

Abstract
Purpose
To evaluate the possibility of genetic involvement in retinopathy of prematurity (ROP). Although ROP is most often associated with low birthweight and low gestational age, these factors do not necessarily predict the severity of ROP. The possible involvement of other factors, including genetic variants, has been considered. Familial exudative vitreoretinopathy (FEVR) is a hereditary vitreoretinal disorder with clinical manifestations similar to those of ROP. Three genes involving the wingless/int1 (Wnt) receptor signaling pathway—FZD4 for frizzled 4, LRP5 for low-density lipoprotein receptor-related protein 5, and ND for Norrie disease protein—are associated with the development of FEVR.
Methods
In the present study, 17 Japanese patients with advanced ROP were screened for these three candidate genes of FEVR. Genomic DNA from each patient was subjected to PCR and direct sequencing of the ND, FZD4, and LRP5 genes.
Results
One patient had a heterozygous mutation in the 5′ untranslated region of the ND gene. Another had a leucine insertion in the signal peptide of LRP5. None showed any mutation in FZD4.
Conclusions
These findings suggest that genetic changes in the Wnt receptor signaling pathway associate to the development of advanced ROP.
Introduction
Retinopathy of prematurity (ROP) is a vasoproliferative disorder of the eye affecting premature neonates and is a leading cause of visual loss in children even in developed countries. However, the pathogenesis of advanced ROP is not fully understood. It is known to occur in association with environmental factors, such as preterm gestational age and early oxygen exposure, but the same factors can lead to differing ROP phenotypes [1]. ROP regresses spontaneously in some patients but progresses rapidly to a severe stage in others. In addition, the incidence of advanced ROP varies among different ethnic groups [2]. These facts suggest that various internal factors, especially genetic variants, may be involved in the development of ROP [3].
Familial exudative vitreoretinopathy (FEVR) is a hereditary vitreoretinal disorder with clinical manifestations similar to those of ROP. FEVR is known to have three inheritance patterns: autosomal dominant (AD) [4]; autosomal recessive (AR) [5]; and X-linked recessive (XL) [6]. Four genes have been identified as candidates for FEVR: mutations of the Norrie disease (ND) gene have been found in XL-FEVR (the ND gene encodes the Norrie disease protein [NDP], Norrin) [7-9]; mutations of the FZD4 gene encoding frizzled 4 have been observed in AD-FEVR [10,11]; and the LRP5 gene encoding low-density lipoprotein receptor-related protein 5 (LRP5) is reported to show mutations in both AD-FEVR [11] and AR-FEVR [12]. Each molecule encoded by the above three genes participates in the wingless/integrated (Wnt) receptor signaling pathway. Norrin acts as a ligand in the Wnt receptor-β-catenin signaling transduction pathway by binding to frizzled 4 [13]. In the eye, Norrin plays roles in the recognition signal for targeting neuronal and retinal connections and in angiogenesis during retina development [14]. Frizzled 4 belongs to the frizzled family of Wnt receptors, and an FZD4 knockout mouse model has demonstrated the importance of this pathway in angiogenesis and retinal development [10]. LRP5 is a member of the low-density lipoprotein receptor family. In Wnt signaling pathways, LRP5 forms a complex with frizzled 4, which acts as a functional receptor pair to activate the canonical Wnt-β-catenin pathway [15]. LRP5-deficient mice show a delay in hyaloid vessel regression, suggesting that LRP5 dysfunction can cause abnormal vascularization during retinal development [16]. Recently, another gene, TSPAN12, which is involved in the Norrin-β-catenin signaling pathway, has been found to be responsible for AD-FEVR [17-19]. Because of the resemblance between ROP and FEVR, genetic changes in the Wnt receptor signaling pathway during retinal development are considered to be likely risk factors for advanced ROP. This idea is supported by several recent studies involving screening for ND and FZD4 gene mutations in ROP [20-25].
