

Abstract
The catalytic potential for H2S biogenesis and homocysteine clearance converge at the active site of cystathionine β-synthase (CBS), a pyridoxal phosphate-dependent enzyme. CBS catalyzes β-replacement reactions of either serine or cysteine by homocysteine to give cystathionine and water or H2S, respectively. In this study, high-resolution structures of the full-length enzyme from Drosophila in which a carbanion (1.70 Å) and an aminoacrylate intermediate (1.55 Å) have been captured are reported. Electrostatic stabilization of the zwitterionic carbanion intermediate is afforded by the close positioning of an active site lysine residue that is initially used for Schiff base formation in the internal aldimine and later as a general base. Additional stabilizing interactions between active site residues and the catalytic intermediates are observed. Furthermore, the structure of the regulatory “energy-sensing” CBS domains, named after this protein, suggests a mechanism for allosteric activation by S-adenosylmethionine.
Keywords: crystallography, hydrogen sulfide
Hydrogen sulfide (H2S), the most recently discovered gasotransmitter (1, 2), is generated by two enzymes in the transsulfuration pathway, cystathionine β-synthase (CBS) and γ-cystathionase, via multiple reactions (3). These enzymes play a key role in homocysteine homeostasis, and mutations in CBS are the single most common cause of hereditary hyperhomocysteinemia (4). Cysteine, the product of the transsulfuration pathway, is the limiting reagent for glutathione biosynthesis and ∼50% of the cysteine in the hepatic glutathione pool is derived via this route (5). Flux through the transsulfuration pathway is governed in part by the allosteric effector S-adenosylmethionine (AdoMet), which binds to the C-terminal regulatory domain of human CBS (hCBS) (6). The latter contains a tandem repeat of “CBS domains” (CBSDs), a secondary structure motif named after this protein and found across all domains of life (7). CBSDs are postulated to function as energy-sensing modules (6).
The catalytic core of CBS is comprised of an N-terminal heme-binding module followed by a pyridoxal phosphate (PLP) domain, which houses the active site. The role of the heme in CBS is enigmatic, and both structural (8) and regulatory (9) functions have been ascribed to it. Changes in the heme coordination environment are transmitted to the PLP site some 20 Å away and lead to inhibition of CBS activity via a shift from the ketoenamine to the enolimine tautomer of the PLP (10). Although the crystal structure of the catalytic core of hCBS is known (11, 12), the architecture of the full-length protein is not. In this study, we report the structures of full-length Drosophila CBS (dCBS) in the substrate-free form (at 1.80-Å resolution) and in which two reaction intermediates, a carbanion (1.70 Å) and an aminoacrylate species (1.55 Å), have been captured (Table 1). The structures reveal how the protein stabilizes the reactive intermediates via coloumbic interactions with active site residues. Furthermore, our structures reveal the juxtaposition of the regulatory CBSDs and the catalytic core in an activated conformation of the protein, providing insights into the mechanism of allosteric regulation by AdoMet.
Table 1.
Data collection and refinement statistics (molecular replacement)
| Protein | dCBS | dCBS + carbanion | dCBS + aminoacrylate |
| Data collection | |||
| Space group | C2221 | C2221 | C2221 |
| Cell dimensions | |||
| a, b, c, Å | 92.9, 138.2, 75.2 | 92.9, 137.9, 75.1 | 93.2, 138.8, 75.1 |
| α, β, γ, ° | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 50-1.80 (1.86-1.80) | 50-1.70 (1.76-1.70) | 50-1.55 (1.61-1.55) |
| Rsym (%) | 8.0 (40.3) | 6.3 (48.2) | 10.0 (54.6) |
| I/σI | 19.3 (4.0) | 16.4 (3.6) | 15.4 (2.1) |
| Completeness (%) | 95.9 (72.2) | 98.9 (98.1) | 93.8 (67.1) |
| Redundancy | 8.0 (5.8) | 5.4 (5.3) | 6.6 (3.4) |
| Refinement | |||
| Resolution, Å | 50-1.80 | 50-1.70 | 50-1.55 |
| No. reflections | 42,962 | 52,465 | 66,337 |
| Rwork/Rfree | 0.159/0.192 | 0.179/0.203 | 0.148/0.188 |
| No. atoms | |||
| Protein | 3,843 | 3,868 | 3,899 |
| Ligands | 58 | 65 | 64 |
| Water | 471 | 598 | 712 |
| B factors | |||
| Protein | 21.4 | 21.8 | 15.2 |
| Heme | 17.7 | 20.1 | 13.4 |
| PLP | 11.5 | 18.5 | 11.7 |
| Serine/aminoacrylate | — | 22.5 (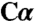 ; 22,8) ; 22,8) |
17.5 |
| Water | 30.1 | 32.5 | 32.6 |
| rmsd | |||
| Bond lengths, Å | 0.004 | 0.011 | 0.008 |
| Bond angles, ° | 0.847 | 1.277 | 1.179 |
| Protein Data Bank code | 2PC2 | 2PC4 | 2PC3 |
Results
Structure of Full-Length dCBS.
The dCBS monomer (residues 7–510) is composed of two modules connected by a long linker (Fig. 1 A and B): The N-terminal module contains the heme- (red) and PLP- (blue) binding domains, and the C-terminal module contains a pair of CBSDs (green). Whereas the heme domain (residues 1–45) lacks secondary structure, the PLP domain (residues 46–350) exhibits a previously described fold (13) containing two subdomains (A and B) that bind PLP at the interface. The N-terminal module of dCBS is very similar to the corresponding module of hCBS (11, 12) (Fig S1). The long linker (αL, residues 351–377) sandwiched between the N- and C-terminal modules includes a helix and contacts residues from both modules. There are slight differences in the secondary structures of the individual CBSDs; although both have the characteristic β-α-β-β-α fold, CBSD1 exhibits a β-α-β-β-α-α-β fold whereas CBSD2 has an α-β-α-β-β-α fold (Fig. 1). The CBSDs do not contact the catalytic core except via the linker (Fig. 1B). Instead, the relative orientation of the N- and C-terminal modules is dictated by the formation of the tight dimer described below.
