

Abstract
The influence of assay method on single dose cyclosporine (CsA) pharmacokinetics was studied in nine dogs receiving either i.v. or oral CsA. Samples were drawn from hepatic, portal, and systemic veins at various times after the dose and CsA levels were determined by radioimmunoassay (RIA) and high-performance liquid chromatography (HPLC). Blood concentration-time data were analyzed by nonlinear least-squares regression, using two-compartment models. RIA/HPLC ratios for all samples were greater than one, and did not change significantly over time. The mean RIA/HPLC ratios for samples drawn from all three veins were higher after oral than i.v. doses of the drug (P<0.05). Area under the concentration-time curve (AUC) was higher and systemic clearance (CIs) lower than calculated on the basis of RIA results, regardless of the route of administration. AUC calculated for CsA metabolites (RIA-HPLC) was highest in the portal vein after an oral dose of CsA. Bioavailability was 20.4% and 27.0% when estimated using HPLC and RIA data, respectively. The mean CsA metabolite index (CMI), when calculated for hepatic, portal, or systemic vein, was greater when the drug was administered orally. The mean hepatic extraction ratio (HER) of the parent drug and for CsA metabolites was approximately 23% in i.v. and p.o. studies. These results suggest that the gastrointestinal tract may play a role in the metabolism of CsA when the drug is administered orally. In addition, if CsA metabolites not measured by HPLC have either toxic or immunosuppressive properties, the RIA assay may be more useful for monitoring patients.
The introduction of cyclosporine (CsA)5 in the immunosuppressive regimen has proven to be the single most important component of success in organ transplantation over the last few years. The drug does not exhibit the bone marrow toxic side effects of immunosuppressive agents used previously, and it exerts selective and reversible inhibition of stimulated T lymphocyte proliferation, apparently by blocking the production of interleukin-2 (1). These advantages are partially offset by the several toxic side effects that have been encountered with increased clinical use of CsA (2, 3). Furthermore, it is often difficult to maintain stable therapeutic blood levels of the drug. Thus, there is considerable interest in the pharmacokinetics and metabolism of CsA (4–7).
Two techniques are available to determine blood, plasma, and serum levels of the drug: the radioimmunoassay (RIA) technique detects the parent drug plus some of the metabolites, and the high performance liquid chromatography (HPLC) technique detects the parent drug only. As might be expected, CsA blood levels determined with the RIA technique are consistently higher than the corresponding values determined by HPLC.
The objective of the present study was to understand the significance of the difference between HPLC and RIA measurements of the drug, particularly as it relates to the pharmacokinetics of CsA. The results of the study suggest that there is significant metabolism of the drug by the gastrointestinal tract when administered orally.
MATERIALS AND METHODS
Experimental procedures
Adult mongrel dogs (weighing 18–22 kg) were used in these studies. The animals were anesthetized and incubated, and the portal vein, a hepatic vein (through the right external jugular vein), and a peripheral vein were cannulated The dogs were allowed to recover from the surgery for 24–48 hr. On the morning of the study, after an overnight fast, a venous blood sample for baseline CsA levels was withdrawn. The animals were then given an i.v. bolus (5 mg/kg) or an oral p.o. dose (17.5 mg/kg) of CsA (these are the daily dosages of CsA used clinically by the authors). The oral dose was administered slowly with a syringe, directly into the dog’s mouth. No other drugs were given either before or during CsA administration. Then 2 ml of blood was collected in heparinized tubes for CsA blood level determination from each i.v. line at the following times after administration of the drug: 10 and 20 min (only for i.v. studies), 0.5, 1, 2, 4, 6, 8, and 24 hr. The samples were stored immediately at −20°C until further processing. Five i.v. and five p.o. studies were conducted.
Cyclosporine assay
CsA concentration was determined by HPLC, using a modification of the method of Sawchuk and Cartier (8). The column temperature was maintained at 70°C. Flow rate of the mobile phase, acetonitrile/methanol/distilled water (49/22/29 by volume) was 1.0 ml/min. Column effluent was monitored at 214 nm. Peak heights were measured manually, and the ratio of CsA to the internal standard cyclosporin D was used to calculate drug concentration. CsA concentration was measured by RIA according to the Sandoz kit instructions.