In the present study, we investigated whether the three genes responsible for FEVR are associated with advanced ROP and whether any association is related to ethnicity. This report discusses our findings regarding the Wnt signaling pathway and its effects on retinal development.
Methods
Patients
Seventeen premature infants (12 boys and 5 girls) were enrolled in this study via a protocol approved by the internal review boards of the National Center for Child Health and Development (Tokyo, Japan) and of the Nippon Medical School (Tokyo, Japan); informed consent was obtained from the parents of all of the subjects. Each patient was of a gestational age of less than 32 weeks, had a birthweight of less than 1,500 g, had undergone eye surgery, and had retinal findings consistent with advanced ROP (Table 1). The stage of ROP was determined by trained ophthalmologists according to the international classification of retinopathy of prematurity [26].
Table 1. List of patients enrolled in this study.
| Case number | Sex | Gestational age (week) | Birthweight (g) | Stage of ROP right/left | Type of ROP | Surgery received | Operation |
|---|---|---|---|---|---|---|---|
| 1 |
M |
27 |
1,084 |
4A/4A |
classic ROP |
both eyes |
buckling |
| 2 |
M |
25 |
972 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 3 |
F |
26 |
906 |
4A/4A |
AP-ROP |
both eyes |
vitrectomy |
| 4 |
M |
23 |
596 |
4A/4A |
AP-ROP |
both eyes |
vitrectomy |
| 5 |
F |
29 |
1212 |
4A/3 |
AP-ROP |
right eye |
vitrectomy |
| 6 |
M |
23 |
512 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 7 |
F |
23 |
642 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 8 |
M |
24 |
670 |
4A/5 |
AP-ROP |
both eyes |
vitrectomy |
| 9 |
M |
23 |
542 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 10 |
F |
24 |
588 |
4A/3 |
AP-ROP |
both eyes |
vitrectomy |
| 11 |
M |
23 |
747 |
4A/3 |
AP-ROP |
right eye |
vitrectomy |
| 12 |
M |
27 |
998 |
4A/4A |
AP-ROP |
both eyes |
vitrectomy |
| 13 |
M |
23 |
458 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 14 |
M |
23 |
560 |
4B/4A |
AP-ROP |
both eyes |
vitrectomy |
| 15 |
M |
23 |
676 |
5/3 |
classic ROP |
right eye |
vitrectomy |
| 16 |
F |
23 |
708 |
5/5 |
AP-ROP |
both eyes |
vitrectomy |
| 17 | M | 24 | 576 | 5/5 | AP-ROP | both eyes | vitrectomy |
Stage of ROP was classified based on ICRP. AP-ROP; aggressive posterior ROP.
Genetic analysis
Genomic DNA was isolated from peripheral blood and amplified with PCR. Each pair of oligonucleotide primers is provided in Table 2 and Table 3. To design the primers and identify the gene variations, we used reference complementary cDNA sequences of ND (NM 000266.3), FZD4 (NM 012193.1), and LRP5 (NM 002335.1) obtained from GenBank (National Center for Biotechnology Information). The ND gene had three exons, and three pairs of primers were used to amplify each exon. The primer pairs for exons 1 and 2 covered the 5′ untranslated region (UTR) and coding region, and the primer pair for exon 3 amplified the coding region. An FZD4 gene with two coding regions was amplified with seven primers (Table 2). Primers 1A forward and 1B reverse were used to detect exon 1, including the 5′ UTR and coding region. The internal primer BF was used to detect the coding region. Exon 2 had a long coding region so it was amplified in two overlapping segments, A and B. LRP5 consisted of 23 exons and encoded 1,615 amino acids. Each exon was amplified with the primer pairs listed in Table 3. The primer pair for exon 1 amplified the coding region and part of the 5′ UTR. Two pairs were used to detect exon 23 including the 3′ UTR.