Fig. 1.

Structure of full-length dCBS. (A) Secondary structure and sequence of residues observed in the dCBS structure (9–506). (B) Two views of the dCBS monomer; N-terminal heme-binding domain (red), PLP domain (blue), C-terminal CBSDs (green), and linker αL (pink). (C) Two views of the dCBS dimer. The heme and PLP cofactors are shown in ball-and-stick representation. Monomers 1 and 2 are shown in dark and light shades, respectively.
Drosophila CBS is a symmetric dimer in the crystal (Fig. 1C), consistent with the solution behavior of the enzyme. In contrast, full-length hCBS exists in multiple oligomeric states ranging from dimers to 16-mers (14, 15). The dCBS monomers have a large dimer interface (∼3,369 Å2) (16) with extensive contributions from both modules (N-terminal ∼1,927 Å2; C-terminal ∼1,442 Å2). The subunit interface is mainly hydrophobic with only 11 hydrogen bonds (seven and four for the N- and C-terminal modules, respectively) and no salt bridges between the two monomers. The linker is also involved in dimerization. In the C-terminal module, hydrogen bonds are formed from H372 and H374 in the linker to the backbone carbonyls of S439* and V438* in the partner subunit (CBSD1*) (Fig. S2). The CBSDs dimerize in a head-to-tail fashion (Figs. 1 and 2), and a pair of four-helix bundles forms the interface (αCBSD11 and αCBSD12 interact with αCBSD22∗ and αCBSD21∗, and αCBSD11∗ and αCBSD12∗ with αCBSD22 and αCBSD21).
Fig. 2.

Structure of the regulatory domain and location of human pathogenic mutations. (A) Two views of the C-terminal CBSD dimer of CBSD1/CBSD2 with CBSD1*/CBSD2*. The dimer is formed in a head-to-tail fashion (CBSD2 interacts with CBSD1*, and CBSD1 interacts with CBSD2*). The two putative AdoMet-binding sites (A and B) as well as their symmetrically equivalent (A* and B*) sites are indicated with arrows. (B) Map of pathogenic mutations in the CBSDs of dCBS. The mutations with no known phenotype are displayed in yellow (dCBS numbering: P389L, A402N, K407Q, L425P, N460C, G487K, A495D, V500S; hCBS numbering: P422L, T434N, R439Q, L456P, R491C, Q526K, V534D, L539S), and activating mutations are indicated with red (dCBS numbering: I403T, V399, D412N, T435L; hCBS numbering I435T, C431, D444N, S466L).
Stabilization of Reactive Intermediates.
In addition to characterizing the apoenzyme, we also crystallized full-length dCBS with each of two substrates, serine and cysteine (Table 1). Substrate binding induces a general collapse of the active site pocket and positions the active site S116 residue in proximity of the substrate. Conformational differences are restricted to the PLP subdomain B, with only small differences in the overall structure (rmsd of 0.73 Å for all 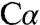 atoms). The most significant change is a shift toward the PLP cofactor of loop LB, which includes S116 and the adjacent β strands (βB2 and βB3) (Fig. S3).
atoms). The most significant change is a shift toward the PLP cofactor of loop LB, which includes S116 and the adjacent β strands (βB2 and βB3) (Fig. S3).
In the CBS-catalyzed reaction, the first substrate (serine or cysteine) binds the PLP cofactor and displaces an active site lysine (K88 in dCBS) to form an external aldimine (Fig. 3). Subsequent deprotonation of the substrate 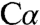 -H results in a carbanion intermediate stabilized both by the protonated Schiff base and via delocalization through the extended π conjugation system of the pyridine ring. We have successfully captured two reaction intermediates in full-length dCBS: a carbanion (intermediate 1) and an aminoacrylate species (intermediate 2). At neutral pH (7.0), intermediate 1 is observed, and electron density for the βOH group is clearly present and reveals how the
-H results in a carbanion intermediate stabilized both by the protonated Schiff base and via delocalization through the extended π conjugation system of the pyridine ring. We have successfully captured two reaction intermediates in full-length dCBS: a carbanion (intermediate 1) and an aminoacrylate species (intermediate 2). At neutral pH (7.0), intermediate 1 is observed, and electron density for the βOH group is clearly present and reveals how the 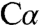 -H bond is activated and the carbanion intermediate is stabilized (Fig. 4 A and B). At lower pH (6.5), electron density for the βOH (or βSH) group is missing, and an aminoacrylate species, intermediate 2, is observed (Fig. 4 C and D).
-H bond is activated and the carbanion intermediate is stabilized (Fig. 4 A and B). At lower pH (6.5), electron density for the βOH (or βSH) group is missing, and an aminoacrylate species, intermediate 2, is observed (Fig. 4 C and D).
Fig. 3.

Scheme depicting the chemical structures of intermediates in the CBS-catalyzed reaction.
Fig. 4.

Characterization of dCBS reaction intermediates. Cross-eyed stereo electron density map (A) and ball and stick model (B) showing the active site geometry of dCBS with serine bound at 1.7-Å resolution and of dCBS with aminoacrylate bound at 1.55-Å resolution (C and D). The blue contours at 1.5σ represent electron density from a weighted 2Fobs - Fcalc map whereas the green contours at 3σ represent positive electron density from a Fobs - Fcalc composite-omit map where the K88 and substrate intermediate (carbanion in A and aminoacrylate in B) were omitted. Atom color coding is as follows: gray, carbon; blue, nitrogen; red, oxygen; orange, phosphorous. The carbon atoms of PLP and the bound substrates are displayed as black spheres. Distances in B between the 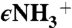 of K88 and the Ca, N atoms of bound serine, and the C4A atom of PLP are 2, 2.5, and 3.0 Å, respectively, whereas the corresponding distances in D are 3.8, 3.9, and 4.0 Å, respectively.
of K88 and the Ca, N atoms of bound serine, and the C4A atom of PLP are 2, 2.5, and 3.0 Å, respectively, whereas the corresponding distances in D are 3.8, 3.9, and 4.0 Å, respectively.
