

Abstract
The reprogramming of DNA-binding specificity is an important challenge for computational protein design that tests current understanding of protein–DNA recognition, and has considerable practical relevance for biotechnology and medicine1–6. Here we describe the computational redesign of the cleavage specificity of the intron-encoded homing endonuclease I-MsoI7 using a physically realistic atomic-level forcefield8,9. Using an in silico screen, we identified single base-pair substitutions predicted to disrupt binding by the wild-type enzyme, and then optimized the identities and conformations of clusters of amino acids around each of these unfavourable substitutions using Monte Carlo sampling10. A redesigned enzyme that was predicted to display altered target site specificity, while maintaining wild-type binding affinity, was experimentally characterized. The redesigned enzyme binds and cleaves the redesigned recognition site ~10,000 times more effectively than does the wild-type enzyme, with a level of target discrimination comparable to the original endonuclease. Determination of the structure of the redesigned nuclease- recognition site complex by X-ray crystallography confirms the accuracy of the computationally predicted interface. These results suggest that computational protein design methods can have an important role in the creation of novel highly specific endonucleases for gene therapy and other applications.
The nucleotide sequence specificity of DNA-binding proteins can not be deduced directly from amino acid sequence because the packing, hydrogen-bonding and electrostatic interactions responsible for nucleotide-specific recognition are dependent on the three-dimensional structure of the protein–DNA complex11,12. While a number of canonical amino acid–nucleotide interaction motifs are observed in protein–DNA interfaces13, they are not in general predictable from sequence information alone. Hence, in place of a simple ‘recognition code’, an atomic-level model of the protein–DNA interface is likely to be necessary to fully capture the basis of recognition specificity. To understand the specificity of naturally occurring DNA-binding proteins, and to design new specificities, we have developed a computational model that explicitly treats the packing, hydrogen-bonding, solvation and electrostatic interactions that underlie protein–DNA interactions9,14. Here we describe the use of this model to redesign the specificity of the I-MsoI homing endonuclease.
I-MsoI, which belongs to the LAGLIDADG family of homing endonucleases, is a 170-residue homodimeric enzyme that cleaves long target sites (20–24 base pairs (bp)) with considerable specificity7,15,16. The homing endonucleases provide an excellent model system for understanding protein–DNA interaction specificity, as well as starting points for engineering of novel specificities for targeted genomics applications, including gene therapy4,5. Crystal structures of the enzymes bound to their recognition sites reveal a rich assortment of side-chain–nucleotide contacts within the DNA major groove, which provide many possibilities for the redesign of specificity16,17.
We first tested our model of the DNA–protein interface by repacking the sidechains at the native I-MsoI interface. Nearly all protein–DNA contacts and most sidechain dihedral angles in the crystal structure were reproduced (Supplementary Fig. S1, Supplementary Table S1). To benchmark the sampling and evaluation of alternative amino acid identities required for protein design, clusters of amino acids were redesigned around each native base pair. All direct hydrogen-bonding contacts between protein and nucleotide bases present in the wild-type complex were preserved, showing that the model captures the important aspects of the naturally occurring interface. To redesign I-MsoI DNA cleavage specificity, we began by screening in silico for base changes predicted to disrupt binding by the wild-type enzyme (Supplementary Figs S2, S3). The amino acids in the vicinity of each of the base-pair substitutions predicted to disrupt binding were then redesigned, and the designs were ranked on the basis of the predicted affinity of the designed protein for the new site, and the predicted decrease in affinity of the native enzyme for the new site (Supplementary Fig. S4).
The design with the largest predicted change in specificity consisted of the base-pair substitution −6C · G to −6G · C in the ‘left’ DNA half-site, and a similar change in the symmetry-related ‘right’ half-site from +6A · T to +6C · G (Supplementary Fig. S5; numbers are the distance in base pairs from the centre of the recognition site). At both positions in the wild-type complex, the key interactions of the base pair are a hydrogen bond of the purine ring with Lys 28 and a water-mediated contact with Thr 83 (Fig. 1a). Converting either base pair to a G · C is predicted to disrupt binding (+3.2 kcal) by the loss of the direct hydrogen-bonding interaction and the resulting desolvation of Lys 28 (Fig. 1b).
Figure 1. Comparison of the predicted interactions in cognate and non-cognate binding complexes, illustrating the designed specificity switch.