Table 2. Primer sequence for ND and FZD4 gene amplification.
| Name | Primer sequence (5′> 3′) | Product size (bp) |
|---|---|---|
|
NDP-1 |
F: CGCCTGATTGATATATGACTGCAATGGC |
322 |
| R: GCTCGGTTTGGAAAGAAGCGATTTCCT | ||
|
NDP-2 |
F: TTCTGGGTAAATAATTCTGGGG |
471 |
| R: GTTTCTGAGGGAAATGCTCTCCTCACA | ||
|
NDP-3 |
F: TAAGGTTGTGGCATGCCCACAGAGTAA |
690 |
| R: CAGAAGATGTCCCAGGAAAAGCTGGGCTTT | ||
|
FZD4-1A |
F: ATAATTTTAGCGCCGCGAGCCTCCAG |
771 |
| R: GAAATCACTTTTCCAGGAGAGCTGTCTCC | ||
|
FZD4-1B |
F: CAAACTGGGGGTGTCTGCCAGAGCA |
475 |
| R: GAAATCACTTTTCCAGGAGAGCTGTCTCC | ||
|
FZD4-2A |
F: GGGAGCATTTGGTCAAACTTCCAAGTC |
757 |
| R: GAGTGTCAGAATAACCCACCAAATGGAGCT | ||
| FZD4-2B | F: CCTGTTCTCATCCAAGAAGGACTTAAGAA |
781 |
| R: TTCAAAATGAAGAAAGCATGGAGGCTGACT |
Table 3. Primer Sequence for LRP5 gene amplification.
| Name | Forward sequence (5′→3′) | Reverse Sequence (5′→3′) | Product size (bp) |
|---|---|---|---|
| Exon 1 |
TCCTCCCCGTCGTCCTG |
ATTGTCCGAGCAACCCG |
269 |
| Exon 2 |
CTTAGCCAGTGGCCCTCA |
AGAGAGAGATGGTGACACT |
496 |
| Exon 3 |
TCTGTGTTAGCTGCTTCTCTT |
CCAGGACTGCGTGGGTA |
259 |
| Exon 4** |
GATGGCTCCTCCACCCCGCT |
GCGCCCCAGCCGGCACT |
250 |
| Exon 5** |
CTCATTCAGAAACAAGTGACGGTCCTC |
GTCCCGTCCCACCGCCT |
216 |
| Exon 6 |
CTGCTGCAGGCCCTTGA |
TCTCCCTCTCGCCTGTG |
506 |
| Exon 7 |
GTCATGGACTTCTGCTTCTT |
TGGCCTCCTGGATCAAAC |
235 |
| Exon 8 |
TGGCCCATCCAGACCTAT |
CAAGTCTGCATGGCTGAG |
298 |
| Exon 9 |
GCATTCATTGTGTGGCTTG |
AAGCCTTTGAGGCAGGA |
446 |
| Exon 10 |
CTTTTCCTCCTCACCTGCT |
GGTGAACACAAGGACGC |
289 |
| Exon 11 |
AGACTCACTGAGCCTGC |
GCCCTCCATGACCAGAAG |
261 |
| Exon 12 |
CCTTTGCTGACACCGTG |
GAAGCTCCTTTCAGCGT |
440 |
| Exon 13 |
CCTGCAGCCCTGTCTTT |
GCCTTGGGAAGCACACC |
271 |
| Exon 14 |
CTCAGGAGTCTTGGTTTCTTT |
GCATTCGGCAGAAGACAC |
308 |
| Exon 15 |
CCCACACCCGTCCTTCA |
GGGTGTCTGCGGTTAGG |
263 |
| Exon 16 |
GAGGTCAGCACTGCTCA |
GGTCGGGTTTAGAGGCCA |
301 |
| Exon 17 |
AGAGCCTGACCTCTGTTT |
TACCTGTCCATCACCCCAA |
206 |
| Exon 18 |
GGCTGCGTGTGATGTTC |
GGTCTTGGCAGAGCCTTGA |
307 |
| Exon 19 |
AGCCTCTCTGAGTGCAT |
TAAACTCCACGTTCCTGGG |
208 |
| Exon 20 |
GGCCACCTCTTTCTGTTT |
AGATCATTCCATATCTCAGGCTC |
294 |
| Exon 21** |
GAGTCTCGTGGGTAGTGGGA |
AGAAAGCAAGGCATGCCTCAGAG |
245 |
| Exon 22 |
CTGGCGAGGCTCTAAGT |
CCCAATGGCCATGGAGG |
174 |
| Exon 23A* |
CTCCTCTGTGTGTGTCCC |
TCAGGATGAGTCCGTGC |
286 |
| Exon 23B* | CTACTTCCATCTCTTCCCGC | CGAAAGAATGGCAGTTCTGTT | 271 |
Primer sequences obtained from Gong et al. [16] ** Exon 23 was amplified in two overlapping segments, A and B.