The electron density in the structure with intermediate 1 bound suggests an unprecedented geometry for the serine 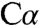 , which is close to the ϵ-amino group of K88. To obtain high-quality electron density unbiased by preliminary assumptions about the substrate structure, the bound substrate and side chain of K88 were excluded from crystallographic refinement until the remainder of the structure was complete and refined. Several alternatives for serine and K88 were then modeled and refined. This analysis is described in detail in Materials and Methods, and the results are graphically summarized in Fig. S4. Briefly, in intermediate 1, K88, which forms a Schiff base with the PLP cofactor (internal aldimine) in the absence of a substrate, is unequivocally located directly below the serine
, which is close to the ϵ-amino group of K88. To obtain high-quality electron density unbiased by preliminary assumptions about the substrate structure, the bound substrate and side chain of K88 were excluded from crystallographic refinement until the remainder of the structure was complete and refined. Several alternatives for serine and K88 were then modeled and refined. This analysis is described in detail in Materials and Methods, and the results are graphically summarized in Fig. S4. Briefly, in intermediate 1, K88, which forms a Schiff base with the PLP cofactor (internal aldimine) in the absence of a substrate, is unequivocally located directly below the serine 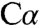 and close both to the α-imino group of the external aldimine and to C4A. The electron density map on the basis of the model lacking the bound serine and the K88 side chain (Fig. 4A and Fig. S4A) shows continuous density between the ϵ-amino group of K88 and the
and close both to the α-imino group of the external aldimine and to C4A. The electron density map on the basis of the model lacking the bound serine and the K88 side chain (Fig. 4A and Fig. S4A) shows continuous density between the ϵ-amino group of K88 and the  of serine, making unambiguous placement of atoms in the electron density difficult. In principle, the
of serine, making unambiguous placement of atoms in the electron density difficult. In principle, the  can be modeled as a trigonal planar (sp2) or tetrahedral (sp3) center, and both geometries were tested in refinement experiments.
can be modeled as a trigonal planar (sp2) or tetrahedral (sp3) center, and both geometries were tested in refinement experiments.
Two models were tested with a tetrahedral  for L-serine. One resulted in unacceptable bonding geometry at
for L-serine. One resulted in unacceptable bonding geometry at 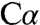 and also required a covalent bond between the ϵ-amino group of K88 and the
and also required a covalent bond between the ϵ-amino group of K88 and the 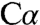 atom (∼1.4 Å), which is chemically unfeasible. The other model resulted in residual difference density at
atom (∼1.4 Å), which is chemically unfeasible. The other model resulted in residual difference density at 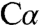 and the β-OH group (Fig. S4C). In contrast, the
and the β-OH group (Fig. S4C). In contrast, the 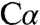 modeled with a trigonal planar geometry resulted in a good fit to density and good bonding geometry (Fig. 4A and Fig. S4B). A trigonal planar serine intermediate would have carbanionic character and is also consistent with PLP chemistry. In PLP enzymes that catalyze racemization, β-replacement, or transamination reactions, the first common step is proton abstraction. We have captured the intermediate resulting from heterolytic
modeled with a trigonal planar geometry resulted in a good fit to density and good bonding geometry (Fig. 4A and Fig. S4B). A trigonal planar serine intermediate would have carbanionic character and is also consistent with PLP chemistry. In PLP enzymes that catalyze racemization, β-replacement, or transamination reactions, the first common step is proton abstraction. We have captured the intermediate resulting from heterolytic 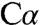 -H bond cleavage and charge separation. In this zwitterionic species, the proton is on the ϵ-amino group of K88, leaving a negative charge on the external aldimine. The
-H bond cleavage and charge separation. In this zwitterionic species, the proton is on the ϵ-amino group of K88, leaving a negative charge on the external aldimine. The  group of K88 is 2.1 Å from
group of K88 is 2.1 Å from 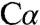 , 2.5 Å from the imino nitrogen of the Schiff base, and 3.0 Å from PLP’s C4A. The proximity of the positively charged K88 ϵ-amino group could be essential for shielding and stabilizing the resulting negative charge in carbanionic intermediate 1. Additionally, the S116 hydroxyl group also contacts the serine
, 2.5 Å from the imino nitrogen of the Schiff base, and 3.0 Å from PLP’s C4A. The proximity of the positively charged K88 ϵ-amino group could be essential for shielding and stabilizing the resulting negative charge in carbanionic intermediate 1. Additionally, the S116 hydroxyl group also contacts the serine  (3.1 Å) from the (anti)side opposite from the K88 ϵ-amino group.
(3.1 Å) from the (anti)side opposite from the K88 ϵ-amino group.
The carbanionic nature of intermediate 1 and the arrangement of the protein residues and PLP cofactor in the active site pocket provide further insight into the chemistry of the CBS reaction. Upon binding of serine, the scissile 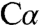 -H bond is initially orthogonal to the plane of the PLP ring and thus oriented for overlap of the nascent p orbital with the conjugated π-system of the Schiff base or pyridine ring. This stereoelectronic control strategy is deployed by PLP enzymes to selectively labilize one of the bonds to the
-H bond is initially orthogonal to the plane of the PLP ring and thus oriented for overlap of the nascent p orbital with the conjugated π-system of the Schiff base or pyridine ring. This stereoelectronic control strategy is deployed by PLP enzymes to selectively labilize one of the bonds to the  atom, as originally proposed by Dunathan (17). The structure of intermediate 1 supports a direct role for K88 in proton abstraction from
atom, as originally proposed by Dunathan (17). The structure of intermediate 1 supports a direct role for K88 in proton abstraction from 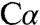 needed to activate substrate for subsequent β-elimination.
needed to activate substrate for subsequent β-elimination.
