

Abstract
The oncogenic fusion protein RET/PTC3 (RP3) that is expressed in papillary thyroid carcinoma (PTC) and thyroid epithelia in Hashimoto’s thyroiditis activates Nuclear Factor-kappa B (NF-κB) and induces pro-inflammatory gene expression; however, the mechanism of this activation is unknown. To address this, we expressed RP3 in murine embryonic fibroblasts (MEFs) lacking key classical and non-canonical NF-κB signaling components. In wild-type MEFs, RP3 upregulated CCL2, CXCL1, GM-CSF and TNF expression and activated classical but not non-canonical NF-κB. RP3 activated NF-κB in IKKβ−/− MEFs but not IKKα- or NEMO-deficient cells and activation was inhibited by a peptide that blocks NEMO binding to the IKKs. RP3 increased the levels of NF-κB-inducing kinase (NIK) and did not activate NF-κB in NIK-deficient MEFs. Notably, NIK stabilization was not accompanied by TRAF3 degradation demonstrating that RP3 disrupts normal basal NIK regulation. Dominant negative NIK blocked RP3-induced NF-κB activation and an RP3 signaling mutant (RP3Y588F) did not stabilize NIK. Finally, examination of PTC specimens revealed strong positive staining for NIK. We therefore conclude that RP3 activates classical NF-κB via NIK, NEMO and IKKα. Importantly, our findings reveal a novel mechanism for oncogene-induced NF-κB activation via stabilization of NIK.
Keywords: Hashimoto’s thyroiditis, IKK, NIK, NF-κB, RET/PTC, Thyroid Cancer
Introduction
Aberrant NF-κB activation occurs in many cancers (Karin, 2006) where it is implicated in tumorigenesis by promoting the expression of anti-apoptotic genes and pro-inflammatory cytokines that cumulatively influence cell survival and tumor progression (Basseres & Baldwin, 2006; Karin, 2006). The NF-κB transcription factor family contains five members named NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA (p65), RelB, and c-Rel that form homo- or heterodimers which are normally held inactive in the cytosol by a group of proteins named the inhibitors of κB or IκBs (Hayden & Ghosh, 2008). Upon stimulation, the IκBs become rapidly phosphorylated, ubiquitinated, and degraded by the 26S proteasome. This releases NF-κB that translocates to the nucleus to promote target gene transcription (Hayden & Ghosh, 2008). In many tumor cells, this normal regulation is dysfunctional, leading to sustained nuclear localization of NF-κB (Basseres & Baldwin, 2006; Karin, 2006).
NF-κB activation is regulated by the IκB kinase (IKK) complex that contains two catalytic subunits, IKKα (IKK1) and IKKβ (IKK2), and a regulatory subunit named NF-κB essential modulator (NEMO) (Hayden & Ghosh, 2008). Deletion of these revealed two separate pathways of NF-κB activation. The classical NF-κB pathway, requires NEMO-and IKKβ-mediated phosphorylation of IκBs leading to nuclear translocation of NF-κB typified by p65:p50 (Hayden & Ghosh, 2008). This pathway is activated by most immune and inflammatory stimuli and is critical for inflammation, immunity, cell survival and proliferation. Aberrant classical NF-κB activation has been extensively described in cancer (Basseres & Baldwin, 2006; Karin, 2006).
The second mechanism is the noncanonical pathway that requires IKKα activation by NIK independently of NEMO and IKKβ (Senftleben et al., 2001; Xiao et al., 2001). In resting cells, NIK is normally degraded via ubiquitination involving TRAF2 and TRAF3 (He et al., 2007; Vallabhapurapu et al., 2008; Zarnegar et al., 2008). Following stimulation, TRAF3 is degraded and NIK levels accumulate and activate IKKα. IKKα then phosphorylates p100 associated with RelB leading to p100 processing to p52 and release of a p52:RelB heterodimer (He et al., 2007; Senftleben et al., 2001; Vallabhapurapu et al., 2008; Xiao et al., 2001; Zarnegar et al., 2008). This NF-κB species regulates lymphoid organogenesis and B cell maturation (Dejardin et al., 2002) and aberrant noncanonical signaling occurs in various cancers (Annunziata et al., 2007; Keats et al., 2007; Nishina et al., 2009; Wharry et al., 2009). Recent studies revealed that mutations in NIK or its regulators stabilize NIK and activate both NF-κB pathways in cancer cells (Annunziata et al., 2007; Keats et al., 2007).
The RET proto-oncogene encodes a receptor tyrosine kinase absent in normal thyroid tissue (Bunone et al., 2000). However, in thyroid malignancies, RET translocates to form fusions of its kinase domain attached to one of several constitutively active gene partners (Bunone et al., 2000). These translocations constitute a family of fusion proteins termed the RET/PTCs of which RET/PTC1 and RET/PTC3 (RP3) are the most prevalent (Ciampi & Nikiforov, 2007). The RET/PTCs occur in papillary thyroid carcinoma (PTC) and intriguingly RET/PTC expression is observed in PTC associated with autoimmune thyroiditis as well as autoimmune thyroiditis with no detectable cancer (Rhoden et al., 2006; Sheils et al., 2000; Wirtschafter et al., 1997). Hence the RET/PTC oncogenes may provide a molecular link between autoimmune inflammatory thyroiditis and thyroid cancer (Eisenlohr & Rothstein, 2006; Muzza et al., 2009).
The un-rearranged RET receptor activates classical NF-κB (Hayashi et al., 2000; Ludwig et al., 2001) and constitutive NF-κB activity occurs in thyroid cancer cell lines (Pacifico et al., 2004; Visconti et al., 1997) and tissue samples of papillary, follicular and anaplastic thyroid cancer (Pacifico & Leonardi, 2009). More importantly, ectopic RP3 expression activates NF-κB and induces pro-inflammatory cytokines and chemokines including GM-CSF, and CCL2 that are targets of the classical NF-κB pathway (Pufnock & Rothstein, 2009; Russell et al., 2003). In addition, RET/PTC1 induces a proinflammatory gene program characterized by classical NF-κB-dependent chemokines, cytokines and growth factors (Borrello et al., 2005). While these studies establish that RET and the RET/PTCs activate classical NF-κB, several lines of evidence indicate that these proteins might also regulate noncanonical NF-κB signaling. Thus, RET null mice fail to develop Peyer’s Patches indicating defective lymphoid organogenesis although this phenotype has not been linked to defects in noncanonical NF-κB signaling (Veiga-Fernandes et al., 2007). More directly, RET/PTC1 expression in thyrocytes induces the noncanonical NF-κB-dependent gene CXCL12 (Borrello et al., 2005). Despite this accumulated evidence, the precise mechanism of NF-κB activation by the RET/PTCs remains unknown.
To address this question we expressed RP3 in mouse embryonic fibroblasts (MEFs) lacking key components of the classical and noncanonical NF-κB pathways and determined the effects of RP3 on NF-κB activation and gene expression. We demonstrate that RP3 activates classical NF-κB via a novel pathway involving NIK, NEMO and IKKα. Importantly, our findings reveal an unanticipated mechanism for oncogene-induced NF-κB activation via stabilization of NIK.