Data analysis
CsA concentration-time data were fitted with DRUGFUN, a nonlinear least-squares regression program available on the PROPHET system (9). All CsA concentration data were weighted as the reciprocal of the measured value squared (1/Y2). Pharmacokinetic parameters were then calculated by fitting the data to two-compartment models, BOLUS 2 for the i.v. data, and KA2 for the p.o. data, which assumes a first-order absorption of the drug. This analysis provides estimates of the volume of distribution of the central compartment (Vd); the initial (t½α) and terminal (t½β) half-lives; the distribution rate constant from tissues to blood (K21); systemic clearance (Cls); lagtime from ingestion to oral absorption; and area under the concentration-time curve (AUC). Bioavailability was calculated by equation 1:
| (1) |
To estimate the concentration of CsA metabolites, the difference between RIA and HPLC drug levels was calculated. This quantity represents an underestimation of metabolite concentration because crossreactivity of the metabolites with the RIA antibody is variable. An index of CsA metabolite production was calculated using equation 2:
| (2) |
The hepatic extraction ratio (HER) of the parent drug was calculated using the AUC values derived from portal (AUCpv) and hepatic (AUChv) venous blood samples:
| (3) |
In this equation, only HPLC determinations were used. The AUC of CsA metabolites (AUC-MET) was calculated by subtracting the AUC derived from the HPLC measurements from the AUC derived from RIA measurements of CsA blood levels. The HER of the CsA metabolites (HER·MET) was calculated by the equation:
| (4) |
where AUC·METpv and AUC·METhv were the AUC·MET in the portal and hepatic vein blood, respectively. The paired t test was used for comparison of RIA/HPLC ratios, calculated pharmacokinetic parameters, and CMI.
RESULTS
CsA blood levels were consistently higher by RIA than HPLC (Figs. 1, 2). Consequently, the RIA/HPLC ratios were always greater than one. These ratios did not change significantly over the time of the i.v. studies. Furthermore, the mean RIA/HPLC ratios were significantly higher after p.o. than i.v. administration of the drug (Table 1). These differences were apparent in systemic, portal, and hepatic blood.
Figure 1.
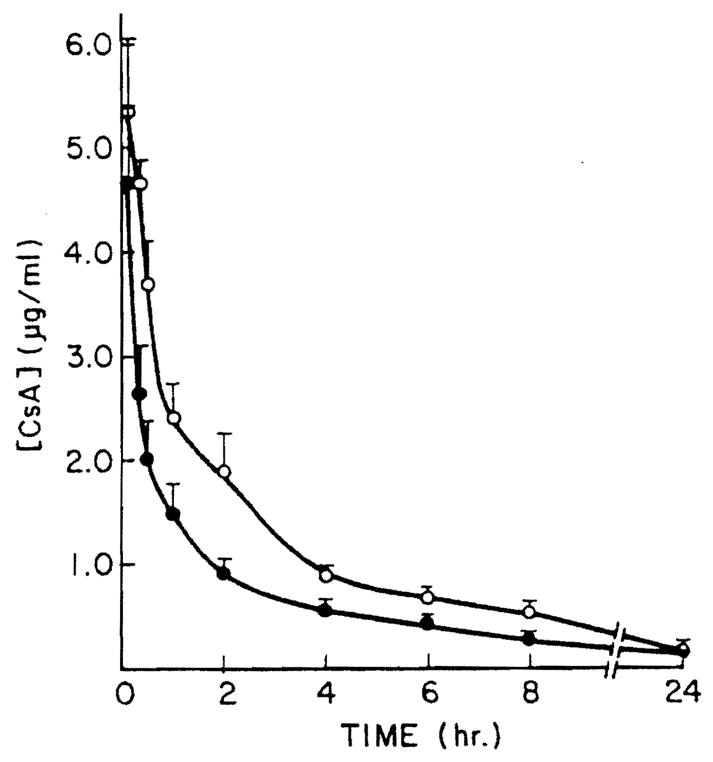
Mean hepatic vein CsA blood concentration after i.v. administration of 5 mg/kg CsA. Each point is the mean of CsA determinations in five animals, with bars indicating standard error. (– –○– –) RIA determinations (– –●––) HPLC determination.
Figure 2.