a, Wild-type I-MsoI, −6C · G (wild type). A water molecule present in the original structure16 is shown. b, Wild-type I-MsoI, −6G · C. c, I-MsoI-K28L/T83R, −6C · G. d, I-MsoI-K28L/T83R, −6G · C. In parts c and d, the van der Waals surfaces of Leu 28 and +6C are shown in grey. Figures were generated using the molecular graphics program PyMOL (Delano Scientific). WT, wild type; DES, designed; blue strands, protein backbone; beige spheres and sticks, DNA backbone; other spheres, constant nucleotides; dashed lines, hydrogen bonds.
Compensation of the base-pair substitution by redesign of the surrounding amino acids yielded the low energy solution K28L, T83R (−4.2 kcal versus wild type). As shown in Fig. 1d, Arg 83 is predicted to make two hydrogen bonds to the guanine nucleotide of the introduced G · C base pair, a sidechain–base interaction motif important in naturally occurring protein–DNA interfaces13,18. The Leu 28 mutation decreases the binding energy of the designed enzyme to wild-type DNA by eliminating a purine-specific hydrogen-bond. Furthermore, Leu 28 in the designed enzyme makes a favourable non-polar packing contact with the C5 of cytosine of the designed DNA (Fig. 1d). This leucine may also contribute to specificity against a purine at this position by unfavourably burying polar surface area of nitrogen N7 (Fig. 1c). The predicted binding energies of the cognate and non-cognate complexes are given in Table 1.
Table 1.
Predicted binding energies*
| Target sites | I-MsoI | I-MsoI-K28L-T83R |
|---|---|---|
| −6C · G, +6A · T | 0.0 (0.0) | +1.6 (+1.58) |
| −6G · C, +6C · G | +3.2 (+2.34) | −4.2 (−1.13) |
Relative binding energies of designed cognate and non-cognate complexes were computed as (energy of complex)−(energy of isolated DNA + isolated protein), with the value for the wild-type complex subtracted to facilitate comparison. The corresponding values for the hydrogen-bonding contribution to the total energy are shown in parentheses.
Competitive in vitro cleavage assays15,19 were performed to assess the specificity of the wild-type and designed enzymes by directly comparing the relative activity of the enzyme on each recognition site. Specific site cleavage, in the presence of both the cognate and non-cognate sites, in addition to 8.3 kilobases (kb) of non-specific vector sequence, is independently observed as the conversion of the linearized plasmid band into two smaller fragments of unique size. As is evident in the upper panel in Fig. 2, 100 nM wild-type I-MsoI cleaves a substantial fraction of the wild-type target site, but little or no cleavage of the designed site occurs at concentrations as high as 6.4 μM. In contrast, cleavage of the designed site by the designed enzyme (Fig. 2, lower panel) is observed at an enzyme concentration of 200 nM, whereas the original site is cleaved only at enzyme concentrations of 3.2 μM and above. To determine the specificity of binding independent of catalysis, gel electrophoretic mobility shift assays of cognate and non-cognate DNA–protein complexes were performed, and a similar switch in specificity was observed (Table 2). The shift in sequence specificity suggested by these results (at least 4,000-fold by competitive cleavage assay, and ~13,000-fold by gel shift assay) is considerably greater than that shown to be required for changes in phenotype in in vivo gene elimination assays using altered homing endonucleases19,20.
Figure 2. Switch in nuclease cleavage specificity.

Equimolar amounts of linearized plasmid DNAs containing wild-type (WT) or designed (DES) I-MsoI cleavage sites were digested by serial dilutions of wild-type or designed I-MsoI endonuclease, and analysed by gel electrophoresis. The switch in sequence specificity is defined as (wild type vs. DES/wild type vs. WT) × (designed vs. WT/designed vs. DES), where quantities in parentheses indicate the lowest enzyme concentration at which significant cleavage of the site is observed. Here, the wild-type enzyme favours the WT site by >27-fold, the designed enzyme favours the DES site by ~25-fold, and hence the specificity switch is greater than 27 × 25 (>4,000-fold).
Table 2.