Results
RP3 activates NF-κB and induces pro-inflammatory gene expression in MEFs
To study the mechanism of RP3-induced NF-κB activation we retrovirally transduced MEFs with either the MIGR1 control vector, or MIGR1 containing RP3 (MIGR1-RP3) (Pufnock & Rothstein, 2009; Russell et al., 2003). Immunoblotting of cell lysates using anti-RET antibody revealed RP3 expression restricted to the RP3-transduced cells (Figure 1a). To determine whether ectopic RP3 expression could activate NF-κB, control and RP3-transduced MEFs were transiently transfected with an NF-κB luciferase reporter plasmid and transcription was assessed by luciferase assay. Consistent with prior studies using thyrocytes (Pufnock & Rothstein, 2009; Russell et al., 2003), NF-κB activity was enhanced in MEFs expressing RP3 compared with control cells (Figure 1b). Furthermore, DNA binding of two distinct NF-κB complexes was markedly enhanced in RP3-expressing cells (Figure 1c).
Figure 1.
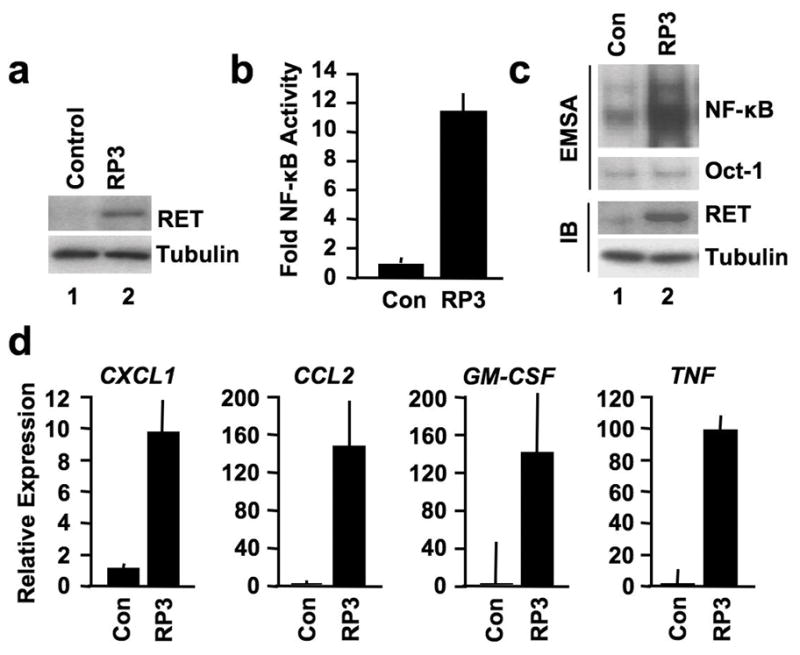
RP3 activates NF-κB and induces pro-inflammatory gene expression in MEFs. (a) Lysates from MEFs retrovirally transduced with either MIGR vector alone (Control) or MIGR expressing RP3 were immunoblotted using the antibodies indicated (right). (b) Control and RP3-expressing MEFs were transiently transfected with the NF-κB-dependent reporter pBIIx-firefly luciferase (FFL) together with β-actin renilla luciferase (RL). Twenty-four hours later, NF-κB activity was determined by dual luciferase assay in each stable line as the ratio of the relative luciferase units (FFL:RL). Results are expressed as fold increase normalized to the Control cells. (c) Nuclear extracts from Control (Con) and RP3-expressing MEFs were prepared for EMSA (top two panels). Assays were performed using either a consensus NF-κB binding site probe or an Oct1 probe as a loading control as shown (right). Whole cell lysates from the same cells were immunoblotted (IB) using the antibodies indicated (right). (d) Expression of the NF-κB-dependent genes CXCL1, CCL2, GM-CSF and TNF in Control and RP3-expressing MEFs was measured by qRT-PCR. RQ levels for each gene were normalized to levels in untreated Control MEFs.
RET/PTC fusions, including RP3, induce pro-inflammatory gene expression in thyroid cells (Borrello et al., 2005; Russell et al., 2003). We therefore asked if RP3 affected known NF-κB target genes in MEFs. As shown in Figure 1d, expression of CXCL1, CCL2, TNF and GM-CSF, were significantly enhanced in MEFs expressing RP3 compared with control cells. As these are classical NF-κB targets, we questioned whether RP3 could also induce expression of the noncanonical NF-κB-dependent gene CXCL12 (Dejardin et al., 2002; Madge et al., 2008); however, this was not induced by RP3 (Figure S1). Thus, ectopic RP3 expression activates NF-κB and induces expression of classical NF-κB-dependent pro-inflammatory genes.
RP3 activates classical NF-κB
NF-κB activation requires signal-induced phosphorylation of the IKKs at specific residues within their activation domains (Hayden & Ghosh, 2008). To determine if RP3 induced phosphorylation of the IKKs we immunoblotted Control and RP3-MEF lysates using an antibody specific for epitopes on phosphorylated IKKs (anti-p-IKKα/β). As shown in Figure 2a, two immunoreactive bands were detected in RP3-expressing cell lysates (top panel) suggesting that RP3 induces the phosphorylation of both IKKα and IKKβ.
Figure 2.

RP3 activates the classical NF-κB pathway. (a and b) Whole cell lysates from control (Con) and RP3-expressing MEFs were immunoblotted using the antibodies specified on the right of each panel. (c) The same lysates were immunoblotted using either anti-p100/p52 (top two panels) or anti-phospho-p100 (p-p100) as shown. (d) Wild-type MEFs were either untreated (& minus;) or incubated with anti-LTβR (αLTβR) for 8 hours (+) then lysates were immunoblotted using anti-p100/p52 (top two panels) or anti-tubulin. (e) Cytoplasmic (Cyto.) and nuclear (Nuc.) extracts from control and RP3-expressing MEFs were immunoblotted using the antibodies indicated (right). Samples were immunoblotted using abti-tubulin and anti-Histone H3 (H3) antibodis to control for cytoplasmic and nuclear integrity and protein loading. (f) Nuclear extracts from Control (Con), RP3-expressing MEFs and MEFs stimulated with TNF (10ng/ml; 15 min) were prepared for EMSA either a consensus NF-κB binding site probe or an Oct1 probe as a loading control. The positions of two DNA-binding complexes are indicated (C1 and C2). (g) The nuclear extracts from RP3-expressing and TNF-stimulated MEFs shown in (f) were used for supershift analysis. Samples were incubated with either a non-specific isotype-matched control antibody (ns) or anti-NF-κB antibodies as shown (top). The positions of the NF-κB complexes (C1 and C2) and the supershifted bands (SS) are indicated.