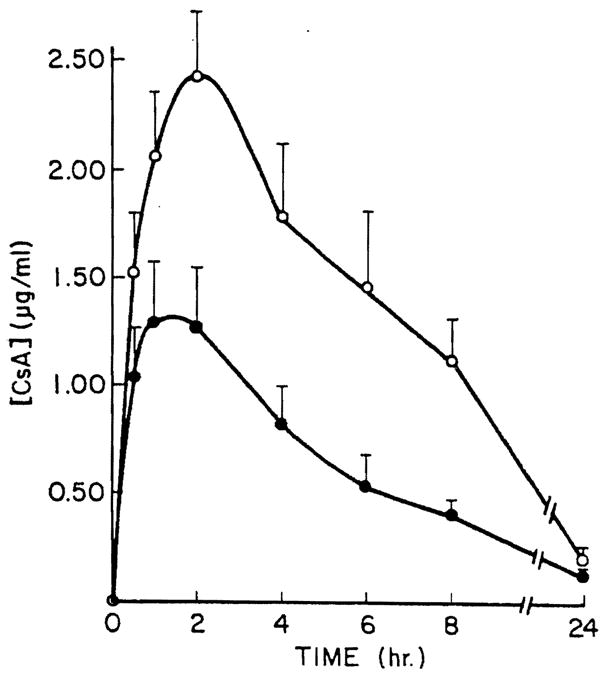
Mean hepatic vein CsA blood concentration was determined at various times after p.o. administration of 17.5 mg/kg CsA. Bars indicate standard error of the mean of four values (– –○– –) RIA determination; (– –●– –)HPLC determination.
Table 1.
RIA/HPLC ratios of blood CsA concentration after a single i.v. or p.o. dose of the drug
| Systemic vein | Portal vein | Hepatic vein | |
|---|---|---|---|
| i.v. | 1.48 | 1.51 | 1.73 |
| p.o. | 2.12 | 2.06 | 2.13 |
The RIA and HPLC concentration-time data from i.v. and p.o. studies were fitted to two-compartment models (Tables 2 and 3). Several differences were observed in the pharamcokinetic parameters. In both the i.v. and p.o. studies, the AUC by HPLC was significantly lower than the AUC by RIA. The Cls in the i.v. studies based on HPLC blood levels were higher than those based on RIA. Bioavailability in four experiments, calculated on the basis of the HPLC data, was 20.4% (range 9.6–30.8) and 27.0% (range 21.9–32.6) when estimated using RIA CsA blood levels.
Table 2.
Pharmacokinetic parameters after a single i.v. dose of CsA (samples drawn from a systemic vein, CsA concentration determined using RIA and HPLC)
| Vd (l/kg) | t½(hr) | t½(hr) | Cls (ml/min/kg) | AUC (μg· min/ml) | |
|---|---|---|---|---|---|
| HPLC: x̄ | 1.65 | 0.91 | 8.54 | 7.06 | 859 |
| SE | 0.15 | 0.09 | 0.73 | 0.87 | 151 |
| RIA: x̄ | 1.48 | 0.01 | 16.2 | 4.40 | 1315 |
| SE | 0.28 | 0.004 | 6.7 | 0.43 | 137 |
Table 3.
Pharmacokinetic parameters after a single p.o. dose of CsA (samples drawn from a systemic vein; CsA concentration determined using RIA and HPLC)
| Lagtime (hr) | t½ (hr) | AUC (μg·min/ml) | |
|---|---|---|---|
| HPLC: x̄ | 0.54 | 6.84 | 752 |
| SE | 0.14 | 1.11 | 104 |
| RIA: x̄ | 0.29 | 9.05 | 1697 |
| SE | 1.11 | 1.11 | 283 |
The mean CMI in systemic blood were 36% and 54% for CsA i.v. and p.o. administration studies, respectively (P<0.05). Similarly, in portal and hepatic blood, the CMI was higher after p.o. than i.v. administration of the drug (Table 4). The highest mean AUC·MET was observed in the portal vein after oral administration of CsA.
Table 4.
AUC·MET and CMI after an i.v. or p.o. dose of CsA (blood samples obtained from hepatic, portal, and systemic veins at various times after the dose)
| Systemic vein |
Portal vein |
Hepatic vein |
||||
|---|---|---|---|---|---|---|
| AUC·MET | CMI (%) | Aue·MET | CMI (%) | Aue·MET | CMI (%) | |
| i.v. | 452 | 36 | 459 | 41 | 463 | 43 |
| p.o. | 945 | 54 | 1124 | 56 | 868 | 53 |
The mean HER of the parent drug were 23% and 25% in the i.v. and p.o. studies, respectively. Total hepatic blood flow in conscious dogs has been estimated at about 30.9 ml/min/kg (10). Using this value and the calculated HER, a mean hepatic clearance of 7.11 ml/min/kg after i.v. administration of CsA may be calculated. This value is virtually identical to the mean CIs of 7.06 ml/min/kg obtained using the pharmacokinetic parameters based on the systemic blood samples (Table 2). The calculated HER for the CsA metabolites were 22% after i.v. and 23% after p.o. administration of the drug.