Like intermediate 1, the aminoacrylate intermediate also exhibits sp2 hybridization at 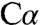 (Fig. 4 C and D). In the crystal, it was trapped under slightly more acidic conditions that could have favored protonation of the βOH leaving group. In this intermediate, the
(Fig. 4 C and D). In the crystal, it was trapped under slightly more acidic conditions that could have favored protonation of the βOH leaving group. In this intermediate, the  group of K88 is distant from
group of K88 is distant from  and interacts instead with two oxygen atoms of the PLP phosphate (at 2.9 and 3.5 Å). In intermediate 2, Cβ is poised for nucleophilic attack by the thiolate of homocysteine. The presence of this aminoacrylate intermediate (λmax = 460 nm), which formed transiently, is supported by stopped-flow studies (Fig. 5). No other intermediate was observed when the stopped-flow studies were performed at a higher pH (8.5), indicating that our crystallization of two intermediates at slightly different pH values was fortuitous. Spectroscopic detection of the aminoacrylate species in heme-containing dCBS was accomplished by use of difference electronic absorption spectroscopy as described in Materials and Methods. Although reaction intermediates have been characterized in the non-heme CBS from yeast (18), the presence of the heme obscures the electronic spectrum of the PLP cofactor, making enzyme-monitored kinetic analyses more challenging (19, 20). Conversion of intermediate 1 to the aminoacrylate species in intermediate 2 could be facilitated by activation of the βOH leaving group of substrate by Y277 and S116.
and interacts instead with two oxygen atoms of the PLP phosphate (at 2.9 and 3.5 Å). In intermediate 2, Cβ is poised for nucleophilic attack by the thiolate of homocysteine. The presence of this aminoacrylate intermediate (λmax = 460 nm), which formed transiently, is supported by stopped-flow studies (Fig. 5). No other intermediate was observed when the stopped-flow studies were performed at a higher pH (8.5), indicating that our crystallization of two intermediates at slightly different pH values was fortuitous. Spectroscopic detection of the aminoacrylate species in heme-containing dCBS was accomplished by use of difference electronic absorption spectroscopy as described in Materials and Methods. Although reaction intermediates have been characterized in the non-heme CBS from yeast (18), the presence of the heme obscures the electronic spectrum of the PLP cofactor, making enzyme-monitored kinetic analyses more challenging (19, 20). Conversion of intermediate 1 to the aminoacrylate species in intermediate 2 could be facilitated by activation of the βOH leaving group of substrate by Y277 and S116.
Fig. 5.

Spectroscopic evidence for formation of an aminoacrylate intermediate. Difference spectra (dCBS•Ser-dCBS) obtained by stopped-flow spectroscopy reveal the presence of the 460-nm absorbing aminoacrylate intermediate.
Mechanistic Implications of Allosteric Regulation via the CBSDs.
The structure of full-length dCBS provides a structural context for understanding hCBS regulation by AdoMet. Although dCBS does not bind (and thus is not regulated by) this allosteric effector, we can nevertheless identify two potential sites for AdoMet binding in hCBS on the basis of structural alignments with other AdoMet-binding CBSD proteins, sequence differences between dCBS and hCBS (68% homology, 51% sequence identity) (Fig. S5), and the location of pathogenic mutations in hCBS that impair AdoMet regulation. These comparisons suggest two possible binding sites for AdoMet at the β-sheet-lined cleft between CBSDs 1 and 2 (Fig. 2A). The head-to-tail arrangement of the two CBSDs brings two symmetrically equivalent AdoMet-binding sites (sites A and B) face to face. This apposition precludes simultaneous binding of two AdoMet molecules in an extended conformation because of steric clashes but allows binding in a bent conformation (Fig. S6) as in an AdoMet-binding CBSD protein from Methanococcus jannaschii (21). Of the two sites, AdoMet is more likely to bind to site A, which is proximal to D412 (dCBS numbering), the location of a patient mutation (D444N). This mutation impairs AdoMet binding, increasing Kact for AdoMet ∼100-fold, but increases basal activity, suggesting that the mutation “locks” hCBS in an activated conformation simulating the AdoMet-bound state (22) (Fig. 2B). Moreover, sequence homology between dCBS and hCBS is lower at site A than B, providing further support for the assignment of site A as the locus of AdoMet binding in hCBS. Unlike hCBS, which is activated ∼2-fold by AdoMet, the basal activity of dCBS is very high, suggesting that it is locked in an activated state. We propose that AdoMet binding to hCBS induces the activated conformation by increasing the CBSD dimerization interface (Fig. S7). Furthermore, we speculate that the activated and therefore AdoMet-insensitive state of dCBS reflects a greater demand on transsulfuration flux for provision of cysteine and is an adaptation to the amino acid-poor diet of the fly.
Discussion
The first structure of an “activated” full-length CBS presented herein reveals the relative orientations of the regulatory and catalytic modules, suggests the basis of allosteric regulation of hCBS, and rationalizes its loss by patient mutations. Furthermore, the elusive carbanion species, a common first chemical intermediate in the vast majority of PLP enzymes, was captured. The dual use of K88 as a general base for  proton abstraction and for stabilization of the resulting PLP-bound carbanion intermediate was also revealed. The second reactive intermediate captured in the dCBS structure, the aminoacrylate species, has been previously observed in the structures of other PLP enzymes (23–26).
proton abstraction and for stabilization of the resulting PLP-bound carbanion intermediate was also revealed. The second reactive intermediate captured in the dCBS structure, the aminoacrylate species, has been previously observed in the structures of other PLP enzymes (23–26).
There are three resonance forms of the carbanion intermediate 1 (Fig. 3) in which the negative charge is formally at 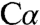 or C4A of PLP with ylide-type electrostatic stabilization provided by the Schiff base or delocalized through the extended conjugated ring system of PLP to give the quinonoid species. Computational studies suggest that the Schiff base could play a more important role in carbanion stabilization than the pyridine ring (27, 28). The resonance form of the carbanion intermediate in which the negative charge is localized on
or C4A of PLP with ylide-type electrostatic stabilization provided by the Schiff base or delocalized through the extended conjugated ring system of PLP to give the quinonoid species. Computational studies suggest that the Schiff base could play a more important role in carbanion stabilization than the pyridine ring (27, 28). The resonance form of the carbanion intermediate in which the negative charge is localized on 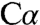 would be stabilized via electrostatic interactions with the
would be stabilized via electrostatic interactions with the 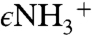 group of K88. The placement of Q191, Y277, and S318 in the catalytic chamber could provide additional stabilization, and, structurally, the short distance between
group of K88. The placement of Q191, Y277, and S318 in the catalytic chamber could provide additional stabilization, and, structurally, the short distance between 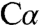 and the ϵNH2 group is consistent with the presence and stabilization of this resonance form. The placement of S318 (solution pKa of ≥15) near the pyridine nitrogen (solution pKa of ∼5) raises questions about the protonation state of the ring nitrogen and its ability to stabilize a quinonoid species, which is not observed by stopped-flow spectroscopy. In tryptophan synthase, a member of the β-replacement subfamily, which also has a serine residue proximal to the pyridine nitrogen, a quinonoid intermediate is observed but only under alkaline conditions (pH > 9) or in the presence of metals such as CsCl (29). The presence of serine contrasts with the presence of a carboxylate side chain in the transaminases, which enhances the “electron sink” character of the pyridine ring and favors formation of the quinonoid intermediate (30).