PCR was performed with LA Taq polymerase (Takara Bio, Otsu, Japan) according to the manufacturer’s recommended protocol. For ND and the A set of FZD4, 10X LA PCR™ buffer II (Takara Bio) was used. GC buffer I (Takara Bio) was used for the other primer pairs. The amplification conditions for ND and FZD4 involved 30 cycles of denaturation at 98 °C for 30 s, annealing at 58 °C for 30 s, and extension at 72 °C for 1 min. For LRP5, amplification was performed using 30 cycles of denaturation for 30 s at 98 °C, followed by 30 s at 61 °C. The PCR products were purified with a PCR clean-up kit (Macherey-Nagel, Düren, Germany) and sequenced with BigDyeTM Terminator version 3.1 (Applied Biosystems, Foster city, CA) on an ABI PRISM® 310 Genetic Analyzer (Applied Biosystems).
Results
Of the 17 infants, 12 were boys and five were girls. Birthweights ranged from 458 to 1,212 g with a mean (±standard deviation [SD]) of 732±221 g; gestational ages ranged from 23 to 29 weeks with a mean (±SD) of 24.3±1.9 weeks. Of the cases of ROP in the patients' 34 eyes, four were classified as ROP stage 3, 13 as stage 4A, one as stage 4B, and 16 as stage 5. Fifteen of the patients showed aggressive posterior (AP) ROP, and the others had classical ROP. Fourteen had undergone surgery on both eyes, and the other three on one eye. Vitrectomy had been performed on 16 eyes and buckling on one eye (Table 1).
Sequencing of three genes was performed. On ND gene screening, one patient (#10) showed a heterozygous A>G mutation in the 5′ UTR of exon 1 at position 237. This mutation was not found in 51 unaffected Japanese subjects. In one patient (#15) LRP5 gene analysis revealed a heterozygous 3-bp insertion within the CTG repeat region of exon 1, which introduced another leucine insertion in the polyleucine region within the signal peptide (Figure 1). This insertion was confirmed with antisense sequence (data not shown). This insertion was not found in 28 unaffected Japanese subjects. No FZD4 mutations were found in any of the patients.
Figure 1.
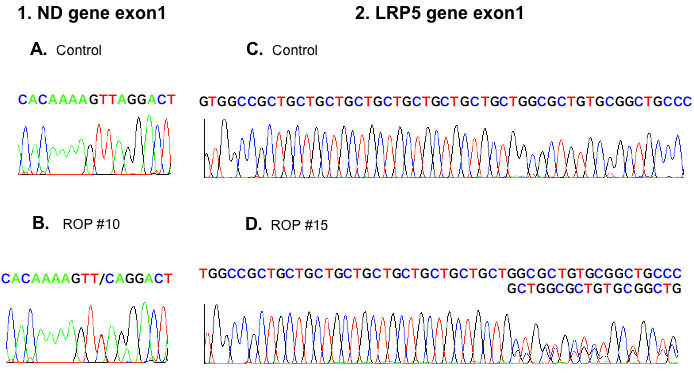
Sequence chromatograms of mutations in retinopathy of prematurity. Left: The chromatogram of ND gene exon 1. It shows the antisense sequence between nucleotide 231 and 245. Retinopathy of prematurity (ROP) #10 shows a heterozygous mutation at 237 T->C. Right: The chromatogram of LRP5 gene exon 1. C, D indicate a sense sequence including CTG repeat regions. The text below each normal chromatogram represents a mutated sequence. ROP #15 shows a heterozygous 3-bp insertion (CTG) that introduced another leucine insertion in the polyleucine region. A, C indicate a sequence of an unaffected individual. B indicates a sequence of ROP #10. D indicates sequence of ROP #15.