A critical signaling event during classical NF-κB activation is site-specific IκBα phosphorylation at Serines 32 and 36 (Hayden & Ghosh, 2008). As shown in Figure 2b, using an antibody that recognizes IκB phosphorylated at Ser32 (anti-p-IκBα) we detected phospho-IκBα in the RP3-expressing MEFs but not in control cells. A further post-translational modification critical for classical NF-κB transcriptional activity is phosphorylation of p65 at Ser536 (Hayden & Ghosh, 2008) and we found this modification using anti-phospho-S536 (anti-p-p65) only in RP3-expressing cells (Figure 2b). Together, these findings demonstrate that RP3 induces two major phosphorylation events in the classical NF-κB pathway. Notably, the level of IκBα was markedly enhanced in RP3 cells (Figure 2b). As the gene encoding IκBα is a target of classical NF-κB (Hayden & Ghosh, 2008), its increased expression supports the conclusion that RP3 activates classical NF-κB in MEFs.
Noncanonical NF-κB signaling involves phosphorylation and processing of p100 to p52 (Senftleben et al., 2001; Xiao et al., 2001). Immunoblotting using anti-p100/p52 or anti-phospho-p100 showed increased phospho-p100 and p52 levels in RP3-expressing cells compared with control MEFs (Figure 2c). However, RP3 upregulated p100 expression suggesting that increased p52 is a consequence of enhanced basal p100 levels rather than signal-induced p100 processing. In this regard, treatment of MEFs with an anti-LTβ receptor antibody that activates the noncanonical pathway led to the expected decrease of p100 and concomitant increase in p52 (Figure 2d). Like IκBα, p100 is regulated by classical NF-κB (Hayden & Ghosh, 2008) and this accounts for its increased expression in cells in which the classical but not the noncanonical pathway is activated. Thus we conclude that RP3 does not activate the noncanonical pathway (i.e. promote p100 processing to p52), but instead, its effects resemble those of TNF that increases p100 levels and concomitantly increases p52 via basal processing (Derudder et al., 2003).
We next explored the nature of the NF-κB proteins in the nucleus of RP3-expressing cells and found that nuclear p65 was increased compared with control cells (Figure 2e). In contrast, the level of nuclear p50 was lower than control MEFs (Figure 2e). Furthermore, the amounts of nuclear p52 and RelB in RP3-transduced cells were no different from the levels in control MEFs although both were increased in the cytoplasm of RP3-transduced cells. Comparison of the nuclear NF-κB complexes induced by either RP3 or TNF by EMSA demonstrated significant similarities such that both stimuli upregulated the DNA-binding of two distinct complexes (Figure 2f). Supershift analysis revealed that the lower complex (C2) induced by both stimuli was completely upshifted by anti-p50 but not by anti-p65, p52, or RelB (Figure 2g) demonstrating that C2 is a p50 homodimer. In contrast, the upper band (C1) was upshifted by only p65 indicating a p65 homodimer. A small residual band was observed in the p65 supershift that we conclude to be non-specific, as it was not shifted by any of the antibodies including anti-c-Rel (not shown). These findings demonstrate that RP3 induces a similar NF-κB complex profile as TNF and thereby strongly support the notion that RP3 activates classical but not noncanonical NF-κB.
NF-κB activation is NEMO and IKKα-dependent
Classical NF-κB activation typically requires IKKβ (Hayden & Ghosh, 2008); however, we demonstrated recently that IKKα is sufficient for IL-1-induced classical signaling (Solt et al., 2007). To establish which IKK subunits are required for RP3-induced NF-κB activation, we stably transduced IKKα−/− and IKKβ−/− MEFs with RP3. Western blotting confirmed that the MEFs lacked the appropriate kinases and that RP3 was expressed in the transduced cells (Figure 3a and b). NF-κB activation was no greater in RP3-expressing IKKα−/− MEFs than basal activity in control cells (Figure 3a, S2); however, similar to WT cells (Figure 1b and Figure S2), NF-κB DNA-binding was markedly enhanced in RP3-expressing IKKβ−/− MEFs (Figure 3b and Figure S2). Consistent with these findings, NF-κB transcriptional activity was increased in WT and IKKβ−/− MEFs expressing RP3, whereas RP3 had no effect on transcription in IKKα-deficient cells (Figure 3c).
Figure 3.
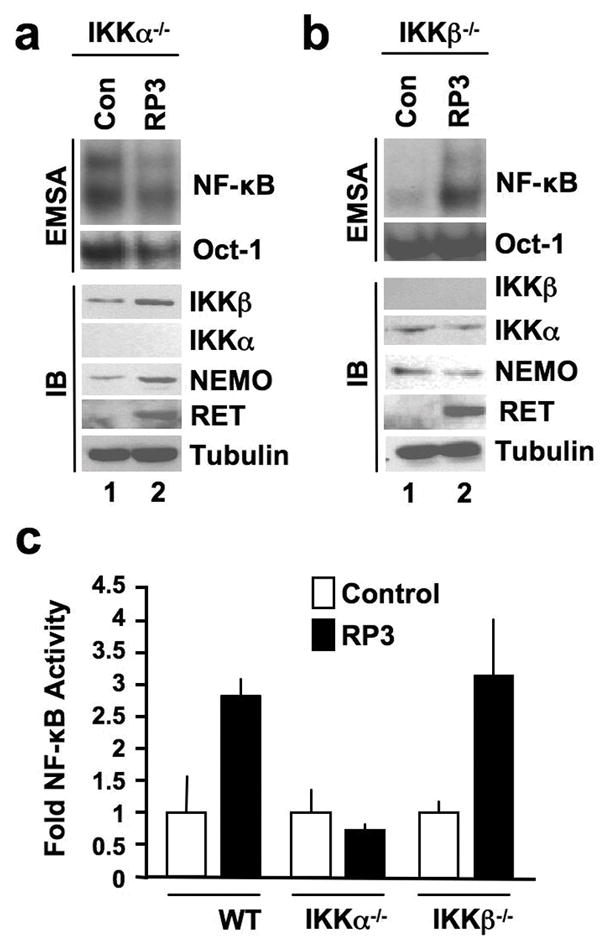
RP3-induced NF-κB activation requires IKKα but not IKKβ. Nuclear extracts from (a) IKKα−/− and (b) IKKβ−/− MEFs retrovirally transduced with either MIGR vector alone (Con) or MIGR expressing RP3 were prepared for EMSA (top two panels). Assays were performed using either a consensus NF-κB binding site probe or an Oct1 probe as a loading control as shown (right). Whole cell lysates from the same cells were immunoblotted (IB) using the antibodies indicated (right). (c) Control (Con) and RP3-expressing wild-type (WT), IKKα−/− and IKKβ−/− MEFs were transiently transfected with pBIIx-firefly luciferase (FFL) and β-actin renilla luciferase (RL). Twenty-four hours later, NF-κB activity was determined by dual luciferase assay in each stable line as the ratio of the relative luciferase units (FFL:RL). Results are expressed as fold increase normalized to the Control cells for each MEF line.