Experimental binding affinities*
| Target sites | Protein |
|
|---|---|---|
| I-MsoI | I-MsoI-K28L-T83R | |
| −6C · G, +6A · T | 61 ± 15 nM | 6.1 ± 1.3 μM |
| −6G · C, +6C · G | >25 μM | 192 ± 30 nM |
Binding affinities for wild-type and designed I-MsoI as determined by gel electromobility shift. Errors represent 67% confidence intervals.
To verify the atomic-level accuracy of the computationally predicted design, the crystallographic structure of the complex was determined to 2.0 Å resolution (Supplementary Fig. S6). A difference map showing the electron density around the redesigned amino acids superimposes well with the predicted structure, confirming the accuracy of the design (Fig. 3). A water molecule is also evident at the site, but it does not appear to contribute to nucleotide-binding specificity.
Figure 3. Crystal structure of the designed enzyme–DNA complex.
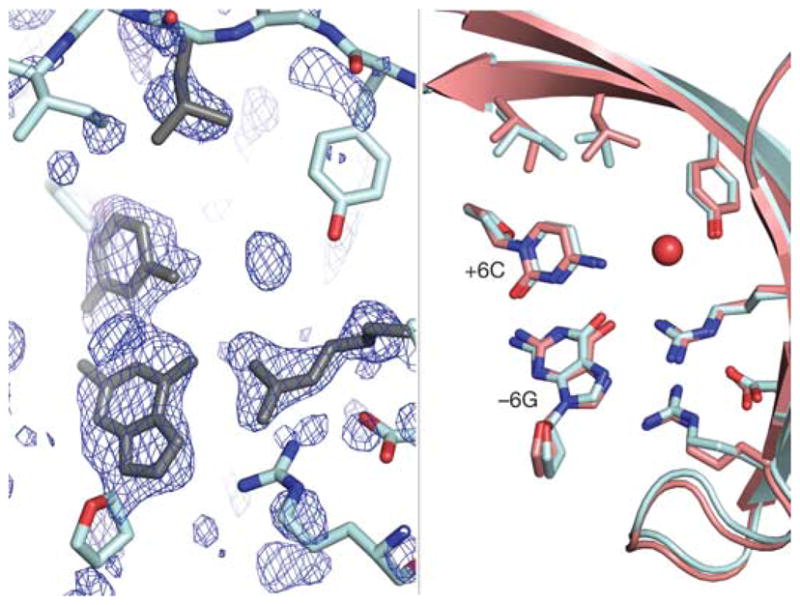
Left, Fo−Fc electron-density map of the redesigned region calculated from a refinement model lacking the redesigned side chains and bases (cyan). The computational design model (grey) fits well into the unassigned density (blue mesh, +2.2σ). Right, superposition of the design model (salmon) and the refined crystal structure (cyan) confirms the accuracy of the design. A new coordinated water molecule (red sphere) is also apparent.
Our results represent a significant advance in the redesign of protein–DNA interaction specificity, and demonstrate the efficacy of explicit, atomic-level modelling of the protein–DNA interface for the re-engineering of specificity. While the subject of this work is a homing endonuclease, the method should be generalizable to any protein–DNA interface redesign problem: for example, the reprogramming of transcription factor binding specificity. The computational approach described here can be improved further by modelling protein and DNA backbone flexibility and water-mediated interactions. Remaining inaccuracies in the potential function can potentially be compensated by focused exploration around promising computational designs using experimental selection methodologies.
The engineering of artificial gene-specific reagents from naturally occurring DNA-binding proteins is of great interest for a variety of targeted genetic applications. Site-specific zinc-finger nucleases (ZFNs), generated as chimaeras of non-specific endonuclease domains fused to zinc-finger domains, can stimulate gene-specific homologous recombination2,3 and have recently been shown to promote the repair of a disease-associated mutation in cultured cells6. The homing endonucleases are an alternative molecular system for the creation of gene-specific DNA cleavage enzymes4 that have also been shown to elicit gene repair by homologous recombination in murine hepatocytes21. These proteins are inherently more site-specific in their DNA cleavage activities than are ZFNs, and have the added advantage that site binding and cleavage are tightly coupled in the same protein domain, perhaps minimizing off-target cleavage. The redesign of existing homing endonuclease proteins to confer novel DNA target specificities remains challenging using methods of directed evolution5,20,22. The use and refinement of the computational modelling and design strategies described here should facilitate such efforts, and allow us to approach our long-term goal of designing novel proteins able to recognize and cleave any desired DNA site with high specificity for targeted genomics applications.