Discussion
A genetic involvement in advanced ROP has been suspected for a decade. In the present study we performed genetic screening of three molecules involved in the Wnt signaling pathway in 17 Japanese infants with advanced ROP (screening patients with uniform ethnic backgrounds is advantageous because genetic polymorphisms due to racial differences can be avoided). One patient had a point mutation in the 5′ UTR of the ND gene, and another had a 3-bp insertion resulting in one amino acid insertion in the signal peptide of the LRP5 gene. These findings suggest an association between genetic factors and the development of ROP.
The three genes chosen for investigation in this study are responsible for FEVR and are involved in retinal development through the Wnt signaling pathway. The ND gene mutation that causes XL-FEVR has been identified. Because the incidence of severe ROP is higher among males than among females, the abnormality of the X chromosome, which includes the ND gene, might be one of factors that influences the severity of ROP [27]. Several studies have demonstrated ND gene variants in ROP [20-22]. In this study, one patient showed a heterozygous single base-pair alternation in the 5′ UTR at position 237 with an A to G nucleotide change. Because the 5′ UTR is responsible for transcription regulation and translation efficiency, this mutation may have altered gene regulation. An inability to activate the Wnt receptor-β-catenin pathway results in the early abrogation of neurosensory and vascular development [14]. Several reports have described genetic variants in the 5′ and 3′ UTRs of the ND gene in patients with ROP, but most mutations have been detected within the coding region in patients with Norrie disease [28]. ROP and Norrie disease have different phenotypes and time of onset as well as speed of disease progression; while Norrie disease causes bilateral retinal malformation, mental retardation, and deafness, the main features of ROP are abnormal retinal vascular development. It has been speculated that incomplete ND proteins with amino acid changes result in malformation of the ocular tissues, whereas inappropriate gene expression affects retinal vascular conformation.
Several reports have referred to the role of the FZD4 gene in ROP [23-25]. Four missense mutations have been reported in severe ROP. Extensive screening of the 5′ UTR and coding region in our study failed to detect any genetic variants, and further investigation is required to clarify the role of the FZD4 gene in advanced ROP.
Although several mutations of LRP5 have been found in FEVR, this study is, to our knowledge, the first to perform genetic screening in ROP. We screened the entire coding region and 3′ UTR of LRP5. Due to the presence of a pseudogene on chromosome 22 that has high homology with the LRP5 5′ UTR, it is difficult to isolate the entire 5′ UTR with PCR [16]. We detected a heterozygous polymorphism in one patient—a 3-bp insertion at the CTG repeat region in exon 1, leading to the addition of a leucine amino acid to the polyleucine residue within the signal peptide. Therefore, the genetic variant in our case with the insertion of a single leucine in the signal peptide might lead to insufficient translocation during protein processing and affected retinal development. In functional assays for the LRP5 signal peptide variants, one leucine insertion in the polyleucine residue impairs Norrin signal transduction [29]. The same group Chung et al. [29] has identified that approximately 10% of German and Turkish unaffected subjects have a heterozygous allele with 10 leucines in the LRP5 signal peptide residue. In contrast, none of the 28 unaffected subjects had this insertion in our study. This may be due to ethnic background differences. PCSK9, a gene implicated in cholesterol metabolism, is known to have polymorphism within signal peptide polyleucine stretches among normal subjects. The heterozygous carriers of a 10-leucine allele had lower low-density lipoprotein cholesterol concentration compared to homozygous carriers of a 9-leucine allele [30]. Taken together, we speculate that the genetic change in our case may have influenced retinal development under premature circumstances.
