing vesicular structures (≥10 µm) at 96 hpi (Fig. 5A, bottom panels).
FIG 5 .

BADinUL71STOP-infected cells display altered vAC morphology. Fibroblasts were infected at a multiplicity of 1 PFU/cell with either BADwt or BADinUL71STOP virus and processed for immunofluorescence 96 h later. DAPI-stained nuclei are blue in merged images. (A) Compared to BADwt-infected cells (top panels), BADinUL71STOP-infected cells accumulate pUL99- and pUL55-containing vesicular structures that are enlarged (middle panels). A minor population of BADinUL71STOP-infected cells (<10%) display grossly enlarged vesicular structures that are ≥10 µm in diameter (bottom panels). Results are representative of experiments conducted in duplicate with two isolates of BADinUL71STOP. (B and C) Compared to BADwt-infected cells, the lysosomal marker LAMP1 (red) is aberrantly sorted into vesicular structures at the periphery of the vAC in BADinUL71STOP-infected cells. Arrows mark colocalization of pUL99 (B) (green) or pUL55 (C) (green) and LAMP1. LAMP1 was detected with either a rabbit polyclonal antibody (B) or a mouse monoclonal antibody (C). Results are representative of experiments conducted in triplicate with two separate isolates of BADinUL71STOP. (D) BADinUL71STOP-infected cells have enlarged, acidified structures in the perinuclear cytoplasmic region. At 96 hpi, live cells were stained with LysoTracker Green. Bars, 10 µm.
Since the morphology of the vAC was altered during BADinUL71STOP infection, we hypothesized that the localization of vAC resident proteins relative to cellular markers of the endocytic/exocytic network may also change. To test this idea, we monitored the localization of pUL99 and pUL55 along with cellular markers of the endocytic/exocytic network. The localization of the late endosomal/lysosomal marker, LAMP1, was markedly altered during mutant infection (Fig. 5B). In cells infected with a pUL71GFP-expressing virus (see Fig. S3E in the supplemental material) or BADwt virus (Fig. 5B, top panels), little colocalization with virus-induced vesicular structures was observed. In BADinUL71STOP-infected cells, LAMP1 was depleted from the center of the vAC and concentrated within the pUL99-containing vesicles. While noticeable in all pUL99 vesicles, this localization was most evident in the extremely enlarged vesicles (Fig. 5B, bottom panels). Since this initial experiment utilized a rabbit polyclonal antibody and rabbit IgG has been reported to localize spuriously to the vAC (27), the localization was confirmed using a mouse antibody to LAMP1 (Fig. 5C). Finally, since LAMP1 is a lysosomal marker, we suspected that the vesicular structures may be acidified, so we labeled infected cells with LysoTracker green. Numerous, small puncta were evident in the vAC of BADwt-infected cells (Fig. 5D, left panels), and the LysoTracker green-stained structures were noticeably larger after infection with BADinUL71STOP (Fig. 5D, right panels). These structures, including examples ≥10 µm, were similar in size and localization to the enlarged viral vesicles observed during mutant virus infection (Fig. 5A and B), consistent with the interpretation that they are acidified.
UL71 mutant virus preparations are less infectious than wild-type virus preparations.
Since the vACs of BADinUL71STOP-infected cells were abnormal and since pUL71 is present in virions (2), we hypothesized that mutant virus might be less infectious than wild-type virus. To test this idea, we quantified viral genomes from titered stocks of mutant and wild-type virus by quantitative PCR (qPCR). BADinUL71STOP virus stocks contained >10-fold more viral genomes per infectious unit than wild-type virus stocks (see Fig. S4A in the supplemental material).