To determine if NEMO is required for RP3-induced NF-κB activation, we stably transduced NEMO−/− MEFs with RP3 or MIGR1 vector alone (Figure 4a). Similar to IKKα−/− cells, RP3 did not induce NF-κB DNA-binding in NEMO-deficient MEFs above that observed in control cells (Figure 4a). Furthermore, RP3 did not activate NF-κB transcriptional activity in the absence of NEMO demonstrating that NEMO is required for the RP3-induced NF-κB activation (Figure 4b). We next asked if direct association of NEMO with the IKKs is required for RP3-induced NF-κB activation. To address this, we used a cell permeable NEMO binding domain (NBD) peptide that prevents NEMO binding to IKKα and IKKβ (May et al., 2000). As shown in Figure 4c, treatment of RP3-expressing MEFs with the wild-type peptide (NBDWT) but not an inactive mutant control (NBDMUT) inhibited NF-κB DNA-binding activity. Consistent with this, NBDWT reduced CCL2 expression in RP3-expressing MEFs (Figure 4d). These accumulated findings lead us to conclude that RP3 activates classical NF-κB via a NEMO- and IKKα-dependent mechanism.
Figure 4.
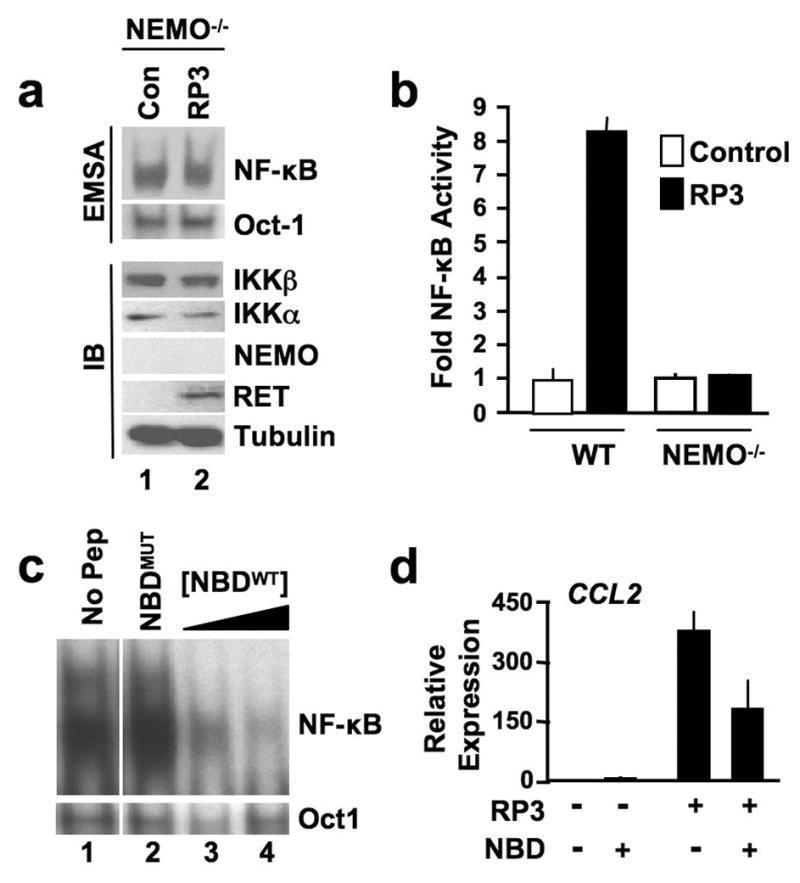
RP3-induced NF-κB activation requires NEMO. (a) Nuclear extracts from NEMO−/− MEFs transduced with either MIGR vector alone (Con) or MIGR expressing RP3 were prepared for EMSA (top two panels). Assays were performed using either an NF-κB binding site probe or an Oct1 probe as shown (right). Whole cell lysates from the same cells were immunoblotted (IB) using the antibodies indicated (right). (b) Control (Con) and RP3-expressing wild-type (WT) and NEMO−/− MEFs were transiently transfected with pBIIx-firefly luciferase and β-actin renilla luciferase. NF-κB activity was determined and the results are expressed as described in Figure 3c. (c) Wild-type MEFs stably transduced with RP3 were incubated for 8 hrs with either vehicle alone (1% DMSO; No Pep), 100μM of the mutant NBD peptide NBDMUT or 50 and 100μM of the WT peptide (NBDWT). NF-κB activation in nuclear extracts was determined by EMSA using a consensus NF-κB binding site probe or an Oct1 probe as a loading control. (d) Control and RP3-expressing MEFs were incubated with either vehicle alone (1% DMSO; -) or NBDWT peptide (100μM) and expression of CCL2 was measured by qRT-PCR. RQ levels were normalized to levels in untreated Control MEFs.
Ectopic RP3 expression stabilizes NIK
Recently, aberrant NIK expression was shown to activate classical NF-κB in certain tumor cells (Annunziata et al., 2007; Keats et al., 2007). We therefore questioned if RP3 affects the levels of NIK or the proteins that regulate NIK expression, TRAF2 and TRAF3 (He et al., 2007; Vallabhapurapu et al., 2008). We found that RP3-expressing MEFs contained elevated levels of NIK protein compared with control cells in which NIK was either absent or minimally expressed (Figure 5a). Surprisingly, unlike signal-induced NIK stabilization that requires TRAF3 degradation (He et al., 2007; Vallabhapurapu et al., 2008), TRAF3 levels were slightly increased in RP3-transduced MEFs compared with control cells. Quantitative RT-PCR demonstrated similar levels of NIK transcription between RP3 and control cells (Figure S3); thus, RP3 stabilizes NIK protein levels without causing TRAF3 degradation.
Figure 5.

RP3 activates NF-κB by stabilizing NIK. (a) Lysates from Control or RP3-expressing WT MEFs were immunoblotted using the antibodies indicated (right). (b) Control (Con) and RP3-expressing wild-type (WT) and NIK−/− MEFs were transiently transfected with pBIIx-firefly luciferase and β-actin renilla luciferase. NF-κB activity was determined and the results are expressed as described in Figure 3c. (c) Control (Con) and RP3-expressing MEFs were transiently transfected with the NF-κB-dependent reporter pBIIx-firefly luciferase (FFL) together with β actin renilla luciferase (RL). Cells were also either mock transfected (−) or transiently transfected with dominant negative NIK (NIKDN; +). Twenty-four hours later, NF-κB activity was determined by dual luciferase assay as described in Figure 3c. Lysates were immunoblotted using the antibodies specified (left).
To establish whether NIK is necessary for RP3-induced NF-κB activation we transduced NIK−/− MEFs with RP3 or the empty MIGR1 vector. We assessed NF-κB activation by luciferase reporter assay and as shown in Figure 5b, RP3 did not activate NF-κB in NIK−/− cells. Furthermore, transient overexpression of dominant negative NIK (DN NIK), inhibited NF-κB in RP3-expressing WT MEFs (Figure 5c). Consistent with the ability of RP3 to stabilize endogenous NIK, the amount of DN NIK was enhanced in RP3 cells (Figure 5c). Overall, the findings in Figure 5 demonstrate that RP3 expression stabilizes NIK levels independently of TRAF3 degradation and that NIK is required for RP3-induced NF-κB activation.