In conclusion, through analysis of genes involving the Wnt receptor signal pathway, we have identified two genetic variants in advanced ROP. Our results suggest that abnormality in the Wnt signaling pathway during retinal development may associate to severe ROP. Although the mutation and polymorphism are implicated in a small number of cases, the risk factors for advanced ROP might be polygenetic. Additionally, the comparison of genetic change between severe and mild cases can help clarify the etiology of ROP; however, a mild case sample was not available to us. Unfortunately, we are unable to determine whether these genetic changes are de novo or inherited because the parents declined to be tested.
Extensive genetic analysis with an increased sample number and genes, including TSPAN12, should lead to a better understanding of the pathogenesis of ROP.
Acknowledgments
We are grateful to the patients and their families who graciously agreed to participate in this study. Also thanks to our clinical fellows, Dr. Reiko Tsukada and Dr. Yukiko Ito, and Ms. Tamae Tanji and Ms. Fumiko Kato for their technical support. This work was supported by grant from Japan Scientific Research. (Grant number: 18591938)
References
- 1.Seiberth V, Linderkamp O. Risk factors in retinopathy of prematurity. A multivariate statistical analysis. Ophthalmologica. 2000;214:131–5. doi: 10.1159/000027482. [DOI] [PubMed] [Google Scholar]
- 2.Saunders RA, Donahue ML, Christmann LM, Pakalnis AV, Tung B, Hardy RJ, Phelps DL. Racial variation in retinopathy of prematurity. The Cryotherapy for Retinopathy of Prematurity Cooperative Group. Arch Ophthalmol. 1997;115:604–8. doi: 10.1001/archopht.1997.01100150606005. [DOI] [PubMed] [Google Scholar]
- 3.Holmström G, van Wijngaarden P, Coster DJ, Williams KA. Genetic susceptibility to retinopathy of prematurity: the evidence from clinical and experimental animal studies. Br J Ophthalmol. 2007;91:1704–8. doi: 10.1136/bjo.2007.117283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gow J, Oliver GL. Familial exudative vitreoretinopathy. An expanded view. Arch Ophthalmol. 1971;86:150–5. doi: 10.1001/archopht.1971.01000010152007. [DOI] [PubMed] [Google Scholar]
- 5.Shastry BS, Trese MT. Familial exudative vitreoretinopathy: further evidence for genetic heterogeneity. Am J Med Genet. 1997;69:217–8. doi: 10.1002/(sici)1096-8628(19970317)69:2<217::aid-ajmg19>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 6.Plager DA, Orgel IK, Ellis FD, Hartzer M, Trese MT, Shastry BS. X-linked recessive familial exudative vitreoretinopathy. Am J Ophthalmol. 1992;114:145–8. doi: 10.1016/s0002-9394(14)73977-7. [DOI] [PubMed] [Google Scholar]
- 7.Chen ZY, Battinelli EM, Fielder A, Bundey S, Sims K, Breakefield XO, Craig IW. A mutation in the Norrie disease gene (NDP) associated with X-linked familial exudative vitreoretinopathy. Nat Genet. 1993;5:180–3. doi: 10.1038/ng1093-180. [DOI] [PubMed] [Google Scholar]
- 8.Shastry BS, Hejtmancik JF, Plager DA, Hartzer MK, Trese MT. Linkage and candidate gene analysis of X-linked familial exudative vitreoretinopathy. Genomics. 1995;27:341–4. doi: 10.1006/geno.1995.1052. [DOI] [PubMed] [Google Scholar]
