

Abstract
We investigate the effects of two structurally similar small cyclic molecules: salicylic acid and perillic acid on a zwitterionic model lipid bilayer, and show that both molecules might have biological activity related to membrane thinning. Salicylic acid is a nonsteroidal antiinflammatory drug, some of the pharmacological properties of which arise from its interaction with the lipid bilayer component of the plasma membrane. Prior simulations show that salicylate orders zwitterionic lipid membranes. However, this is in conflict with Raman scattering and vesicle fluctuation analysis data, which suggest the opposite. We show using extensive molecular dynamics simulations, cumulatively >2.5 μs, that salicylic acid indeed disorders membranes with concomitant membrane thinning and that the conflict arose because prior simulations suffered from artifacts related to the sodium-ion induced condensation of zwitterionic lipids modeled by the Berger force field. Perillic acid is a terpenoid plant extract that has antiinfective and anticancer properties, and is extensively used in eastern medicine. We found that perillic acid causes large-scale membrane thinning and could therefore exert its antimicrobial properties via a membrane-lytic mechanism reminiscent of antimicrobial peptides. Being more amphipathic, perillic acid is more potent in disrupting lipid headgroup packing, and significantly modifies headgroup dipole orientation. Like salicylate, the membrane thinning effect of perillic acid is masked by the presence of sodium ions. As an alternative to sodium cations, we advocate the straightforward solution of using larger countercations like potassium or tetra-methyl-ammonium that will maintain electroneutrality but not interact strongly with, and thus not condense, the lipid bilayer.
Introduction
Nonsteroidal antiinflammatory drugs (NSAIDs) like salicylate (SAL) exert their analgesic action by inhibition of cyclooxygenase, the enzyme responsible for the production of prostaglandin (1). However, several other effects of SAL, such as inhibition of sulfate transport (2), the genesis of potentially fatal gastric lesions (3), and temporary hearing loss (4), may be attributed to direct interactions with the lipid bilayer component of the cellular plasma membrane (5). Additionally, the study of the effect of drugs on membrane properties is gaining significance as more evidence accumulates that the function of transmembrane proteins such as the GABA receptor can be influenced by membrane elasticity (6). Several investigations have therefore explored the interactions of various NSAIDs with bilayers composed of either model or natural lipid mixtures. In most cases, it was found that NSAIDs in general (7), and SAL in particular (8–11), increased the fluidity of model membranes which manifested as, for example, in a lowering of the area compressibility modulus of the membrane in giant vesicles (8) or a lowering and broadening of main phase transition of lipids in calorimetric measurements (12). For SAL, confocal Raman microscopy experiments suggested that SAL decreased lipid tail order parameters (11), with a concomitant increase of area per molecule in surface-pressure area isotherms (11).
Molecular dynamics (MD) simulations are now routinely used for molecular-level analysis of investigations of the dynamic, electrical, structural, and mechanical properties of model membranes, and processes occurring close to the membrane such as transport across ion channels, conformational analysis of membrane-associated proteins, and the binding of small molecules and peptides to membranes. The interactions of SAL with a (DMPC) 1,2-dimyristoyl-sn-glycero-3-phosphocholine lipid bilayer were investigated using extensive and careful MD simulations in 2005 (13). Despite the sufficiently long time- and length-scales of the simulations, there was disagreement with the Raman microscopy and monolayer experiments (11). Simulations showed that SAL ordered DMPC acyl tails and slightly decreased the area per lipid.
Composite drug-membrane or peptide-membrane systems are often charged, and it is a common practice in MD simulations to use monovalent counterions to keep the simulation cell electrostatically neutral or to add monovalent salt in the form of NaCl (typically 150 mM) in the aqueous phase to mimic ionic conditions near living cell membranes. The simulations published by Song et al. (13) used the Berger force field for lipids along with the Na+ ion as a counterion. Long-time simulations (>>20 ns) of ions with both anionic and zwitterionic lipids using the Berger force field show that sodium ions condense lipid bilayers resulting in an increase in the bilayer thickness and lipid tail order parameters, and a decreased area per lipid molecule as a result of strong binding of Na+ to the glycerol backbone of the phospholipids (14–20). In an effort to reconcile MD simulations with available experimental data, we ran extensive SAL-DMPC simulations with the larger potassium (K+) ion, which did not bind to the bilayer interface as strongly as Na+. Use of the larger cation enabled us to distinguish the effect of the cation from that of the drug molecule. Indeed, our results are in excellent agreement with experiments with regard to the interactions of SAL with a zwitterionic bilayer.