Earlier studies established that activated RET autophosphorylates tyrosine 1062 forming a Shc docking site that mediates activation of downstream signals (Hayashi et al., 2000). Moreover, an RP3 mutant containing a tyrosine to phenylalanine substitution at the equivalent residue (RP3Y588F) did not activate NF-κB or induce inflammatory gene expression in thyrocytes or fibrosarcoma cells (Pufnock & Rothstein, 2009; Russell et al., 2003). To determine if this same RP3Y588F mutant could stabilize NIK and activate NF-κB in MEFs, we retrovirally transduced WT cells with RP3Y588F. Compared with RP3, RP3Y588F did not stabilize NIK levels above those in control MEFs (Figure 6a) and consistent with previous reports (Pufnock & Rothstein, 2009; Russell et al., 2003), RP3Y588F did not activate NF-κB (Figure 6b). Furthermore, although RP3Y588F partially induced CCL2 expression, this was significantly less than the levels induced by RP3 (Figure 6c). Thus we conclude that RP3-induced NIK stabilization and NF-κB activation requires autophosphorylation of Y588.
Figure 6.

RP3Y588F does not stabilize NIK or activate NF-κB. (a) Lysates from Control (Con), RP3- and RP3Y588F-expressing MEFs were immunoblotted using the antibodies indicated (right). (b) Control (Con), RP3- and RP3Y588F-expressing MEFs were transiently transfected with pBIIx-firefly luciferase and β-actin renilla luciferase. NF-κB activity was determined by dual luciferase assay and the results are expressed as described in Figure 3c. (c) Expression of CCL2 in Control (Con) RP3- and RP3Y588F-expressing MEFs was measured by qRT-PCR and RQ levels were normalized to levels in untreated Control MEFs.
NIK is expressed in thyroid carcinoma tissue
To establish whether NIK is expressed in human papillary thyroid carcinoma specimens, we immunohistochemically stained tissue blocks from patients with well differentiated RET positive PTC (Figure 7). Staining for RET was similar to that detailed earlier (Nibu et al., 2005) and appeared homogeneous throughout the tumor tissue whereas vascular endothelial cells (EC) were negative (Figure 7b). As previously described (Pacifico & Leonardi, 2009; Pacifico et al., 2004; Visconti et al., 1997), a high number of tumor cells stained positively for nuclear p65 indicating activation of classical NF-κB (Figure 7c). EC did not display any nuclear staining demonstrating that NF-κB activation is limited to the tumor cells. Staining with anti-NIK revealed strong positive immunoreactivity in tumor tissues whereas EC were uniformly negative (Figure 7d). Consistent with previous studies demonstrating that NIK shuttles between the cytoplasm and nucleus (Birbach et al., 2004), NIK was expressed in both compartments in neoplastic cells. These immunohistochemical data therefore demonstrate active NF-κB and NIK expression in PTC cells but not in adjacent nonmalignant EC within the tumor tissue.
Figure 7.
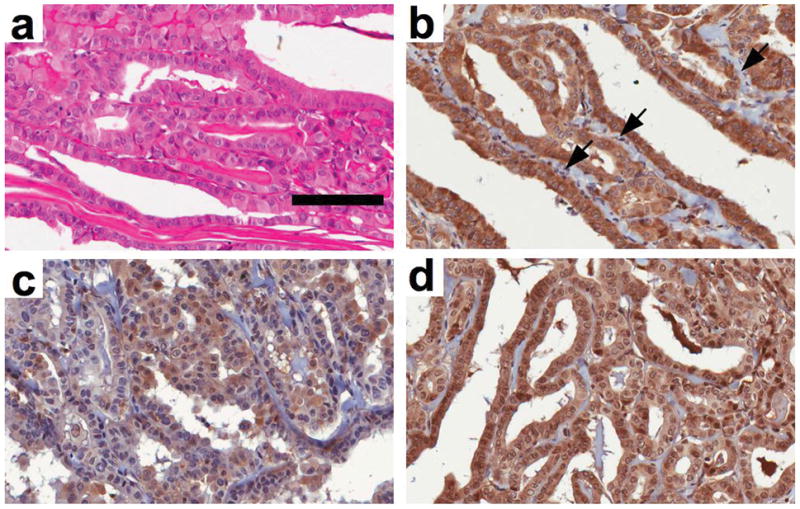
NIK is expressed in papillary thyroid carcinoma. Tissue blocks of well-differentiated PTC were H&E (a) or immunohistochemically stained using anti-RET (b) anti-p65 (c) or anti-NIK (d). Vascular endothelial cells within the tumor are indicated with arrows in (b). The scale bar in (a) is 100μm and all panels are shown at 40x magnification.
Discussion
Understanding how oncogenes dysregulate normal signaling mechanisms to convert cells from quiescent tissue components into invasive lesions is critical for developing effective anti-cancer strategies. In this study we show that the oncogenic fusion protein RP3 aberrantly activates NF-κB signaling by stabilizing the normally absent or minimally expressed NIK protein. Most importantly, our findings provide the first evidence of an oncogene modulating NIK protein levels to activate classical NF-κB
Although NF-κB activation has been reported in thyroid malignancies (Pacifico & Leonardi, 2009) the mechanisms controlling aberrant NF-κB in thyroid cancer are poorly understood. Ectopic RP3 expression in MEFs induced classical NF-κB signaling and nuclear accumulation of p65, but did not activate noncanonical NF-κB. Supershifts demonstrated that RP3 also induced the DNA-binding of p50 homodimers although the overall amount of nuclear p50 was decreased in RP3-expressing cells. Notably, the nuclear complexes induced by RP3 were very similar to those activated by TNF that only activates the classical NF-κB pathway. Previously, RET/PTCs were reported to induce a pro-inflammatory gene program hallmarked by classical NF-κB-dependent genes (Borrello et al., 2005; Russell et al., 2003). Consistent with this, RP3 induced expression of CXCL1, CCL2, GM-CSF and TNF in MEFs. Furthermore, the amounts of IκBα and p100 that are targets of classical NF-κB (Hayden & Ghosh, 2008), were increased in RP3-expressing cells. Together, these findings support the notion that RP3 mediates pro-inflammatory effects through activation of the classical NF-κB pathway.
Classical NF-κB activation typically requires NEMO and IKKβ but not IKKα (Hayden & Ghosh, 2008). It is therefore notable that RP3 activates NF-κB in IKKβ−/− but not IKKα−/− MEFs indicating that IKKβ cannot substitute for IKKα in transducing the RP3 signal. Our study therefore identifies RP3 as a member of a subset of classical NF-κB inducers that can utilize NEMO and IKKα but do not require IKKβ. This finding contradicts the data that RET-induced NF-κB activation requires IKKβ (Ludwig et al., 2001). However, RET is a transmembrane protein whereas RP3 mostly localizes to the cytoplasm (Monaco et al., 2001). Consequently, the distinct structures, stoichiometry and localization of RP3 and RET may contribute to the separate signals activated by each protein.
Extensive efforts are focused on developing selective IKKβ inhibitors to block aberrant classical NF-κB activation (Gilmore & Herscovitch, 2006). However, since IKKα is crucial for RP3-induced NF-κB activation, we predict that targeting IKKβ would not block the effects of RP3. Recent studies revealed that small molecule IKKβ inhibition incompletely inhibits classical NF-κB in B cell lymphoma cells and that concomitant ablation of IKKα is required for effective blockade (Lam et al., 2008). Consequently, in situations such as RP3 expression in disease, NF-κB will be activated via NEMO and IKKα and thus, inhibiting NEMO function might be a more effective strategy to ablate pathological NF-κB activity. Supporting this notion, the cell-permeable NBD peptide that disrupts NEMO function inhibited RP3-induced NF-κB and gene expression.