- 9.Shastry BS, Hejtmancik JF, Trese MT. Identification of novel missense mutations in the Norrie disease gene associated with one X-linked and four sporadic cases of familial exudative vitreoretinopathy. Hum Mutat. 1997;9:396–401. doi: 10.1002/(SICI)1098-1004(1997)9:5<396::AID-HUMU3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Robitaille J, MacDonald ML, Kaykas A, Sheldahl LC, Zeisler J, Dubé MP, Zhang LH, Singaraja RR, Guernsey DL, Zheng B, Siebert LF, Hoskin-Mott A, Trese MT, Pimstone SN, Shastry BS, Moon RT, Hayden MR, Goldberg YP, Samuels ME. Mutant frizzled-4 disrupts retinal angiogenesis in familial exudative vitreoretinopathy. Nat Genet. 2002;32:326–30. doi: 10.1038/ng957. [DOI] [PubMed] [Google Scholar]
- 11.Toomes C, Bottomley HM, Jackson RM, Towns KV, Scott S, Mackey DA, Craig JE, Jiang L, Yang Z, Trembath R, Woodruff G, Gregory-Evans CY, Gregory-Evans K, Parker MJ, Black GC, Downey LM, Zhang K, Inglehearn CF. Mutations in LRP5 or FZD4 underlie the common familial exudative vitreoretinopathy locus on chromosome 11q. Am J Hum Genet. 2004;74:721–30. doi: 10.1086/383202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jiao X, Ventruto V, Trese MT, Shastry BS, Hejtmancik JF. Autosomal recessive familial exudative vitreoretinopathy is associated with mutations in LRP5. Am J Hum Genet. 2004;75:878–84. doi: 10.1086/425080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–95. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- 14.Rehm HL, Zhang DS, Brown MC, Burgess B, Halpin C, Berger W, Morton CC, Corey DP, Chen ZY. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J Neurosci. 2002;22:4286–92. doi: 10.1523/JNEUROSCI.22-11-04286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–8. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- 16.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marcelino J, Suwairi W, Heeger S, Sabatakos G, Apte S, Adkins WN, Allgrove J, Arslan-Kirchner M, Batch JA, Beighton P, Black GC, Boles RG, Boon LM, Borrone C, Brunner HG, Carle GF, Dallapiccola B, De Paepe A, Floege B, Halfhide ML, Hall B, Hennekam RC, Hirose T, Jans A, Jüppner H, Kim CA, Keppler-Noreuil K, Kohlschuetter A, LaCombe D, Lambert M, Lemyre E, Letteboer T, Peltonen L, Ramesar RS, Romanengo M, Somer H, Steichen-Gersdorf E, Steinmann B, Sullivan B, Superti-Furga A, Swoboda W, van den Boogaard MJ, Van Hul W, Vikkula M, Votruba M, Zabel B, Garcia T, Baron R, Olsen BR, Warman ML, Osteoporosis-Pseudoglioma Syndrome Collaborative Group. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–23. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 17.Nikopoulos K, Gilissen C, Hoischen A, van Nouhuys CE, Boonstra FN, Blokland EA, Arts P, Wieskamp N, Strom TM, Ayuso C, Tilanus MA, Bouwhuis S, Mukhopadhyay A, Scheffer H, Hoefsloot LH, Veltman JA, Cremers FP, Collin RW. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:240–7. doi: 10.1016/j.ajhg.2009.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poulter JA, Ali M, Gilmour DF, Rice A, Kondo H, Hayashi K, Mackey DA, Kearns LS, Ruddle JB, Craig JE, Pierce EA, Downey LM, Mohamed MD, Markham AF, Inglehearn CF, Toomes C. Mutations in TSPAN12 cause autosomal-dominant familial exudative vitreoretinopathy. Am J Hum Genet. 2010;86:248–53. doi: 10.1016/j.ajhg.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- 20.Haider MZ, Devarajan LV, Al-Essa M, Kumar HA. C597→A polymorphism in the Norrie disease gene is associated with advanced retinopathy of prematurity in premature Kuwaiti infants. J Biomed Sci. 2002;9:365–70. doi: 10.1007/BF02256593. [DOI] [PubMed] [Google Scholar]