We observed no difference in the events leading up to viral assembly and egress between BADwt- and BADinUL71STOP-infected cells during infections normalized for the input of infectious units (Fig. 2C and D). To determine the point in the infectious cycle at which the mutant is blocked and a higher genome dosage is required to complete the preassembly events of infection normally, we assayed several early events during infections normalized for the input of either infectious units or viral genomes. We assayed the binding of virions to cells at 4°C and observed nearly identical levels of cell-associated, viral genomes when the infections were normalized for equal input of viral genomes (see Fig. S4B in the supplemental material). However, when infections were normalized for equal input of infectious units, we observed 10-fold-more bound BADinUL71STOP genomes (see Fig. S4B in the supplemental material), illustrating that mutant virions, while less infectious, are competent for binding to fibroblasts. Entry was assayed by measuring nuclear delivery of the virion protein pUL83 at 6 hpi, and nearly identical levels of pUL83-positive nuclei were generated during infections normalized for the input of infectious units. When we normalized for the input of viral genomes, 10-fold-fewer pUL83-positive nuclei in BADinUL71STOP-infected cells were produced (Fig. S4C). To corroborate these observations, expression of the IE1 immediate-early protein was assayed by immunofluorescence at 24 hpi. We observed nearly identical levels of IE1-positive nuclei during infections normalized for the input of infectious units, but when infections were normalized for the input of viral genomes, we observed 10-fold-fewer IE1-positive nuclei in BADinUL71STOP-infected cells (Fig. S4D).
Virions isolated from UL71 mutant-virus-infected cells are aggregated and associated with membrane fragments.
Having determined that BADinUL71STOP virus stocks are less infectious than wild-type virus stocks, we explored the possibility that the mutant virions were assembled improperly. Cell-free and total (cell-free plus cell-associated) virus particles were partially purified by centrifugation through a glycerol tartrate gradient (Fig. 6A). Both preparations from BADwt displayed a characteristic set of bands (28), corresponding to noninfectious enveloped particles (NIEPs) (above band A), virions (band A), and dense bodies (band B). In contrast, cell-free virion preparations from BADinUL71STOP infections displayed no discernible bands, while total virion preparations displayed a single, “hybrid” band of intermediate migration (band D). We isolated the material in bands A and B from the wild-type virus gradient, and three fractions from the mutant gradient, fractions C, D, and E. Fraction D contains the only visible band, and fractions C and E correspond to the regions where virions and dense bodies would be found in wild-type virus gradients. We investigated the abundance of viral DNA and viral infectivity in each of these fractions (Fig. 6B). DNase I-insensitive viral DNA was isolated from equal volumes of each sample and quantified by qPCR. Predictably, the majority of BADwt viral DNA was found in fraction A (virions), but a large amount was also found in fraction B (dense bodies). Modest amounts of viral DNA were detected in fractions C and E, which as noted above, correspond to the predicted migration of BADinUL71STOP virions and dense bodies. The vast majority of BADinUL71STOP viral DNA, however, was detected in hybrid band D. The migration of infectivity mirrored that of viral DNA, with the majority of the BADwt infectivity in band A and BADinUL71STOP infectivity in band D (Fig. 6B).
FIG 6 .

Purified BADinUL71STOP particles are aggregated and associated with membranes. (A) BADinUL71STOP particles sediment differently than wild-type virions. Virus particles from the supernatants of infected cultures or from cells plus supernatants were concentrated and then resolved by centrifugation through a glycerol tartrate gradient. (B) BADinUL71STOP genomes and infectivity sediment differently than those of wild type. Infectivity was measured for each isolated band species by TCID50 assay, and viral genomes were quantified by qPCR using UL123-specific primers. (C) The BADinUL71STOP virion band contains elevated levels of representative virion proteins. Proteins in the indicated factions were assayed by Western blotting for capsid (pUL85), tegument (pUL83 and pUL99), and envelope (pUL55) proteins. Loading was normalized to viral genomes. (D) Electron micrographs of particles in gradient fractions. BADwt virions, BADwt dense bodies, and BADinUL71STOP virion aggregates were isolated from indicated gradient fractions and visualized by transmission electron microscopy. The areas marked by green boxes in the top panels are shown at higher magnification in the bottom panels.