DISCUSSION
The pharmacokinetic analysis of CsA concentration-time data show that in both i.v. and p.o. studies the AUC is higher and Cls lower when calculated on the basis of RIA CsA blood concentration. The clinical relevance of this difference is not obvious. Yee et al. (7) suggest that differences in Cls measurements resulting from different assay techniques can significantly affect dosage recommendations. However, targeted CsA levels are different depending on whether RIA or HPLC is used to monitor CsA therapy. In this study, for example, the mean Cls calculated on the basis of HPLC was 7.06 ml/min/kg; if the selected average CsA target blood concentration at steady state is 400 ng/ml by HPLC, then the required CsA i.v. dose, calculated as target concentration times clearance, would be 4.1 mg/kg/24 hr. Since the RIA/HPLC ratio observed in this study was 1.5, the equivalent RIA-targeted CsA average blood concentration would be 600 ng/ml. The required dose of CsA to maintain this blood concentration, calculated using the Cls derived from RIA studies (4.40 ml/min/kg), would be 3.8mg/kg/24 hr. It is suggested, therefore, that either HPLC-derived or RIA-derived pharmacokinetic parameters may be used to estimate CsA intravenous dosages, as long as the optimal target CsA blood level has been determined by the same technique as the one used to make dosage recommendations. However, RIA measures both parent drug and metabolites, which introduces additional variability, so extra caution should be used when calculating pharmacokinetic parameters using RIA data. For example, the average t½β after an i.v. dose appears to be twofold higher when calculated using RIA than by HPLC (Table 2), but the standard error for the RIA value is so large that the difference between the RIA and HPLC t½β values is not statistically significant.
For the oral drug, the required dose is calculated as target concentration times clearance divided by bioavailability. Using a target concentration of 400 ng/ml and the HPLC pharmacokinetic data, the required dose would be 19.9 mg/kg/24 hr. For RIA monitoring of an oral dose, the target should be 2.1 times the HPLC target, because the average RIA/HPLC ratio for a p.o. dose is 2.1 (Table 2). Using a target value of 840 ng/ml (2.1×400) and the RIA bioavailability, the calculated dose is 19.7 mg/kg/24 hr, virtually identical to the dose suggested by HPLC pharmacokinetics.
We suggest that either RIA or HPLC pharmacokinetics can be used to calculate dose requirements with quite similar results, provided that appropriate target values are used and that the RIA/HPLC ratio is known for the particular situation. To achieve the desired concentrations of parent, immunosuppressive drug, the RIA target in healthy dogs must be 1.5 times the HPLC target for i.v. administration of the drug, and 2.1 times for p.o. This suggested difference in target RIA values for i.v. versus p.o. drug administration appears to be a new observation which may have clinical utility. The reason that a higher RIA target is needed for oral drug is the presence of a larger quantity of metabolites when drug is given p.o.
The clinical usefulness of RIA pharmacokinetics as a tool to achieve desired levels of parent CsA depends on the interpatient and intrapatient variability in the RIA/HPLC ratio, as well as the size of the acceptable range of parent CsA levels. Especially if metabolites not measured by HPLC have either toxic or immunosuppressive properties, the RIA determination may be particularly helpful. Regardless of which assay method is being used, CsA blood levels must continue to be monitored periodically throughout the time the patient is receiving the drug.
In this work, the difference between RIA and HPLC CsA concentrations was used as a tool to explore the patterns of CsA metabolism after i.v. and p.o. administration of the drug. It appears that a greater quantity of metabolites is present in the blood when CsA is given orally; the RIA/HPLC ratios were significantly higher in the p.o. than in the i.v. studies. Furthermore, in the p.o. studies, the AUC·MET was highest in the portal vein, suggesting that after oral administration of CsA, a part of the drug is metabolized by the gastrointestinal flora and mucosa. The resulting metabolites and the absorbed parent drug are carried to the liver by the portal blood. From the calculated HER of the parent drug and metabolites, it appears that CsA and its metabolites are eliminated by the liver at an almost equal rate whether the drug is administered i.v. or orally. The larger quantity of metabolites detected in the systemic and hepatic blood after oral CsA administration seems to be of gastrointestinal origin. When the drug is given orally, the additional metabolites of gut origin cannot be immediately excreted by the liver and are delivered to systemic circulation. These metabolites are responsible for the higher RIA/HPLC ratios described in the p.o. studies.