METHODS
Computational design
Models of every single base-pair substitution in the I-MsoI–target site complex were generated, and a Monte Carlo search procedure was used to sample alternative conformations and identities of the surrounding amino acid side chains. Protein positions were repacked or redesigned if at least one arginine rotamer placed at the position could make contact to the substituted DNA. Side-chain rotamer conformations were taken from the Dunbrack library23, and supplemented with extra rotamers generated by varying χ1 and χ2 independently by plus or minus one standard deviation of the principal distribution for the rotamer. For still finer sampling of the conformations of long polar side chains, additional rotamers were generated by similarly perturbing χ3 and χ4; these were retained for the full combinatorial search if they had favourable hydrogen-bonding energies with the DNA bases. The physically realistic all atom force field used to guide the Monte Carlo search is composed of a Lennard–Jones-based treatment of packing, an orientation dependent hydrogen-bonding potential, a generalized Born-based treatment of electrostatic solvation energy24, a PDB-derived side-chain torsional potential, and amino acid dependent reference energies which represent average residue energies in the unbound and unfolded state. The lowest energy structures identified in the Monte Carlo search were further optimized by continuous minimization of side-chain and DNA torsion angles using the Powell method14,25. The in silico protein–DNA complexes produced by sequence design followed by minimization were ranked on the basis of the predicted affinity of the designed enzyme for the new site, and the predicted loss in affinity of the wild-type enzyme for the new site (Supplementary Fig. S4).
Experiments
Full details of the experimental methods are given in Supplementary Information; a summary is given below.
Expression and purification of I-MsoI was performed as previously described16. Substrates for competitive cleavage were as follows: oligonucleotide duplexes corresponding to I-MsoIWT (5′-GCAGAACGTCGTGAGACAGTTCCG-3′; bold font indicates positions that were changed in the design) and I-MsoIDES (5′-GCAGAAGGTCGTGAGACCGTTCCG-3′) cleavage sites were incorporated into plasmid vectors of sizes 5.4 kb (wild-type site) and 2.9 kb (designed site). To facilitate product identification, the plasmid substrates were linearized by restriction-enzyme digestion before use in cleavage assays.
Serial twofold dilutions of wild-type and designed protein were added to reaction mixtures containing 50 nM of both linearized cleavage constructs in the presence of magnesium. Reactions proceeded for 1 h at 37 °C. Electrophoretic mobility shift assays to determine binding constants of the cognate and non-cognate complexes were carried out in the presence of calcium, using 5′-radiolabelled DNA duplexes and non-specific competitor DNA. Crystals of I-MsoI-K28L-T83R bound to designed DNA duplex formed in previously described conditions16. Data were collected at the Advanced Light Source. The structure was solved by molecular replacement using the original I-MsoI structure, and refined at 2.0 Å resolution using CNS26. The final refinement statistics (Supplementary Table S2) for I-MsoI-K28L-T83R were Rwork/Rfree = 0.229/0.271.
Supplementary Material
Acknowledgments
We thank J. L. Eklund for assistance with binding assays, and B. W. Shen for assistance with data collection and refinement. This work was supported by fellowships from the Jane Coffin Childs Memorial Fund (J.J.H.), the National Science Foundation (C.M.D.), and grants from the National Institute of Health (R.J.M. and B.L.S.), the Howard Hughes Medical Institute (D.B.), and the Gates Foundation Grand Challenges Program (B.L.S., D.B., R.J.M.).
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions J.J.H. and C.M.D. developed the original protein–DNA interface design methods and code. J.A. made further code and method developments, generated and assessed the computational predictions, and performed mutagenesis, biochemical characterization, and crystallization. D.S. collected and processed the crystallographic data, and aided in protein purification and structure refinement.