To characterize the particles present in the BADinUL71STOP hybrid band, aliquots containing equal numbers of viral genomes were analyzed by Western blotting for capsid (pUL85), tegument (pUL83 and pUL99), and envelope (pUL55) proteins (Fig. 6C). All protein species probed were present in BADinUL71STOP band D, but the protein abundance per unit genome in the mutant particles appeared more similar to wild-type dense bodies rather than wild-type virions.
To further characterize the particles present in the BADinUL71STOP hybrid band, we compared them to BADwt virions and dense bodies by transmission electron microscopy (TEM). BADwt virions and dense bodies (Fig. 6D, left) displayed typical morphology, with virions generally present as individuals or in groups of 3 to 4 particles at maximum and dense bodies appearing as individuals, small clusters, or large, membrane-associated aggregates. The aggregates likely cause the characteristic heterogeneity observed for the dense body band in gradients. The particles comprising the BADinUL71STOP hybrid band D resembled virions and NIEPs, but they existed almost exclusively as aggregations of particles (Fig. 6D, small virion aggregates), or very large, membranous aggregates (Fig. 6D). Consistent with this observation, the BADinUL71STOP hybrid band in the gradient was heterogeneous and more closely resembled the BADwt dense body band than the virion band.
BADinUL71STOP particles accumulate at the periphery of large ICIs.
To investigate the intracellular trafficking of mutant virus particles, sections of infected cells were analyzed by TEM. At 72 hpi, capsids were identified in the nuclei of infected cells (see Fig. S5 in the supplemental material), with little difference between the mutant and wild-type virus. At 96 hpi, however, many cytoplasmic virus particles were visible in cells infected with wild-type virus (Fig. 7A and B, arrows), while almost none were found in cells infected with mutant virus (Fig. 7C and D). Although cytoplasmic virus particles were not visible in cells infected with mutant virus, large, electron-dense intracytoplasmic inclusions (ICIs) were clearly visible in the cytoplasm at 96 hpi (Fig. 7C and D). On average, these inclusions were much larger in mutant-virus-infected cells than in wild-type-virus-infected cells. By 144 hpi, large ICIs were visible in cells infected with wild-type virus (Fig. 8A and B) and in cells infected with mutant virus (Fig. 8C and D). At this very late stage of infection, few wild-type virions were evident in the cytoplasm. In contrast, a large number of cytoplasmic UL71 mutant virus particles were discernible (Fig. 8C and D, arrows), and large numbers of vesicles containing viral particles had accumulated at the periphery of the ICIs.
FIG 7 .

At 96 hpi, BADinUL71STOP-infected cells contain large ICIs but few cytoplasmic virus particles. Cells were infected at a multiplicity of 2 with BADwt (A and B) or BADinUL71STOP (C and D). Arrows indicate cytoplasmic virus particles. Nu, nucleus; ICI, intracytoplasmic inclusion.
FIG 8 .

At 144 hpi, BADinUL71STOP-infected cells contain more cytoplasmic virus particles than BADwt-infected cells, many of which are associated with large ICIs. Cells were infected at a multiplicity of 2 with either BADwt (A and B) or BADinUL71STOP (C and D). Arrows indicate cytoplasmic virus particles. Nu, nucleus; ICI, intracytoplasmic inclusion.
DISCUSSION
The cytoplasmic events of HCMV virion maturation and trafficking are only beginning to be elucidated. Several viral proteins are known to be required for virion assembly and egress (4), and cellular proteins of the endoplasmic reticulum (29), Golgi apparatus (11), endosomal recycling complex (14), multivesicular body (10, 30) and ESCRT (endosomal sorting complex required for transport) (31) cellular trafficking pathways have also been implicated in virion maturation.