As already reported, CsA appears to be a low-to-intermediate extraction drug (11), the clearance of which is therefore more dependent on hepatic intrinsic clearance than on liver blood flow. The most frequently reported CsA bioavailability ranges from 20 to 50% (12, 13). As a consequence, the oral dosages required to attain the same CsA blood levels as after i.v. administration should be 2–5 times the i.v. CsA dose. If the orally administered, absorbed drug was metabolized only by the liver, it might be expected that the amount of metabolites delivered to the systemic circulation would be of the same order of magnitude as after i.v. administration of CsA. These metabolites would be those produced in the liver and not immediately excreted in the bile. The finding that maximal amounts of CsA metabolites are present in the portal vein after oral administration of the drug suggests that, as the liver extracts metabolites and the parent drug at a fixed rate, the CsA metabolites of gastrointestinal origin are partially delivered to the hepatic and systemic blood. Further studies must be conducted to elucidate the mechanism by which these metabolites are generated.
It is therefore apparent that bioavailability calculations obtained from RIA CsA blood concentration data might be unpredictably affected by the presence of the additional metabolites of gut origin. It is well recognized that alterations in liver function can affect CsA pharmacokinetics (14); however, when CsA is given orally, even alterations in the flora and the enzymatic activity of the mucosa of the gastrointestinal tract may significantly affect CsA metabolism and pharmacokinetics.
Acknowledgments
We thank Ms. Patricia Arndt for her excellent secretarial assistance.
Footnotes
This work was supported by research grants from the Veterans Administration and Project Grant AM-29961 from the National Institutes of Health, Bethesda, MD.
Abbreviations used: AUC, area under the concentration/time curve; Cls, systemic clearance; CMI, CsA metabolite index; CsA, cyclosporine; HPLC, high performance liquid chromatography; p.o., oral; Vd, volume distribution.
References
- 1.Elliott JF, Lin Y, Mizel SB, Bleackley RC, Harnish DG, Paetkan V. Induction of Interleukin 2 messenger RNA inhibited by cyclosporin A. Science. 1984;226:1439. doi: 10.1126/science.6334364. [DOI] [PubMed] [Google Scholar]
- 2.Starzl TE. Clinical aspects of cyclosporine therapy: a summation. Transplant Proc. 1982;14(suppl 1):3103. [PMC free article] [PubMed] [Google Scholar]
- 3.Britton S, Palacios R. Cyclosporin A: Usefulness, risks and mechanism of action. Immunol Rev. 1982;65:5. doi: 10.1111/j.1600-065x.1982.tb00425.x. [DOI] [PubMed] [Google Scholar]
- 4.Kahan BD, Ried M, Newburger J. Pharmacokinetics of cyclosporine in human renal transplantation. Transplant Proc. 1983;15:446. [Google Scholar]
- 5.Kekown PA, Stiller CR, Laupacis AL, et al. The effects and side effects of cyclosporine: relationship to drug pharmacokinetics. Transplant Proc. 1983;14:659. [PubMed] [Google Scholar]
- 6.Newburger J, Kahan BD. Cyclosporine pharmacokinetics in man. Transplant Proc. 1983;15(suppl 1):2413. [Google Scholar]
- 7.Yee GC, Kennedy MS, Storb R, Donnal TE. Pharmacokinetics of intravenous cyclosporine in bone marrow transplant patients. Transplantation. 1984;38:511. doi: 10.1097/00007890-198411000-00014. [DOI] [PubMed] [Google Scholar]
- 8.Sawchuck RJ, Cartier LL. Liquid chromatographic determination of cyclosporine A in blood and plasma. Clin Chem. 1981;27:1368. [PubMed] [Google Scholar]
- 9.Holford NHG. Drugfun. In: Perry HM, editor. Prophet public procedures notebook. Cambridge, MA: Bolt, Baranek & Newman; 1982. [Google Scholar]
- 10.Nxumalo JL, Teranaka M, Schenk WG. Hepatic blood flow measurement III. Ann Surg. 1978;187:299. doi: 10.1097/00000658-197803000-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ptachcinski RJ, Venkatamaran R, Burckart GJ, Yang S, Van Thiel DH, Starzl TE. Cyclosporine pharmacokinetics in adult liver transplant patients. Hepatology. 1983;3:829. [Google Scholar]
- 12.Wood AJ, Maurer G, Niederberger W, Beveridge T. Cyclosporine: pharmacokinetics, metabolism and drug interactions. Transplant Proc. 1983;15(suppl 1):2409. [Google Scholar]
- 13.Beveridge T. Pharmacokinetics and metabolism of Cyclosporin A. In: White DJG, editor. Cyclosporin A. New York: Elsevier; 1982. [Google Scholar]
- 14.Burckart G, Starzl T, Williams L, et al. Cyclosporine monitoring and pharmacokinetics in pediatric liver transplant patients. Transplant Proc. 1985;17:286. [PMC free article] [PubMed] [Google Scholar]