Author Information The atomic coordinates of the redesigned I-MsoI endonuclease bound to its cognate DNA have been deposited in the Protein Data Bank with the accession number 2FLD. Reprints and permissions information are available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
References
- 1.Uil TG, Haisma HJ, Rots MG. Therapeutic modulation of endogenous gene function by agents with designed DNA-sequence specificities. Nucleic Acids Res. 2003;31:6064–6078. doi: 10.1093/nar/gkg815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bibikova M, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–297. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 4.Wickelgren I. Molecular biology. Spinning junk into gold. Science. 2003;300:1646–1649. doi: 10.1126/science.300.5626.1646. [DOI] [PubMed] [Google Scholar]
- 5.Stoddard BL. Homing endonuclease structure and function. Q Rev Biophys. 2005;38:1–47. doi: 10.1017/S0033583505004063. [DOI] [PubMed] [Google Scholar]
- 6.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 7.Lucas P, Otis C, Mercier JP, Turmel M, Lemieux C. Rapid evolution of the DNA-binding site in LAGLIDADG homing endonucleases. Nucleic Acids Res. 2001;29:960–969. doi: 10.1093/nar/29.4.960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohl CA, Strauss CE, Misura KM, Baker D. Protein structure prediction using Rosetta. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 9.Havranek JJ, Duarte CM, Baker D. A simple physical model for the prediction and design of protein–DNA interactions. J Mol Biol. 2004;344:59–70. doi: 10.1016/j.jmb.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 10.Voigt CA, Gordon DB, Mayo SL. Trading accuracy for speed: A quantitative comparison of search algorithms in protein sequence design. J Mol Biol. 2000;299:789–803. doi: 10.1006/jmbi.2000.3758. [DOI] [PubMed] [Google Scholar]
- 11.Kono H, Sarai A. Structure-based prediction of DNA target sites by regulatory proteins. Proteins. 1999;35:114–131. [PubMed] [Google Scholar]
- 12.Pabo CO, Nekludova L. Geometric analysis and comparison of protein–DNA interfaces: why is there no simple code for recognition? J Mol Biol. 2000;301:597–624. doi: 10.1006/jmbi.2000.3918. [DOI] [PubMed] [Google Scholar]
- 13.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morozov AV, Havranek JJ, Baker D, Siggia ED. Protein–DNA binding specificity predictions with structural models. Nucleic Acids Res. 2005;33:5781–5798. doi: 10.1093/nar/gki875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seligman LM, et al. Mutations altering the cleavage specificity of a homing endonuclease. Nucleic Acids Res. 2002;30:3870–3879. doi: 10.1093/nar/gkf495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chevalier B, Turmel M, Lemieux C, Monnat RJ, Jr, Stoddard BL. Flexible DNA target site recognition by divergent homing endonuclease isoschizomers I-CreI and I-MsoI. J Mol Biol. 2003;329:253–269. doi: 10.1016/s0022-2836(03)00447-9. [DOI] [PubMed] [Google Scholar]
- 17.Heath PJ, Stephens KM, Monnat RJ, Jr, Stoddard BL. The structure of I–Crel, a group I intron-encoded homing endonuclease. Nature Struct Biol. 1997;4:468–476. doi: 10.1038/nsb0697-468. [DOI] [PubMed] [Google Scholar]
- 18.Seeman NC, Rosenberg JM, Rich A. Sequence-specific recognition of double helical nucleic acids by proteins. Proc Natl Acad Sci USA. 1976;73:804–808. doi: 10.1073/pnas.73.3.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sussman D, et al. Isolation and characterization of new homing endonuclease specificities at individual target site positions. J Mol Biol. 2004;342:31–41. doi: 10.1016/j.jmb.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 20.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc. 2006;128:2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- 21.Gouble A, et al. Efficient in toto targeted recombination in mouse liver by meganuclease-induced double-strand break. J Gene Med. 2006 doi: 10.1002/jgm.879. published online 13 February 2006. [DOI] [PubMed] [Google Scholar]
- 22.Arnould S, et al. Engineering of large numbers of highly specific homing endonucleases that induce recombination on novel DNA targets. J Mol Biol. 2006;355:443–458. doi: 10.1016/j.jmb.2005.10.065. [DOI] [PubMed] [Google Scholar]
- 23.Dunbrack RL, Jr, Cohen FE. Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci. 1997;6:1661–1681. doi: 10.1002/pro.5560060807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Onufriev A, Bashford SD, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized Born model. Proteins. 2004;55:383–394. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 25.Press WH, Flannery BP, Teukolsky SA, Vetterling WT. Numerical Recipes in C: The Art of Scientific Computing. Cambridge Univ. Press; New York: 1992. [Google Scholar]
- 26.Brunger AT, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.