Our results argue that pUL71 is dispensable for events before cytoplasmic envelopment (Fig. 2C and D) but required for correct morphogenesis of the viral assembly compartment (vAC) (6, 8, 9) and its associated vesicular system (Fig. 4 and 5; see Fig. S3 in the supplemental material). In the absence of pUL71, the vAC-associated vesicular structures are enlarged (Fig. 5A), similar to those previously observed during infections with pUL97-deficient virus or pharmacological inhibitors of pUL97 (32, 33). These enlarged structures aberrantly contain LAMP1, a cellular marker of late endosomes/lysosomes (Fig. 5B and C). We observed different LAMP1 localizations in side-by-side analysis of mutant and wild-type viruses. This improper vAC constitution generates progeny that are highly cell associated (Fig. 2B and 6A), and when virus stocks are prepared by sonication of infected cells, mutant virus preparations are less infectious on a per genome basis than wild-type virus preparations (see Fig. S4 in the supplemental material). Despite the effect on egress, the pUL71-deficient mutant remains competent for cell-to-cell spread (Fig. 3B), consistent with the earlier conclusion that envelopment is not required for cell-to-cell spread of HCMV (34), and supporting the view that multiple, distinct virus trafficking pathways exist (26). A subset of these pathways may promote cell-to-cell spread independently from extracellular virion release.
UL71 mutant virus particles isolated by sonication of infected cells displayed a unique migration through glycerol-tartrate density gradients (Fig. 6A). The distribution of mutant viral genomes and infectivity within the gradient followed this novel migration pattern and differed from that observed for wild-type virus preparations (Fig. 6B). Analysis of virion proteins revealed that while all assayed species were in UL71 mutant virus particles, their abundance more closely resembled wild-type virus dense bodies than wild-type virions (Fig. 6C). Visualization of the mutant particles released from cells by sonication revealed that they were aggregated and associated with sheaths of membrane, as was the case for wild-type virus dense bodies (Fig. 6D). These membranes are likely associated with viral proteins, possibly accounting for the similarity in protein abundance for mutant particles and wild-type dense bodies. Although the aggregates of particles with membrane sheaths are produced by mechanical disruption, this observation demonstrates a fundamental difference between wild-type and mutant particles. Wild-type particles were discernible in both cell-free and cell-associated preparations (Fig. 6A), as expected (28), while mutant virus particles were observed only as aggregates in cell-associated preparations (Fig. 6A).
Intracellular trafficking of mutant and wild-type virus particles was monitored by TEM (Fig. 7 and 8; see Fig. S5 in the supplemental material). In contrast to cells infected with the wild-type virus, almost no virus particles were present in the cytoplasm of cells infected with mutant virus at 96 hpi (Fig. 7). This may indicate that, in addition to cytoplasmic trafficking of virus particles, pUL71 contributes to nuclear egress of nucleocapsids. We did not observe localization of pUL71 to the nucleus or nuclear rim (Fig. 4; see Fig. S3 in the supplemental material), which suggests that if pUL71 acts to promote translocation of nucleocapsids into the cytoplasm, it does so indirectly. By 144 hpi, mutant-virus-infected cells contained many more cytoplasmic virus particles than wild-type-virus-infected cells (Fig. 8), presumably because many wild-type virus particles were released from infected cells before this time (Fig. 2A). Conversely, mutant-virus-infected cells developed large, electron-dense ICIs by 96 hpi (Fig. 7). Large ICIs were evident in wild-type-virus-infected cells at 144 hpi (Fig. 8).
Electron-dense ICIs have been observed previously in TEM analyses of HCMV-infected fibroblasts and characterized as lysosomes (35–37). In murine cytomegalovirus (CMV)-infected mice, ICI formation in hepatocytes correlated with a reduction in virus titer relative to the titer from salivary gland cells, where the lysosomal structures were not observed (38). Similar lysosomal structures formed with the same kinetics during HCMV infection of cultured fibroblasts, leading to the proposal that they were a cell type-specific intrinsic defense against infection. These results fit with our observation of large LAMP1-positive vesicles (Fig. 5B and C), as well as LysoTracker green-positive, acidified structures in BADinUL71STOP-infected cells (Fig. 5D).