Intriguingly, RP3 stabilized NIK levels in MEFs and NIK was required for RP3-induced NF-κB. Furthermore, we detected strong positive NIK expression in RET positive PTC tissues demonstrating the potential importance of this stabilization in human disease. NIK is a key regulator of noncanonical NF-κB signaling that is minimally expressed in resting cells and accumulates only following ligation of specific receptors (Hayden & Ghosh, 2008). Contrary to this tight physiological regulation, elevated NIK levels have been reported in multiple myeloma (Annunziata et al., 2007; Keats et al., 2007) and pancreatic cancer cells (Nishina et al., 2009; Wharry et al., 2009). Additionally, mutations in NIK or mutations in the proteins that negatively regulate NIK expression lead to the observed dysregulated NIK expression in multiple myeloma (Annunziata et al., 2007; Keats et al., 2007). It is highly unlikely that RP3 expression itself causes mutations in NIK or in any of the genes regulating NIK expression. Hence, we conclude that RP3 aberrantly modulates the pathways that control NIK stabilization.
NIK is normally maintained absent or at a low level by a TRAF2, TRAF3, and cIAP1/2 complex that ubiquitinates NIK causing its proteasomal degradation (Vallabhapurapu et al., 2008). Ligation of specific receptors results in TRAF3 degradation and NIK accumulation (He et al., 2007; Vallabhapurapu et al., 2008; Zarnegar et al., 2008). Surprisingly, NIK levels were stabilized by RP3 without a reduction in TRAF3 leading us to hypothesize that RP3 interferes with the normal NIK regulatory machinery. However, we did not detect any interactions between RP3 and either TRAF2 or TRAF3 and RP3 did not bind NIK, NEMO or IKKα (Figure S4). One possibility is that RP3 phosphorylates an unknown tyrosine residue or motif on one of the key regulatory proteins, and this “disconnects” NIK from the negative influence of this apparatus. Alternatively, RP3 may target an as yet unknown component of this TRAF2/3 complex or may aberrantly activate a normally unrelated protein that blocks the negative regulation of NIK expression. Recently, Razani and colleagues (Razani et al., 2010) reported that IKKα plays a critical role in downregulating NIK levels following noncanonical NF-κB pathway activation and these workers hypothesized that dysregulation of this mechanism may contribute to aberrant NIK expression in disease. It is therefore intriguing to consider that although RP3 activates classical NF-κB via IKKα, it may also interfere with the negative feedback from IKKα and block its ability to phosphorylate NIK. Determining which, if any, of these scenarios contribute to the ability of RP3 to bypass the normal mechanisms and relieve the negative regulation of NIK will be the subject of intense further work.
Consistent with previous studies, RP3-induced NF-κB activation required the intact signaling function of RP3 as mutating a tyrosine 588 (Y588) in RP3 (Pufnock & Rothstein, 2009; Russell et al., 2003) abrogated NIK stabilization. Phosphorylation of the equivalent residue in RET (Y1062) generates a docking site for adaptors containing SH2 or PTB domains (Wells & Santoro, 2009). This tyrosine phosphorylation leads to downstream signals that activate the RAS/RAF/ERK/MAPK and PI3K/AKT pathways (Hayashi et al., 2000; Lodyga et al., 2009; Santoro et al., 1993; Wells & Santoro, 2009). This same tyrosine residue, Y1062, is also required for RET-induced NF-κB activity (Hayashi et al., 2000). We postulate that constitutive autophosphorylation of Y588 in RP3 engages similar signals that facilitate NIK stabilization by inactivating the TRAF/cIAP complex. One mechanism we explored was the PI3K/AKT pathway; however, although pharmacological inhibition completely blocked RP3-induced AKT phosphorylation, NIK stabilization remained intact (Figure S5). Hence we conclude that RP3-induced NIK stabilization does not involve activation of the PI3K/AKT pathway. Although further work is clearly required, our findings strongly suggest that crosstalk between signals elicited at Y588 and the NIK regulatory machinery, mediates aberrant classical NF-κB activation.
In addition to their role in cancer, RET/PTCs occur in autoimmune thyroiditis with no apparent neoplasia (Muzza et al., 2009). Consequently, RET/PTC signaling might regulate immune cell infiltration in diseases including Hashimoto’s thyroiditis, a hallmark of which is formation of lymphoid follicles in the thyroid. As noncanonical NF-κB signaling regulates lymphoid organogenesis (Hayden & Ghosh, 2008), it is tempting to speculate that RP3-induced NIK stabilization induces this noncanonical lymphoid organogenesis gene program. However, RP3 expression did not activate noncanonical NF-κB signaling or gene expression in MEFs suggesting that the precise cellular context in which NIK is aberrantly expressed might influence noncanonical NF-κB activation. Further investigation to determine whether NIK expression correlates with thyroiditis is required; however, our data support the hypothesis that RP3-induced NIK stabilization plays a role in thyroid malignancy and possibly in the development of autoimmune thyroiditis.
In conclusion, we show that the RP3 stabilizes NIK and activates classical NF-κB via NEMO and IKKα. As NF-κB activation mediates the oncogenic and pro-inflammatory function of RP3 (Russell et al., 2003; Pufnock & Rothstein, 2009; Pacifico & Leonardi, 2010), our demonstration that NIK is absolutely required for RP3-induced NF-κB activation strongly suggests that NIK stabilization regulates these effects of RP3. Further intense studies will be required to address this in detail; however, our findings provide the rational basis for pursuing strategies to block aberrant NIK expression and NF-κB activation in thyroid cancer and in the highly prevalent autoimmune disease Hashimoto’s thyroiditis. As the NBD peptide blocks RP3-induced NF-κB, targeting NEMO is a potentially important therapeutic approach. However, this might ablate normal classical NF-κB activity, and consequently, targeting NIK expression might be a more specific strategy. Identifying the precise mechanisms through which RP3 disconnects NIK from its regulatory complex will be crucial in this regard. Nevertheless, our findings reveal that an oncogene can bypass the negative regulatory signals controlling NIK expression resulting in stabilization of this critical signaling molecule.
Materials and Methods
Antibodies
Anti-RET, IKKα, p50, NIK, TRAF3, TRAF2, RelB, and NEMO were from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-tubulin was from Sigma-Aldrich (St. Louis, MO). Anti-IKKβ, phospho IKKα/β, p65, phospho-p65, IκBα, phospho-IκBα, histone H3, p100, and phospho-p100 were from Cell Signaling Technology (Beverly, MA). Secondary antibodies were from Jackson ImmunoResearch Laboratories, (West Grove, PA). Anti-Lymphotoxin-β Receptor was from BD Biosciences (San Jose, CA).