and the ϵNH2 group is consistent with the presence and stabilization of this resonance form. The placement of S318 (solution pKa of ≥15) near the pyridine nitrogen (solution pKa of ∼5) raises questions about the protonation state of the ring nitrogen and its ability to stabilize a quinonoid species, which is not observed by stopped-flow spectroscopy. In tryptophan synthase, a member of the β-replacement subfamily, which also has a serine residue proximal to the pyridine nitrogen, a quinonoid intermediate is observed but only under alkaline conditions (pH > 9) or in the presence of metals such as CsCl (29). The presence of serine contrasts with the presence of a carboxylate side chain in the transaminases, which enhances the “electron sink” character of the pyridine ring and favors formation of the quinonoid intermediate (30).
Although charge separation following 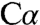 deprotonation has been postulated as a necessary first step in the catalytic mechanism of many PLP enzymes (31), to our knowledge, structural evidence for the resulting carbanion species has eluded characterization. A mixture of a quinonoid species and other intermediates has been reported in the 2.9-Å crystal structure of PLP-dependent serine hydroxymethyltransferase, in which retro-aldol cleavage of the
deprotonation has been postulated as a necessary first step in the catalytic mechanism of many PLP enzymes (31), to our knowledge, structural evidence for the resulting carbanion species has eluded characterization. A mixture of a quinonoid species and other intermediates has been reported in the 2.9-Å crystal structure of PLP-dependent serine hydroxymethyltransferase, in which retro-aldol cleavage of the 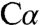 -Cβ bond rather than proton abstraction from
-Cβ bond rather than proton abstraction from 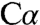 generates a quinonoid intermediate (32). Quinonoid intermediates have also been reported in the structures of tyrosine phenol lyase in the presence of substrate analogs (33) and in tryptophan synthase in the presence of a substrate mimic (34). Although the quinonoid species obtained with a substrate mimic in the tryptophan synthase structure is not directly comparable to the serine-derived intermediate in dCBS, it exhibits a similar
generates a quinonoid intermediate (32). Quinonoid intermediates have also been reported in the structures of tyrosine phenol lyase in the presence of substrate analogs (33) and in tryptophan synthase in the presence of a substrate mimic (34). Although the quinonoid species obtained with a substrate mimic in the tryptophan synthase structure is not directly comparable to the serine-derived intermediate in dCBS, it exhibits a similar 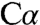 geometry. The active site lysine is 4 Å from the
geometry. The active site lysine is 4 Å from the  , and no other active site residues are in close proximity. In tyrosine phenol lyase, an aspartate group is positioned close to the pyridine nitrogen, poised to stabilize a quinonoid species, and the ϵ-amino group of the active site lysine is 3.7 Å away from
, and no other active site residues are in close proximity. In tyrosine phenol lyase, an aspartate group is positioned close to the pyridine nitrogen, poised to stabilize a quinonoid species, and the ϵ-amino group of the active site lysine is 3.7 Å away from  . The only other carbanion species that has been characterized crystallographically is the α-carbanion/enamine of thiamine diphosphate in transketolase (35). We expect the mechanism of carbanion stabilization by a multipurpose Schiff base-forming lysine to be common to other PLP enzymes that do not depend on the formation of a quinonoid intermediate.
. The only other carbanion species that has been characterized crystallographically is the α-carbanion/enamine of thiamine diphosphate in transketolase (35). We expect the mechanism of carbanion stabilization by a multipurpose Schiff base-forming lysine to be common to other PLP enzymes that do not depend on the formation of a quinonoid intermediate.
Both the canonical β-replacement reaction of serine by homocysteine and the H2S-generating β-replacement of cysteine by homocysteine catalyzed by hCBS are activated by the V-type allosteric effector, AdoMet (3, 36). The therapeutic potential of H2S in animal models of reperfusion injury and inflammation is attracting considerable interest in development of H2S-releasing and -inhibiting compounds (37). A potential strategy for modulating H2S production and for enhancing homocysteine clearance in homocystinuric patients is via the regulatory domain of CBS; however, structural insights into the mechanism of AdoMet activation have been lacking. On the basis of the analysis of the dCBS structure, which represents a form of the enzyme locked in an activated state, we posit that AdoMet binding decreases conformational flexibility and increases access to the active site (Fig. S7). There are several possibilities for how increased rigidity of the regulatory domain might activate CBS. First, by rendering the C-terminal domain less flexible, access to the active site might be less impeded (Fig. S7). In support of this model are normal-mode analyses of motions of the N- and C-terminal modules in the isolated monomer (see Materials and Methods), which suggest that an energetically accessible conformation exists in which the C-terminal module hinders approach to the active site that could, in turn, influence the rate-limiting product release step. Second, by repositioning the linker, AdoMet binding might help stabilize the PLP binding pocket. The linker emerges from a loop between βA6 and αA9 that forms part of the PLP binding pocket. Thus, changes in the relative orientations of the N- and C-terminal modules will translate into changes in the linker region that are propagated to the active site. Truncation of the C-terminal module after the linker (dCBS-∆C144 and hCBS-∆C143) has differential effects in the Drosophila and human enzymes. In hCBS, deletion of the regulatory domain (ΔC143) results in a fourfold increase in kcat, and H/D exchange mass spectrometric analysis reveals focal changes in the kinetics of deuterium incorporation restricted to the segment of the protein immediately upstream of the truncation site (22, 38). In contrast, truncation of dCBS in the C-terminal linker (ΔC144) greatly diminishes enzymatic activity (from 1,200 to 70 μmol cystathionine h-1 mg-1 protein in wild-type and ΔC144 dCBS, respectively). Thus, while the C-terminal domain imposes intrasteric inhibition on hCBS, it activates dCBS, which implies that the arrangement of the CBSDs is likely different in the two species and might help explain why AdoMet regulates hCBS but not dCBS. In dCBS, the regulatory domains from adjacent subunits form a tight dimer, which we postulate represents the activated state of the protein. The influence of dimerization on activation is consistent with the ∼7.5-fold higher basal activity of dCBS. We infer that this tight dimer is absent in hCBS because truncation of the regulatory domain is not inhibitory. Therefore, we propose that AdoMet enhances activity by inducing dimerization or enhancing the dimer interface between the regulatory domains. There is precedent for AdoMet-induced dimerization in a CBSD protein (21), and the proposed mechanism is consistent with the location and phenotypes of a number of pathogenic mutations at the interface (Fig. 2B), which render hCBS unresponsive to AdoMet but elicit higher basal activity, thereby mimicking the activated state.