Our observations, considered in light of the earlier visualization of lysosomal structures (38), are consistent with a kinetic model of degradation, wherein the formation of large lysosomal structures occurs earlier during mutant virus infection than during wild-type virus infection, resulting in enhanced degradation of mutant virus particles. This model suggests that the virus particles associated with the periphery of ICIs in BADinUL71STOP-infected cells (Fig. 8C and D) subsequently enter the ICI where they are degraded. Trafficking to these lysosomal structures during wild-type virus infection is presumably avoided by rapid, efficient egress before their formation occurs. In the case of mutant virus infection, virion trafficking is inefficient and lysosomal formation occurs earlier, resulting in a larger proportion of virus particles being degraded by lysosomes or sequestered on their periphery. Alternatively, these structures could be directly involved in the normal egress of virus particles, as smaller ICIs are discernible by 96 hpi in wild-type-virus-infected cells (Fig. 7). While direct fusion of multivesicular body-like collections of virions at the plasma membrane has been documented (11), it is possible that acidified structures are also utilized for egress. The acquisition of proper membranes would likely be important to efficiently utilize acidified structures for egress, as both pH and lipid composition have been shown to be required for formation of acidified multivesicular liposomes (39). In the case of UL71 mutant virus infection, acquisition of the proper membrane could be impaired by improper vAC morphogenesis (Fig. 5), resulting in inefficient membrane fusion and impaired budding into acidified structures. While we favor the kinetic model of degradation, further experiments are required to rule out the second model.
MATERIALS AND METHODS
Cells and viruses.
MRC5 fibroblasts (ATCC) were cultured in medium supplemented with 10% fetal bovine serum. The wild-type, parental strain for all mutant viruses was the bacterial artificial chromosome (BAC)-derived AD169 strain BADwt (40). BADinGFP (41) and BFXwt-GFP (42) are derivatives of BADwt and a BAC-cloned FIX clinical isolate, respectively, that replicate with wild-type virus kinetics. To generate BADinUL71STOP, a pUL71-deficient mutant virus, we utilized GalK recombineering (23), and BADinUL71GFP was generated using Kan-FRT recombineering (43). Details for their construction can be found in Text S1 and Table S1 in the supplemental material. In each case, gross mutant BAC DNA integrity was confirmed by restriction digestion, and mutations were verified by sequence analysis.
Virus stocks were produced in fibroblasts by pooling cell-free and cell-associated virus, and the titers of the virus stocks were determined by limiting dilution assay. For some experiments, stocks were concentrated by centrifugation through sorbitol cushions. To purify virions, virus stocks were resolved on sodium tartrate gradients (28).
To analyze the contributions of extracellular and intracellular virus to the spread of infection through a monolayer, medium supplemented with 3% (vol/vol) CytoGam (CSL Behring) was added to cells immediately after infection, an amount shown previously to neutralize extracellular virus (44). The cells were subsequently fixed and permeabilized with ice-cold methanol and prepared for microscopy as detailed below. The cells were imaged directly on the plate with a Nikon Eclipse TE2000-U fluorescent microscope, and images were processed with Q-Capture Pro software (45).
Analysis of viral nucleic acids and proteins.
Intracellular viral RNA and DNA and viral DNA in virus stocks were quantified as described previously (46, 47), and details can be found in Text S1 in the supplemental material.