Cells and Tissue Culture
Wild Type (WT), IKKα−/−, and IKKβ−/− MEFs were from Dr. Inder Verma (The Salk Institute) and NEMO−/− cells were from Dr. Michael Karin (UCSD School of Medicine). NIK −/− MEFs were a gift from Amgen Inc, (Thousand Oaks, CA). All cells were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 10% FBS, 2mM L-Glutamine, Penicillin (50 units/ml) and Streptomycin (50 μg/ml).
Generation of Stable Cell Lines
Cloning was performed using cloned Pfu DNA polymerase (Stratagene, La Jolla, CA). cDNA for RET/PTC 3 (Pufnock & Rothstein, 2009; Russell et al., 2003) was subcloned into the retroviral vector MIGR1 (from Dr. Warren Pear, University of Pennsylvania). Plasmids were transiently transfected into Plat-E cells using Fugene 6 (Roche Applied Science, Indianapolis, IN) and medium was harvested 48hrs later. Cells were transduced and sorted as previously described (Solt et al., 2007).
Transfections and Luciferase Reporter Assays
Cells were transfected and NF-κB dual luciferase assays (Promega Corporation, Madison, WI) were preformed as previously described (Solt et al., 2007). Dominant Negative (DN) NIK was cloned from human cDNA and point mutations that render the kinase inactive (Xiao et al., 2001) were inserted by site-directed mutagenesis.
Immunoblotting
Lysis and immunoblotting was performed as described before (Solt et al., 2007; Wharry et al., 2009).
Electrophoretic Mobility Shift Assays (EMSAs)
Nuclear extracts were generated and EMSAs using consensus oligonucleotide probes to detect NF-κB and Oct-1, and supershifts using anti-NF-κB specific antibodies were performed as previously described (Solt et al., 2007).
mRNA isolation and quantitative Real Time PCR
All mRNA isolation and quantitative Real Time PCR analyses were performed as described previously (Solt et al., 2007; Wharry et al., 2009). Details of the primers used are provided in the Supplementary Information.
NBD Peptides
The sequences and application of the wild-type and mutant NBD peptides were described previously (May et al., 2000). Peptides were obtained from the Keck Foundation Biotechnology Resource Laboratory at Yale University (New Haven, CT).
Immunohistochemistry
PTC tissue blocks were provided by Dr. Y. Lv (Department of Pathology, Capital University of Medical Sciences, Beijing, China). Fresh-cut 5-μm sections from four separate PTC samples were used for immunohistochemical staining as described previously (Nellore et al., 2009).
Supplementary Material
Acknowledgments
This work was supported by grants from the National Institutes of Health (RO1HL080612, R21CA135602 and T32CA009683, T32GM07229) and the Mari Lowe Center for Comparative Oncology.
Footnotes
Conflict of Interest
The authors declare no conflict or competing financial interest in this work.
Supplementary Information
Supplementary Information is available at Oncogene’s website.
References
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, et al. Frequent engagement of the classical and alternative NF-κB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell. 2007;12:115–130. doi: 10.1016/j.ccr.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basseres DS, Baldwin AS. NF-κB and IκB kinase pathways in oncogenic initiation and progression. Oncogene. 2006;25:6817–6830. doi: 10.1038/sj.onc.1209942. [DOI] [PubMed] [Google Scholar]
- Birbach A, Bailey ST, Ghosh S, Schmid JA. Cytosolic, nuclear and nucleolar localization signals determine subcellular distribution and activity of the NF-κB inducing kinase NIK. J Cell Sci. 2004;117:3615–3624. doi: 10.1242/jcs.01224. [DOI] [PubMed] [Google Scholar]
- Borrello MG, Alberti L, Fischer A, Degl’innocenti D, Ferrario C, Gariboldi M, et al. Induction of a proinflammatory program in normal human thyrocytes by the RET/PTC1 oncogene. Proc Natl Acad Sci U S A. 2005;102:14825–14830. doi: 10.1073/pnas.0503039102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunone G, Uggeri M, Mondellini P, Pierotti MA, Bongarzone I. RET receptor expression in thyroid follicular epithelial cell-derived tumors. Cancer Res. 2000;60:2845–2849. [PubMed] [Google Scholar]
- Ciampi R, Nikiforov YE. RET/PTC rearrangements and BRAF mutations in thyroid tumorigenesis. Endocrinology. 2007;148:936–941. doi: 10.1210/en.2006-0921. [DOI] [PubMed] [Google Scholar]
- Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-β receptor induces different patterns of gene expression via two NF-κB pathways. Immunity. 2002;17:525–535. doi: 10.1016/s1074-7613(02)00423-5. [DOI] [PubMed] [Google Scholar]
- Derudder E, Dejardin E, Pritchard LL, Green DR, Korner M, Baud V. RelB/p50 dimers are differentially regulated by tumor necrosis factor-α and lymphotoxin-β receptor activation: critical roles for p100. J Biol Chem. 2003;278:23278–23284. doi: 10.1074/jbc.M300106200. [DOI] [PubMed] [Google Scholar]
- Eisenlohr LC, Rothstein JL. Oncogenic inflammation and autoimmune disease. Autoimmun Rev. 2006;6:107–114. doi: 10.1016/j.autrev.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Gilmore TD, Herscovitch M. Inhibitors of NF-κB signaling: 785 and counting. Oncogene. 2006;25:6887–6899. doi: 10.1038/sj.onc.1209982. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kawai K, et al. Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene. 2000;19:4469–4475. doi: 10.1038/sj.onc.1203799. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-κB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- He JQ, Saha SK, Kang JR, Zarnegar B, Cheng G. Specificity of TRAF3 in its negative regulation of the noncanonical NF-κB pathway. J Biol Chem. 2007;282:3688–3694. doi: 10.1074/jbc.M610271200. [DOI] [PubMed] [Google Scholar]
- Karin M. NF-κB in cancer development and progression. Nature. 2006;441:431–436. doi: 10.1038/nature04870. [DOI] [PubMed] [Google Scholar]
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng WJ, et al. Promiscuous mutations activate the noncanonical NF-κB pathway in multiple myeloma. Cancer Cell. 2007;12:131–144. doi: 10.1016/j.ccr.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam LT, Davis RE, Ngo VN, Lenz G, Wright G, Xu W, et al. Compensatory IKKα activation of classical NF-κB signaling during IKKβ inhibition identified by an RNA interference sensitization screen. Proc Natl Acad Sci U S A. 2008;105:20798–20803. doi: 10.1073/pnas.0806491106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodyga M, De Falco V, Bai XH, Kapus A, Melillo RM, Santoro M, et al. XB130, a tissue-specific adaptor protein that couples the RET/PTC oncogenic kinase to PI 3-kinase pathway. Oncogene. 2009;28:937–949. doi: 10.1038/onc.2008.447. [DOI] [PubMed] [Google Scholar]