- 21.Hiraoka M, Berinstein DM, Trese MT, Shastry BS. Insertion and deletion mutations in the dinucleotide repeat region of the Norrie disease gene in patients with advanced retinopathy of prematurity. J Hum Genet. 2001;46:178–81. doi: 10.1007/s100380170085. [DOI] [PubMed] [Google Scholar]
- 22.Shastry BS, Pendergast SD, Hartzer MK, Liu X, Trese MT. Identification of missense mutations in the Norrie disease gene associated with advanced retinopathy of prematurity. Arch Ophthalmol. 1997;115:651–5. doi: 10.1001/archopht.1997.01100150653015. [DOI] [PubMed] [Google Scholar]
- 23.MacDonald ML, Goldberg YP, Macfarlane J, Samuels ME, Trese MT, Shastry BS. Genetic variants of frizzled-4 gene in familial exudative vitreoretinopathy and advanced retinopathy of prematurity. Clin Genet. 2005;67:363–6. doi: 10.1111/j.1399-0004.2005.00408.x. [DOI] [PubMed] [Google Scholar]
- 24.Ells A, Guernsey DL, Wallace K, Zheng B, Vincer M, Allen A, Ingram A, DaSilva O, Siebert L, Sheidow T, Beis J, Robitaille JM. Severe retinopathy of prematurity associated with FZD4 mutations. Ophthalmic Genet. 2010;31:37–43. doi: 10.3109/13816810903479834. [DOI] [PubMed] [Google Scholar]
- 25.Drenser KA, Dailey W, Vinekar A, Dalal K, Capone A, Jr, Trese MT. Clinical presentation and genetic correlation of patients with mutations affecting the FZD4 gene. Arch Ophthalmol. 2009;127:1649–54. doi: 10.1001/archophthalmol.2009.322. [DOI] [PubMed] [Google Scholar]
- 26.International Committee for the Classification of Retinopathy of Prematurity The International Classification of Retinopathy of Prematurity revisited. Arch Ophthalmol. 2005;123:991–9. doi: 10.1001/archopht.123.7.991. Review. [DOI] [PubMed] [Google Scholar]
- 27.Nødgaard H, Andreasen H, Hansen H, Sørensen HT. Risk factors associated with retinopathy of prematurity (ROP) in northern Jutland, Denmark 1990–1993. Acta Ophthalmol Scand. 1996;74:306–10. doi: 10.1111/j.1600-0420.1996.tb00098.x. [DOI] [PubMed] [Google Scholar]
- 28.Wu WC, Drenser K, Trese M, Capone A, Jr, Dailey W. Retinal phenotype-genotype correlation of pediatric patients expressing mutations in the Norrie disease gene. Arch Ophthalmol. 2007;125:225–30. doi: 10.1001/archopht.125.2.225. [DOI] [PubMed] [Google Scholar]
- 29.Chung BD, Kayserili H, Ai M, Freudenberg J, Uzümcü A, Uyguner O, Bartels CF, Höning S, Ramirez A, Hanisch FG, Nürnberg G, Nürnberg P, Warman ML, Wollnik B, Kubisch C, Netzer C. A mutation in the signal sequence of LRP5 in a family with an osteoporosis-pseudoglioma syndrome (OPPG)-like phenotype indicates a novel disease mechanism for trinucleotide repeats. Hum Mutat. 2009;30:641–8. doi: 10.1002/humu.20916. [DOI] [PubMed] [Google Scholar]
- 30.Yue P, Averna M, Lin X, Schonfeld G. The c.43_44insCTG variation in PCSK9 is associated with low plasma LDL-cholesterol in a Caucasian population. Hum Mutat. 2006;27:460–6. doi: 10.1002/humu.20316. [DOI] [PubMed] [Google Scholar]