Proteins within infected cells and in virus particles were monitored by Western blotting (46). Proteins were reacted with primary antibodies (see Table S2 in the supplemental material) and then with horseradish peroxidase (HRP)-conjugated secondary antibodies, which were detected with enhanced chemiluminescence (ECL) reagent (GE Healthcare). To analyze protein localization by immunofluorescence, cells were cultured on glass coverslips, fixed for 15 min with 2% paraformaldehyde, permeabilized for 15 min in phosphate-buffered saline (PBS) containing 0.1% Triton X-100 (PBS-T), and blocked for 1 h in 0.05% PBS-T containing 2% bovine serum albumin (BSA). Proteins were labeled with primary antibodies (see Table S2 in the supplemental material) in 0.05% PBS-T plus 2% BSA for 1 h. The coverslips were washed 3 times with PBS plus 0.05% Tween 20, incubated with 4′,6′-diamidino-2-phenylindole (DAPI)- and Alexa Fluor-conjugated secondary antibodies in 0.05% PBS-T plus 2% BSA for 1 h, washed 3 times with 0.05% PBS-T, and mounted on glass slides with SlowFade Gold anti-fade solution (Invitrogen), and images were captured on a Leica DM5500 microscope. pUL83- and pUL123-positive nuclei were imaged as described previously (46).
Electron microscopy.
Gradient-purified virus particles were immobilized on carbon-coated grids and negatively stained with 0.1% uranyl acetate. To image cells, infected cells and mock-infected cells were fixed in 2.5% glutaraldehyde and 1.6% paraformaldehyde in 100 mM sodium cacodylate, pH 7.4, harvested by scraping, pelleted by centrifugation, cut into ≤1-mm blocks, fixed with 1% osmium tetroxide (aqueous), and stained with 0.5% uranyl acetate. The cells were dehydrated in ethanol, infiltrated with Epon plastic and ethanol, and embedded in plastic. Sections (~60 nm) were cut, stained with uranyl acetate and lead citrate, and viewed using a Zeiss 912AB transmission electron microscope. Images were captured using AMT Capture Engine v5344.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grants CA82396 and CA85786 from the National Cancer Institute to T.S. and a National Science Foundation Graduate Research Fellowship awarded to A.W.
We thank our colleagues for helpful discussions of this work, J. Hwang (Princeton University) for advice with virion purification, D. Stern (Princeton University) for allowing us to use his confocal microscope, and W. Gibson (Johns Hopkins University) for antibody to pUL85.
Footnotes
Citation Womack, A, and T. Shenk. 2010. Human cytomegalovirus tegument protein pUL71 is required for efficient virion egress. mBio 1(5):e00282-10. doi:10.1128/mBio.00282-10.
SUPPLEMENTAL MATERIAL
REFERENCES
- 1. Mocarski E. S., Shenk T., Pass R. F. 2007. Cytomegaloviruses, p. 2701–2772 In Knipe D. M., Howley P. M. (ed.), Fields virology, 5th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Varnum S. M., Streblow D. N., Monroe M. E., Smith P., Auberry K. J., Pasa-Tolic L., Wang D., Camp D. G., II, Rodland K., Wiley S., Britt W., Shenk T., Smith R. D., Nelson J. A. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Murphy E., Shenk T. 2008. Human cytomegalovirus genome. Curr. Top. Microbiol. Immunol. 325:1–19 [DOI] [PubMed] [Google Scholar]
- 4. Gibson W. 2008. Structure and formation of the cytomegalovirus virion. Curr. Top. Microbiol. Immunol. 325:187–204 [DOI] [PubMed] [Google Scholar]
- 5. AuCoin D. P., Smith G. B., Meiering C. D., Mocarski E. S. 2006. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J. Virol. 80:8199–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das S., Vasanji A., Pellett P. E. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Krzyzaniak M., Mach M., Britt W. J. 2007. The cytoplasmic tail of glycoprotein M (gpUL100) expresses trafficking signals required for human cytomegalovirus assembly and replication. J. Virol. 81:10316–10328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sanchez V., Greis K. D., Sztul E., Britt W. J. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sanchez V., Sztul E., Britt W. J. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J. Virol. 74:3842–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cepeda V., Esteban M., Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell. Microbiol. 12:386–404 [DOI] [PubMed] [Google Scholar]