The interactions of ions with lipid bilayers can affect structure, dynamics, and stability of the membrane as well as membrane-associated processes like the membrane potential, insertion of proteins in the membrane, membrane fusion and transport across membranes (14,15). Mostly computer simulations have addressed the interactions between monovalent cations and the lipid bilayer, and there have been only occasional experimental measurements. It is important to accurately model ion-membrane interactions in such systems so that the bilayer properties are reproduced accurately, to quantify the impact of the small molecule on the bilayer. A detailed description of ion-lipid interactions is beyond the scope of this article.
We also ran simulations of deprotonated perillic acid (DPAC) (21): a terpenoid plant extract and a metabolite of the extract from the Perilla frutescens plant species, which is extensively used in food and medicine in Eastern Asia (22). The structure of DPAC is similar to SAL in many respects (Fig. 1). DPAC has a wide range of useful and remarkably diverse clinical properties, which, like SAL, might manifest themselves via interactions with the lipid bilayer. It was therefore of interest to investigate the effects of DPAC on a lipid bilayer. The ordering effects of sodium also mask the membrane-perturbing effects of DPAC. The masking effect of sodium was corrected by the use of a larger tetra methyl ammonium (TMA) ion, which, like K+, did not penetrate into the bilayer. Thus, the two case studies of SAL and DPAC suggest that the presence of Na+ can bias the results of MD simulations of lipid bilayers. It was important to investigate both K+ and TMA+ because it is possible that one of the two may be unsuitable for use in setting up a simulation for specific applications. There was no particular reason of choosing K+ with the SAL simulations and TMA+ with the DPAC simulations. We also ran simulations of the pure DMPC bilayer in the presence of different types of ions.
Figure 1.
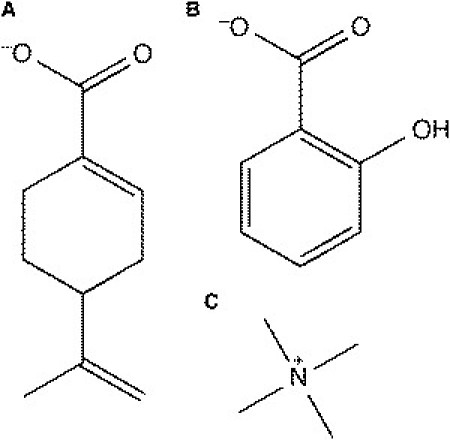
Molecules of interest. (A) Deprotonated perillic acid (DPAC), a cyclic terpene. (B) Salicylate (SAL), an anionic nonsteroidal antiinflammatory drug. (C) Tetra-methyl-ammonium (TMA+) cation.
In the following sections, we describe in detail the interactions between SAL and DPAC and a DMPC lipid bilayer, and show how the molecules could exert their diverse cytotoxic and clinical effects by perturbing the integrity of the lipid bilayer. We also suggest the use of larger countercations to negate the possible artifacts of using Na+.
Methods
Four simulations of a pure DMPC bilayer containing 128 lipids in the presence of pure water, 32 molecules of NaCl, KCl, and TMA-Cl, corresponding to a concentration of ∼180 mM salt, were implemented. Simulations of a larger DMPC bilayer containing 512 lipids without ions were also carried out. A full list of simulations implemented is provided in Table 1.
Table 1.
Overview of simulations performed
| Bilayer | Number of drug molecules | Counterion | Simulation time (ns) | Name |
|---|---|---|---|---|
| Pure DMPC bilayer | ||||
| DMPC | — | None | 220 | DMPC |
| DMPC-BIG | — | None | 220 | |
| DMPC | — | 32 Na+ Cl– | 220 | DMPC-Na+ |
| DMPC | — | 32 K+ Cl– | 220 | |
| DMPC | — | 32 TMA+ Cl– | 220 | |
| DMPC + SAL | ||||
| DMPC | 10, 20 | K+ | 220 | K10, K20 |
| DMPC | 10, 20 | Na+ | 220 | Na10, Na20 |
| DMPC-BIG | 20 | K+ | 160 | BIG-K20 |
| DMPC-BIG | 20 | Na+ | 160 | BIG-Na20 |
| DMPC + DPAC | ||||
| DMPC | 32 | Na+ | 220 | DMPC-Na+ |
| DMPC | 32 | K+ | 220 | DMPC-K+ |
| DMPC | 32 | TMA+ | 220 | DMPC-TMA+ |
The DMPC bilayer has 128 lipids, whereas the DMPC-BIG bilayer has 512 lipids.
Simulations of SAL with a DMPC bilayer at two different concentrations (50 mM and 100 mM) were carried out. The 50 mM systems contained 10 SAL molecules and 10 counterions, whereas the 100 mM systems contained 20 SAL molecules and 20 counterions. These systems will be labeled K10, K20, Na10, and Na20 from now on, with the letters denoting the ion and the trailing number denoting the number of SAL molecules. With 100 mM SAL, simulations were also implemented with a larger lipid bilayer containing 512 lipids. These systems will be called BIG-K20 and BIG-Na20.