In summary, the structure of full-length dCBS provides important insights into how reactive intermediates are stabilized during biogenesis of H2S and clearance of homocysteine. CBS catalyzes the committing step in the conversion of homocysteine to cysteine, the limiting precursor needed for synthesis of the antioxidant glutathione. The quantitative significance of the transsulfuration pathway for redox homeostasis (5) and its impairment in a number of pathological states renders CBS an attractive target for therapeutic intervention. To this end, our structure, by providing insights into the mechanism of allosteric regulation of hCBS by AdoMet, will be useful for rational targeting of the regulatory domain for modulating its activity.
Materials and Methods
Expression and Purification of Wild Type and dCBS-ΔC144.
Full-length and truncated dCBS were expressed as glutathione S-transferase fusions as described previously for hCBS (39). Additional details of the purification are described in SI Methods.
dCBS Activity Assay.
The activity of dCBS was measured by using a radioactive assay reported previously (39).
Stopped-Flow Experiments.
In these experiments, difference spectroscopy was used to monitor transient intermediates formed upon rapid mixing of dCBS with substrates, and additional details are described in SI Methods.
Crystallization and Cryoprotection.
dCBS was concentrated to 15–20 mg/mL in 50 mM Tris pH 8.0. Crystals were grown by the vapor diffusion method by mixing 2 μL of protein solution with 2 μL of reservoir solution in sitting drop plates (Corning) stored at 4 °C. Additional experimental details are described in SI Methods.
Data Collection and Structure Determination.
Diffraction data for substrate-free dCBS, dCBS•L-cysteine, dCBS•L-serine, and dCBS•aminoacrylate were collected at GM/CA-CAT 23-ID-D (APS) on a Mar300 detector and processed with HKL200 (40). Balbes (41) was used to determine initial phases for the substrate-free dCBS structure through molecular replacement using a single monomer of the hCBS N-terminal domain [Protein Data Bank ID code 1JBQ (11)] as a search model. All structures were refined originally with PHENIX (42) including rigid body refinement of the individual domains followed by simulated annealing in torsional space, individual atomic refinement, and restrained individual B-factor adjustment with maximum-likelihood targets. Initial density allowed for the PLP and heme cofactors to be modeled and added. Iterative rounds of manual model building and modification performed with Coot (43), followed by refinement, allowed us to model electron density belonging to the C-terminal CBSD domains as well as the linker between the N-terminal and C-terminal domains. In later rounds of refinement, electron density in the dCBS•L-cysteine, and dCBS•L-serine complexes near the PLP cofactor was modeled and refined with L-serine in its carbanionic form as displayed in Fig. 4A and Fig. S4B. Likewise in later rounds of refinement, electron density in the dCBS•aminoacrylate complex near the PLP cofactor was modeled and refined with aminoacrylate. Library descriptions for the serine-carbanion and PLP and the aminoacrylate and PLP were created with the PRODRG server (44). REFMAC5 (45) was subsequently employed for restrained refinement of all models using isotropic individual B factors. Finally, the geometric quality of the models and their agreement with the structure factors were assessed with MolProbity (46). Pymol (47) was used to create Figs. 1, 2, and 4, and Figs. S1–S4, S6, and S7.
Model Refinement of Serine Intermediates and K88 in the dCBS•L-Ser Binary Complex.
Following molecular replacement and rigid body refinement, a model of the dCBS•L-Ser binary complex was refined with REFMAC5. In this model, L-Ser and the side chain of K88 were omitted. Positive electron density was evident in the vicinity of the PLP cofactor that belongs to the bound substrate and the K88 side chain. No substrate and no side chain for K88 were modeled in this electron density to eliminate phase bias from a potentially incorrect model for serine and K88. Unbiased electron density for serine and K88 was interpreted after completion of refinement of the remainder of the protein and solvent. This final model was used as a starting point for refinement experiments to determine the best fit of serine and K88 to the density (Fig. S4). Three different serine intermediates were modeled: (i) a carbanion with trigonal planar  geometry (Fig. 4 A and B and Fig. S4 D and E), (ii) a serine molecule that has tetrahedral
geometry (Fig. 4 A and B and Fig. S4 D and E), (ii) a serine molecule that has tetrahedral  geometry away from K88 (tetrahedral intermediate 1) (Fig. S4C), and (iii) a serine model that has tetrahedral
geometry away from K88 (tetrahedral intermediate 1) (Fig. S4C), and (iii) a serine model that has tetrahedral 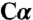 geometry toward K88 (tetrahedral intermediate 2) (Fig. S4 F and G). The side chain of K88 was omitted from some refinement experiments (Fig. S4 A, E, and F) and included in others (Fig. S4 B, C, and G). Serine better fits to the electron density as a carbanion than with tetrahedral
geometry toward K88 (tetrahedral intermediate 2) (Fig. S4 F and G). The side chain of K88 was omitted from some refinement experiments (Fig. S4 A, E, and F) and included in others (Fig. S4 B, C, and G). Serine better fits to the electron density as a carbanion than with tetrahedral 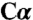 geometry, which resulted in negative electron density (Fig. S4 C, F, and G). Moreover, the tetrahedral intermediate 2 forms a chemically implausible covalent bond with K88 (Fig. S4G).
geometry, which resulted in negative electron density (Fig. S4 C, F, and G). Moreover, the tetrahedral intermediate 2 forms a chemically implausible covalent bond with K88 (Fig. S4G).