- 11. Homman-Loudiyi M., Hultenby K., Britt W., Soderberg-Naucler C. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, Rab 3, trans-Golgi network 46, and mannosidase II. J. Virol. 77:3191–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Severi B., Landini M. P., Govoni E. 1988. Human cytomegalovirus morphogenesis: an ultrastructural study of the late cytoplasmic phases. Arch. Virol. 98:51–64 [DOI] [PubMed] [Google Scholar]
- 13. Tooze J., Hollinshead M., Reis B., Radsak K., Kern H. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163–178 [PubMed] [Google Scholar]
- 14. Krzyzaniak M. A., Mach M., Britt W. J. 2009. HCMV-encoded glycoprotein M (UL100) interacts with Rab11 effector protein FIP4. Traffic 10:1439–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kalejta R. F. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Daikoku T., Ikenoya K., Yamada H., Goshima F., Nishiyama Y. 1998. Identification and characterization of the herpes simplex virus type 1 UL51 gene product. J. Gen. Virol. 79(Pt. 12):3027–3031 [DOI] [PubMed] [Google Scholar]
- 17. Nozawa N., Daikoku T., Koshizuka T., Yamauchi Y., Yoshikawa T., Nishiyama Y. 2003. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J. Virol. 77:3204–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nozawa N., Kawaguchi Y., Tanaka M., Kato A., Kimura H., Nishiyama Y. 2005. Herpes simplex virus type 1 UL51 protein is involved in maturation and egress of virus particles. J. Virol. 79:6947–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marchler-Bauer A., Anderson J. B., Chitsaz F., Derbyshire M. K., DeWeese-Scott C., Fong J. H., Geer L. Y., Geer R. C., Gonzales N. R., Gwadz M., He S., Hurwitz D. I., Jackson J. D., Ke Z., Lanczycki C. J., Liebert C. A., Liu C., Lu F., Lu S., Marchler G. H., Mullokandov M., Song J. S., Tasneem A., Thanki N., Yamashita R. A., Zhang D., Zhang N., Bryant S. H. 2009. CDD: specific functional annotation with the Conserved Domain Database. Nucleic Acids Res. 37:D205–D210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yu D., Silva M. C., Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dunn W., Chou C., Li H., Hai R., Patterson D., Stolc V., Zhu H., Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beghetto E., Paolis F. D., Spadoni A., Del Porto P., Buffolano W., Gargano N. 2008. Molecular dissection of the human B cell response against cytomegalovirus infection by lambda display. J. Virol. Methods 151:7–14 [DOI] [PubMed] [Google Scholar]
- 23. Warming S., Costantino N., Court D. L., Jenkins N. A., Copeland N. G. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chee M. S., Bankier A. T., Beck S., Bohni R., Brown C. M., Cerny R., Horsnell T., Hutchison C. A., III, Kouzarides T., Martignetti J. A., Preddie E., Satchwell S. C., Tomlinson P., Weston K. M., Barrell B. G. 1990. Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immunol. 154:125–169 [DOI] [PubMed] [Google Scholar]
- 25. McMahon T. P., Anders D. G. 2002. Interactions between human cytomegalovirus helicase-primase proteins. Virus Res. 86:39–52 [DOI] [PubMed] [Google Scholar]
- 26. Moorman N. J., Sharon-Friling R., Shenk T., Cristea I. M. 2010. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9:851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Murayama T., Natsuume-Sakai S., Shimokawa K., Furukawa T. 1986. Fc receptor(s) induced by human cytomegalovirus bind differentially with human immunoglobulin G subclasses. J. Gen. Virol. 67(Pt. 7):1475–1478 [DOI] [PubMed] [Google Scholar]
- 28. Irmiere A., Gibson W. 1983. Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 130:118–133 [DOI] [PubMed] [Google Scholar]