Simulations of DPAC with a DMPC bilayer with TMA counterions were carried out at a range of concentrations, with the DPAC/DMPC ratio ranging from 1:128 to 1:4. Simulations at the highest concentrations were implemented with Na+ and K+ counterions as well. In this article, the three simulations with the highest DPAC concentrations will be discussed.
Force-field parameters
The modified Berger force field (23), with parameters adapted from http://moose.bio.ucalgary.ca/ was used for DMPC. Water was represented by a simple point charge (SPC) water model (24). Force-field parameters for SAL used in Song et al. (13) were used, and were obtained via a personal communication from Dr. Nathan A. Baker, Pacific Northwest National Laboratory, 2010. The parameters for DPAC were obtained as described previously (25). In short, the structure was geometry optimized using Gaussian03 (26) at the B3LYP/6-311++G∗∗ level of theory. The electrostatic potentials were sampled for DPAC at a number of points at the HF/6-31G∗ level of theory and the atomic partial charges were calculated using an electrostatic potential fit. The optimized structure had its nonpolar hydrogen atoms combined to the attached carbon atoms to create the united-atoms structure. The atoms were assigned atom types from the corrected GROMOS-87 force field whenever possible. The terminal methylene groups were assigned the atom type DH2, based on prior simulations of isoprene (27). The methine group of the cyclohexene group was given the new atom type CDB, the LJ parameters of which were taken from the CR61 atom type (carbon in six-membered aromatic ring) of the corrected GROMOS-87 force field. LJ parameters for all other atom types were from the corrected GROMOS-87 or Berger force fields. The bonded force-field parameters were obtained from our quantum mechanics calculations, GROMOS-87, and Berger force fields, from Siwko et al. (27) or from analogous atom types. The topology and force-field parameters for TMA were adapted from the parameters of the choline group in the Berger force field.
System construction
The pure DMPC bilayer contained 64 lipids in each leaflet. The bilayer had been preequilibrated and simulated for ∼100 ns. The initial rectangular box sizes were 64.705 Å × 64.998 Å × 107.560 Å. To construct each of the initial configurations with terpenes, SAL and DPAC molecules were placed at a random position in the aqueous phase surrounding the lipid bilayer in a random orientation, and water molecules within a 1.0 Å distance were deleted.
Simulation procedure
All simulations were performed with the GROMACS package version 4.0.4 or 4.0.5 (28–31). An energy minimization using the steepest descent method with all bonds constrained was performed until the maximum force experienced by the system was below 500 kJ mol−1 nm−1. After successful energy minimization, the systems were heated during 10 ps of MD by assigning random velocities to the particles according to a Maxwell distribution at 310 K. Other than the velocity generation, the parameters used for heating were identical to the parameters used for the production run. For the production run, the leap-frog integrator (32) was used with a time step of 2 fs. All bond lengths were constrained using the LINCS algorithm (33,34), and water molecules were constrained with SETTLE (35). Periodic boundary conditions were applied in all directions. A neighbor list with a 10 Å cutoff was used for nonbonded interactions and was updated every 20 fs. The van der Waals interactions were truncated with a cutoff of 10.0 Å and the electrostatic interactions were treated with the particle-mesh Ewald method using default parameters (36,37). The center of mass translation was removed at every step of the simulation.
The NPT statistical ensemble was used. Temperature coupling was performed using the Berendsen thermostat (38) separately for the lipids and the rest of the system with a reference temperature of 310 K, and a time constant of 0.1 ps for both subgroups. A semiisotropic pressure coupling was applied using the Berendsen barostat (38) with a coupling constant of 0.1 ps and a reference pressure of 1.0 bar in all directions. Because the compressibility, κ, for the systems was not known, the value for pure water, 4.5 × 10−5 bar−1, was used. Trajectories were sampled every 10 ps. For calculation of ensemble-averaged properties, the first 100 ns of each simulation were discarded. The analysis was carried out using GROMACS or custom-made programs. Visualization and snapshots were rendered using VMD (39).
Results
We will start this section by describing the effects of SAL and DPAC on a DMPC bilayer, and then show how counterions impact the conclusions drawn from the simulations. In the end, we present some results from the drug-free bilayers in the presence of different salts.
Interactions of SAL with DMPC
The results of the simulations of the larger bilayer patch (512 lipids) and the smaller bilayer patches (128 lipids) were identical in all respects. Thus, there is no finite size effect in the current set of simulations and we present results only from the simulations of the smaller patches.