- Ludwig L, Kessler H, Wagner M, Hoang-Vu C, Dralle H, Adler G, et al. NF-κB is constitutively active in C-cell carcinoma and required for RET-induced transformation. Cancer Res. 2001;61:4526–4535. [PubMed] [Google Scholar]
- Madge LA, Kluger MS, Orange JS, May MJ. Lymphotoxin-α1β2 and LIGHT induce classical and noncanonical NF-κB-dependent proinflammatory gene expression in vascular endothelial cells. J Immunol. 2008;180:3467–3477. doi: 10.4049/jimmunol.180.5.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May MJ, D’Acquisto F, Madge LA, Glockner J, Pober JS, Ghosh S. Selective inhibition of NF-κB activation by a peptide that blocks the interaction of NEMO with the IκB kinase complex. Science. 2000;289:1550–1554. doi: 10.1126/science.289.5484.1550. [DOI] [PubMed] [Google Scholar]
- Monaco C, Visconti R, Barone MV, Pierantoni GM, Berlingieri MT, De Lorenzo C, et al. The RFG oligomerization domain mediates kinase activation and relocalization of the RET/PTC3 oncoprotein to the plasma membrane. Oncogene. 2001;20:599–608. doi: 10.1038/sj.onc.1204127. [DOI] [PubMed] [Google Scholar]
- Muzza M, Degl’innocenti D, Colombo C, Perrino M, Ravasi E, Rossi S, et al. The tight relationship between papillary thyroid cancer, autoimmunity and inflammation: clinical and molecular studies. Clin Endocrinol (Oxf) 2009 doi: 10.1111/j.1365-2265.2009.03699.x. (In Press) [DOI] [PubMed] [Google Scholar]
- Nellore A, Paziana K, Ma C, Tsygankova OM, Wang Y, Puttaswamy K, et al. Loss of Rap1GAP in papillary thyroid cancer. J Clin Endocrinol Metab. 2009;94:1026–1032. doi: 10.1210/jc.2008-1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nibu K, Otsuki N, Nakao K, Sugasawa M, Rothstein JL. RET/PTC fusion gene rearrangements in Japanese thyroid carcinomas. Eur Arch Otorhinolaryngol. 2005;262:368–373. doi: 10.1007/s00405-004-0835-8. [DOI] [PubMed] [Google Scholar]
- Nishina T, Yamaguchi N, Gohda J, Semba K, Inoue J. NIK is involved in constitutive activation of the alternative NF-κB pathway and proliferation of pancreatic cancer cells. Biochem Biophys Res Commun. 2009;388:96–101. doi: 10.1016/j.bbrc.2009.07.125. [DOI] [PubMed] [Google Scholar]
- Pacifico F, Leonardi A. Role of NF-κB in thyroid cancer. Mol Cell Endocrinol. 2010;321:29–35. doi: 10.1016/j.mce.2009.10.010. [DOI] [PubMed] [Google Scholar]
- Pacifico F, Mauro C, Barone C, Crescenzi E, Mellone S, Monaco M, et al. Oncogenic and anti-apoptotic activity of NF-κB in human thyroid carcinomas. J Biol Chem. 2004;279:54610–54619. doi: 10.1074/jbc.M403492200. [DOI] [PubMed] [Google Scholar]
- Pufnock JS, Rothstein JL. Oncoprotein signaling mediates tumor-specific inflammation and enhances tumor progression. J Immunol. 2009;182:5498–5506. doi: 10.4049/jimmunol.0801284. [DOI] [PubMed] [Google Scholar]
- Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, et al. Negative feedback in noncanonical NF-κB signaling modulates NIK stability through IKKα-mediated phosphorylation. Sci Signal. 2010;3:ra41. doi: 10.1126/scisignal.2000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoden KJ, Unger K, Salvatore G, Yilmaz Y, Vovk V, Chiappetta G, et al. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J Clin Endocrinol Metab. 2006;91:2414–2423. doi: 10.1210/jc.2006-0240. [DOI] [PubMed] [Google Scholar]
- Russell JP, Shinohara S, Melillo RM, Castellone MD, Santoro M, Rothstein JL. Tyrosine kinase oncoprotein, RET/PTC3, induces the secretion of myeloid growth and chemotactic factors. Oncogene. 2003;22:4569–4577. doi: 10.1038/sj.onc.1206759. [DOI] [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Grieco M, Berlingieri MT, Vecchio G, Fusco A. The TRK and RET tyrosine kinase oncogenes cooperate with ras in the neoplastic transformation of a rat thyroid epithelial cell line. Cell Growth Differ. 1993;4:77–84. [PubMed] [Google Scholar]
- Senftleben U, Cao Y, Xiao G, Greten FR, Krahn G, Bonizzi G, et al. Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway. Science. 2001;293:1495–1499. doi: 10.1126/science.1062677. [DOI] [PubMed] [Google Scholar]
- Sheils OM, O’Eary JJ, Uhlmann V, Lattich K, Sweeney EC. RET/PTC-1 Activation in Hashimoto Thyroiditis. Int J Surg Pathol. 2000;8:185–189. doi: 10.1177/106689690000800305. [DOI] [PubMed] [Google Scholar]
- Solt LA, Madge LA, Orange JS, May MJ. Interleukin-1-induced NF-κB activation is NEMO-dependent but does not require IKKβ. J Biol Chem. 2007;282:8724–8733. doi: 10.1074/jbc.M609613200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhapurapu S, Matsuzawa A, Zhang W, Tseng PH, Keats JJ, Wang H, et al. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-κB signaling. Nat Immunol. 2008;9:1364–1370. doi: 10.1038/ni.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veiga-Fernandes H, Coles MC, Foster KE, Patel A, Williams A, Natarajan D, et al. Tyrosine kinase receptor RET is a key regulator of Peyer’s patch organogenesis. Nature. 2007;446:547–551. doi: 10.1038/nature05597. [DOI] [PubMed] [Google Scholar]
- Visconti R, Cerutti J, Battista S, Fedele M, Trapasso F, Zeki K, et al. Expression of the neoplastic phenotype by human thyroid carcinoma cell lines requires NF-κB p65 protein expression. Oncogene. 1997;15:1987–1994. doi: 10.1038/sj.onc.1201373. [DOI] [PubMed] [Google Scholar]
- Wells SA, Jr, Santoro M. Targeting the RET Pathway in Thyroid Cancer. Clin Cancer Res. 2009;15:7119–7123. doi: 10.1158/1078-0432.CCR-08-2742. [DOI] [PubMed] [Google Scholar]
- Wharry CE, Haines KM, Carroll RG, May MJ. Constitutive non-canonical NF-κB signaling in pancreatic cancer cells. Cancer Biol Ther. 2009;8:1567–1576. doi: 10.4161/cbt.8.16.8961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtschafter A, Schmidt R, Rosen D, Kundu N, Santoro M, Fusco A, et al. Expression of the RET/PTC fusion gene as a marker for papillary carcinoma in Hashimoto’s thyroiditis. Laryngoscope. 1997;107:95–100. doi: 10.1097/00005537-199701000-00019. [DOI] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, Sun SC. NF-κB-inducing kinase regulates the processing of NF-κB2 p100. Mol Cell. 2001;7:401–409. doi: 10.1016/s1097-2765(01)00187-3. [DOI] [PubMed] [Google Scholar]
- Zarnegar B, Yamazaki S, He JQ, Cheng G. Control of canonical NF-κB activation through the NIK-IKK complex pathway. Proc Natl Acad Sci U S A. 2008;105:3503–3508. doi: 10.1073/pnas.0707959105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.