- 29. Buchkovich N. J., Maguire T. G., Alwine J. C. 2010. Role of the endoplasmic reticulum chaperone BiP, SUN domain proteins, and dynein in altering nuclear morphology during human cytomegalovirus infection. J. Virol. 84:7005–7017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fraile-Ramos A., Pelchen-Matthews A., Risco C., Rejas M. T., Emery V. C., Hassan-Walker A. F., Esteban M., Marsh M. 2007. The ESCRT machinery is not required for human cytomegalovirus envelopment. Cell. Microbiol. 9:2955–2967 [DOI] [PubMed] [Google Scholar]
- 31. Tandon R., AuCoin D. P., Mocarski E. S. 2009. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 83:10797–10807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Azzeh M., Honigman A., Taraboulos A., Rouvinski A., Wolf D. G. 2006. Structural changes in human cytomegalovirus cytoplasmic assembly sites in the absence of UL97 kinase activity. Virology 354:69–79 [DOI] [PubMed] [Google Scholar]
- 33. Prichard M. N., Britt W. J., Daily S. L., Hartline C. B., Kern E. R. 2005. Human cytomegalovirus UL97 kinase is required for the normal intranuclear distribution of pp65 and virion morphogenesis. J. Virol. 79:15494–15502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silva M. C., Schroer J., Shenk T. 2005. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. U. S. A. 102:2081–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luse S. A., Smith M. G. 1958. Electron microscopy of salivary gland viruses. J. Exp. Med. 107:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montplaisir S., Belloncik S., Leduc N. P., Onji P. A., Martineau B., Kurstak E. 1972. Electron microscopy in the rapid diagnosis of cytomegalovirus: ultrastructural observation and comparison of methods of diagnosis. J. Infect. Dis. 125:533–538 [DOI] [PubMed] [Google Scholar]
- 37. Ruebner B. H., Hirano T., Slusser R. J., Medearis D. N., Jr. 1965. Human cytomegalovirus infection. Electron microscopic and histochemical changes in cultures of human fibroblasts. Am. J. Pathol. 46:477–496 [PMC free article] [PubMed] [Google Scholar]
- 38. Ruebner B. H., Hirano T., Slusser R., Osborn J., Medearis D. N., Jr. 1966. Cytomegalovirus infection. Viral ultrastructure with particular reference to the relationship of lysosomes to cytoplasmic inclusions. Am. J. Pathol. 48:971–989 [PMC free article] [PubMed] [Google Scholar]
- 39. Matsuo H., Chevallier J., Mayran N., Le Blanc I., Ferguson C., Faure J., Blanc N. S., Matile S., Dubochet J., Sadoul R., Parton R. G., Vilbois F., Gruenberg J. 2004. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303:531–534 [DOI] [PubMed] [Google Scholar]
- 40. Yu D., Smith G. A., Enquist L. W., Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang D., Bresnahan W., Shenk T. 2004. Human cytomegalovirus encodes a highly specific RANTES decoy receptor. Proc. Natl. Acad. Sci. U. S. A. 101:16642–16647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murphy E., Vanicek J., Robins H., Shenk T., Levine A. J. 2008. Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc. Natl. Acad. Sci. U. S. A. 105:5453–5458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu P., Jenkins N. A., Copeland N. G. 2003. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 13:476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schroer J., Shenk T. 2008. Inhibition of cyclooxygenase activity blocks cell-to-cell spread of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 105:19468–19473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mach M., Osinski K., Kropff B., Schloetzer-Schrehardt U., Krzyzaniak M., Britt W. 2007. The carboxy-terminal domain of glycoprotein N of human cytomegalovirus is required for virion morphogenesis. J. Virol. 81:5212–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Feng X., Schroer J., Yu D., Shenk T. 2006. Human cytomegalovirus pUS24 is a virion protein that functions very early in the replication cycle. J. Virol. 80:8371–8378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mitchell D. P., Savaryn J. P., Moorman N. J., Shenk T., Terhune S. S. 2009. Human cytomegalovirus UL28 and UL29 open reading frames encode a spliced mRNA and stimulate accumulation of immediate-early RNAs. J. Virol. 83:10187–10197 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.