In a final experiment, serine was modeled in its carbanionic form, whereas the K88 side chain was concurrently modeled in two different conformations (Fig. S4D). The first conformation at 70% occupancy corresponds to the K88 position shown in Fig. 4 A and B and Fig. S4 B and C and is directly below the 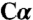 of the modeled serine. The second conformation was modeled at 30% occupancy in the same position as seen in the aminoacrylate intermediate shown in Fig. 4 C and D. The resulting electron density is shown in Fig. S4D where there is no apparent electron density that corresponds to the K88 conformation observed in the presence of the aminoacrylate intermediate.
of the modeled serine. The second conformation was modeled at 30% occupancy in the same position as seen in the aminoacrylate intermediate shown in Fig. 4 C and D. The resulting electron density is shown in Fig. S4D where there is no apparent electron density that corresponds to the K88 conformation observed in the presence of the aminoacrylate intermediate.
Normal-Mode Analysis.
Normal modes were computed by using the web interface (http://lorentz.immstr.pasteur.fr/nomad-ref.php) of the program ElNemo (48). The atomic coordinates of the substrate-free dCBS monomer and dimer were used as a starting model.
Calculation of Dimer Interface.
Dimer interfaces were analyzed by using the Protein, Interfaces, Surfaces, and Assemblies server (16).
Supplementary Material
ACKNOWLEDGMENTS.
We thank Drs. Scott Pletcher and Hadise Kabil (University of Michigan) for sharing the expression construct for dCBS. This work was supported by grants from the National Institutes of Health [HL58984 (to R.B.) and GM048533 (to J.L.S.)].
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The crystallographic coordinates have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3PC2 for the dCBS substrate-free structure, 3PC3 for the dCBS aminoacrylate structure, and 3PC4 for the dCBS carbanion structure).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1011448107/-/DCSupplemental.
References
- 1.Kimura H. Hydrogen sulfide: From brain to gut. Antioxid Redox Signal. 2010;12:1111–1123. doi: 10.1089/ars.2009.2919. [DOI] [PubMed] [Google Scholar]
- 2.Kabil O, Banerjee R. The redox biochemistry of hydrogen sulfide. J Biol Chem. 2010;285:21903–21907. doi: 10.1074/jbc.R110.128363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R. Relative contributions of cystathionine beta-synthase and gamma-cystathionase to H2S biogenesis via alternative trans-sulfuration reactions. J Biol Chem. 2009;284:22457–22466. doi: 10.1074/jbc.M109.010868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kraus JP, et al. Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat. 1999;13:362–375. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Mosharov E, Cranford MR, Banerjee R. The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry. 2000;39:13005–13011. doi: 10.1021/bi001088w. [DOI] [PubMed] [Google Scholar]
- 6.Scott JW, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 8.Cherney MM, et al. Ferrous human cystathionine beta-synthase loses activity during enzyme assay due to a ligand switch process. Biochemistry. 2007;46:13199–13210. doi: 10.1021/bi701159y. [DOI] [PubMed] [Google Scholar]
- 9.Singh S, et al. Modulation of the heme electronic structure and cystathionine beta-synthase activity by second coordination sphere ligands: The role of heme ligand switching in redox regulation. J Inorg Biochem. 2009;103:689–697. doi: 10.1016/j.jinorgbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weeks CL, Singh S, Madzelan P, Banerjee R, Spiro TG. Heme regulation of human cystathionine beta-synthase activity: Insights from fluorescence and Raman spectroscopy. J Am Chem Soc. 2009;131:12809–12816. doi: 10.1021/ja904468w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. Structure of human cystathionine beta-synthase: a unique pyridoxal 5′- phosphate-dependent heme protein. EMBO J. 2001;20:3910–3916. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taoka S, et al. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry. 2002;41:10454–10461. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 13.Hyde CC, Miles EW. Three dimensional structure of the tryptophan synthase alfa 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 14.Sen S, Banerjee R. A pathogenic linked mutation in the catalytic core of human cystathionine beta-synthase disrupts allosteric regulation and allows kinetic characterization of a full-length dimer. Biochemistry. 2007;46:4110–4116. doi: 10.1021/bi602617f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frank N, Kery V, Maclean KN, Kraus JP. Solvent-accessible cysteines in human cystathionine beta-synthase: Crucial role of cysteine 431 in S-adenosyl L-methionine binding. Biochemistry. 2006;45:11021–11029. doi: 10.1021/bi060737m. [DOI] [PubMed] [Google Scholar]
- 16.Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- 17.Dunathan HC. Conformation and reaction specificity in pyridoxal phosphate enzymes. Proc Natl Acad Sci USA. 1966;55:712–716. doi: 10.1073/pnas.55.4.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taoka S, Banerjee R. Stopped-flow kinetic analysis of the reaction catalyzed by the full length yeast cystathionine beta synthase. J Biol Chem. 2002;277:22421–22425. doi: 10.1074/jbc.M202513200. [DOI] [PubMed] [Google Scholar]
- 19.Evande R, Ojha S, Banerjee R. Visualization of PLP-bound intermediates in hemeless variants of human cystathionine beta-synthase: Evidence that lysine 119 is a general base. Arch Biochem Biophys. 2004;427:188–196. doi: 10.1016/j.abb.2004.04.027. [DOI] [PubMed] [Google Scholar]
- 20.Bruno S, et al. Functional properties of the active core of human cystathionine beta-synthase crystals. J Biol Chem. 2001;276:16–19. doi: 10.1074/jbc.C000588200. [DOI] [PubMed] [Google Scholar]
- 21.Lucas M, et al. Binding of S-methyl-5′-thioadenosine and S-adenosyl-L-methionine to protein MJ0100 triggers an open-to-closed conformational change in its CBS motif pair. J Mol Biol. 2010;396:800–820. doi: 10.1016/j.jmb.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 22.Evande R, Boers GHJ, Blom HJ, Banerjee R. Alleviation of intrasteric inhibition by the pathogenic activation domain mutation, D444N, in human cystathionine beta-synthase. Biochemistry. 2002;41:11832–11837. doi: 10.1021/bi026248d. [DOI] [PubMed] [Google Scholar]
- 23.Schneider TR, et al. Loop closure and intersubunit communication in tryptophan synthase. Biochemistry. 1998;37:5394–5406. doi: 10.1021/bi9728957. [DOI] [PubMed] [Google Scholar]
- 24.Clausen T, Kaiser JT, Steegborn C, Huber R, Kessler D. Crystal structure of the cystine C-S lyase from Synechocystis: stabilization of cysteine persulfide for FeS cluster biosynthesis. Proc Natl Acad Sci USA. 2000;97:3856–3861. doi: 10.1073/pnas.97.8.3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schnell R, Oehlmann W, Singh M, Schneider G. Structural insights into catalysis and inhibition of O-acetylserine sulfhydrylase from Mycobacterium tuberculosis crystal structures of the enzyme alpha-aminoacrylate intermediate and an enzyme-inhibitor complex. J Biol Chem. 2007;282:23473–23481. doi: 10.1074/jbc.M703518200. [DOI] [PubMed] [Google Scholar]
- 26.Shimon LJ, et al. Two structures of alliinase fromAlliium sativum L: Apo form and ternary complex with aminoacrylate reaction intermediate covalently bound to the PLP cofactor. J Mol Biol. 2007;366:611–625. doi: 10.1016/j.jmb.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 27.Bach RD, Canepa C, Glukhovstev MN. Influence of electrostatic effects on activation barriers in enzymatic reactions: Pyridoxal 5′-phosphate-dependent decarboxylation of α-amino acids. J Am Chem Soc. 1999;121:6542–6555. [Google Scholar]
- 28.Toney MD. Computational studies on nonenzymatic and enzymatic pyridoxal phosphate catalyzed decarboxylations of 2-aminoisobutyrate. Biochemistry. 2001;40:1378–1384. doi: 10.1021/bi0012383. [DOI] [PubMed] [Google Scholar]
- 29.Mozzarelli A, et al. Effect of pH and monovalent cations on the formation of quinonoid intermediates of the tryptophan synthase alpha(2)beta(2) complex in solution and in the crystal. J Biol Chem. 2000;275:6956–6962. doi: 10.1074/jbc.275.10.6956. [DOI] [PubMed] [Google Scholar]
- 30.Yano T, Kuramitsu S, Tanase S, Morino Y, Kagamiyama H. Role of Asp222 in the catalytic mechanism of Escherichia coli aspartate aminotransferase: the amino acid residue which enhances the function of the enzyme-bound coenzyme pyridoxal 5′-phosphate. Biochemistry. 1992;31:5878–5887. doi: 10.1021/bi00140a025. [DOI] [PubMed] [Google Scholar]
- 31.Eliot AC, Kirsch JF. Pyridoxal phosphate enzymes: Mechanistic, structural, and evolutionary considerations. Annu Rev Biochem. 2004;73:383–415. doi: 10.1146/annurev.biochem.73.011303.074021. [DOI] [PubMed] [Google Scholar]
- 32.Szebenyi DM, Liu X, Kriksunov IA, Stover PJ, Thiel DJ. Structure of a murine cytoplasmic serine hydroxymethyltransferase quinonoid ternary complex: evidence for asymmetric obligate dimers. Biochemistry. 2000;39:13313–13323. doi: 10.1021/bi000635a. [DOI] [PubMed] [Google Scholar]
- 33.Milic D, Demidkina TV, Faleev NG, Matkovic-Calogovic D, Antson AA. Insights into the catalytic mechanism of tyrosine phenol-lyase from X-ray structures of quinonoid intermediates. J Biol Chem. 2008;283:29206–29214. doi: 10.1074/jbc.M802061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barends TR, et al. Structure and mechanistic implications of a tryptophan synthase quinonoid intermediate. ChemBioChem. 2008;9:1024–1028. doi: 10.1002/cbic.200700703. [DOI] [PubMed] [Google Scholar]
- 35.Fiedler E, et al. Snapshot of a key intermediate in enzymatic thiamin catalysis: Crystal structure of the alpha-carbanion of (alpha,beta-dihydroxyethyl)-thiamin diphosphate in the active site of transketolase from Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:591–595. doi: 10.1073/pnas.022510999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen X, Jhee KH, Kruger WD. Production of the neuromodulator H2S by cystathionine beta-synthase via the condensation of cysteine and homocysteine. J Biol Chem. 2004;279:52082–52086. doi: 10.1074/jbc.C400481200. [DOI] [PubMed] [Google Scholar]
- 37.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 38.Sen S, Yu J, Yamanishi M, Schellhorn D, Banerjee R. Mapping peptides correlated with transmission of intrasteric inhibition and allosteric activation in human cystathionine beta-synthase. Biochemistry. 2005;44:14210–14216. doi: 10.1021/bi051046d. [DOI] [PubMed] [Google Scholar]
- 39.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine beta-synthase activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 40.Otwinowski Z, Minor W. Methods in Enzymology. Vol 276. New York: Springer; 1997. Processing of X-ray diffraction data collected in oscillation mode; pp. 307–326. (Macromolecular Crystallography, Part A). [DOI] [PubMed] [Google Scholar]
- 41.Long F, Vagin AA, Young P, Murshudov GN. BALBES: A molecular-replacement pipeline. Acta Crystallogr, Sect D: Biol Crystallogr. 2008;64:125–132. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 44.Schuttelkopf AW, van Aalten DM. PRODRG: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 45.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 46.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeLano WL. The PyMOL Molecular Graphics System. Palo Alto, CA: DeLano Scientific; 2002. [Google Scholar]
- 48.Suhre K, Sanejouand YH. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.