As seen in prior MD simulations with DPPC (1,2-dipalmitoyl-sn-glycero-3-phosphocholine) (13), all SAL molecules bound to the lipid headgroup region. Fig. 2 shows the density of the phosphate group of DMPC and SAL for the K20 and Na20 simulations. The SAL molecules are all located at the lipid-water interface. The bilayer thickness was calculated from the distance between the two maxima in the electron density profiles of the lipids. The phosphate peaks in K20 are slightly wider than those in DMPC, suggesting slightly higher fluctuations of the phosphate groups along the bilayer normal. Thus, insertion of SAL resulted in an overall decrease in the bilayer thickness in the presence of the larger K+ ion. Notice that the phosphate/phosphate thickness is higher in Na20. The distribution of SAL is narrower in Na20, because the bilayer is condensed and fluctuations of the headgroups along the bilayer normal are lower in the presence of Na+, as apparent from the wider distribution of the phosphate peaks in K20.
Figure 2.
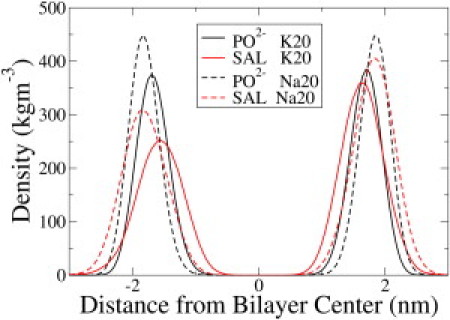
Density profiles of SAL and the phosphate (PO2–) group of the lipids from the K20 and Na20 simulations. The distribution of SAL is not symmetric about the origin because a different number of SAL molecules were placed on the two sides of the bilayer at the beginning of the simulation. The presence of SAL resulted in only a slight thinning of the membrane compared to pure DMPC (not shown).
The projected area per lipid (AL) of the bilayer was obtained by dividing the area of the simulation box in the membrane plane by the number of lipids in one leaflet. The (AL) increased from 64.5 ± 0.39 Å2 in pure DMPC to 66.24 ± 1.02 Å2 in K20. The presence of Na+ in the Na10 and Na20 simulations, however, suppressed this thinning effect, and the bilayer appears thicker (Fig. 2). AL decreased to 60.36 ± 0.33 in Na20. The hydroxyl and carboxyl moieties of SAL interacted preferentially with the choline group of DMPC (Fig. 3). There were no other specific interactions between SAL and DMPC. Fig. S1 in the Supporting Material shows the radial distribution functions drawn between the cation and the SAL molecules in the K20 and Na20 simulations. More potassium ions are associated with SAL in K20 than Na+ in Na20. Although K+ ions by themselves do not associate with the DMPC bilayer (see Fig. 6 later), the presence of anionic SAL molecules pulls K+ from the aqueous phase to the lipid-water interface, where K+ and SAL molecules form ion pairs. A similar effect is not possible for Na+ because Na+ ions remain bound to the glycerol backbone of the lipids, although the small peak at r = 2.2 Å in Fig. 3 suggests occasional Na+-SAL interactions.
Figure 3.
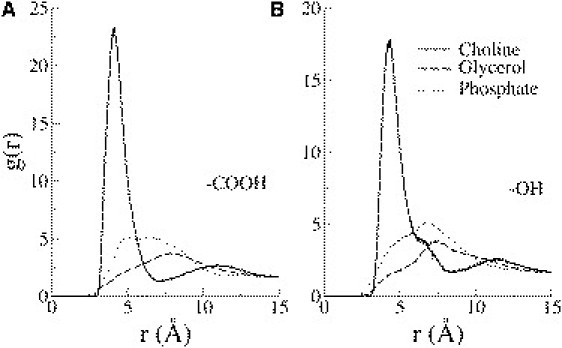
Radial pair correlation functions between the SAL carboxyl (A) and hydroxyl (B) groups with the various lipid headgroup moieties from the K20 simulations. The salicylate functional group interacted closely with the positively charged choline moiety on the lipids. No specific interactions with the phosphate or the glycerol group were apparent.
Figure 6.
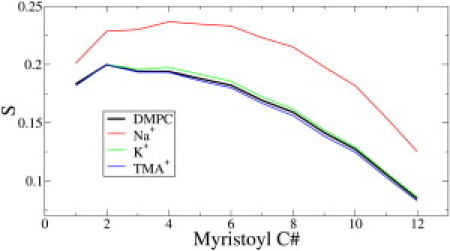
Lipid tail order parameters for the sn-1 acyl chain of DMPC in the presence of different ions. Na+, which binds deepest in the bilayer, significantly increases the order of the tail, while TMA+ and K+ have no effect. The ion concentration is ∼180 mM.
The orientational order parameters of the hydrocarbon tails of the lipids were used to quantify the fluidity of the membrane. The order parameter for a given vector is defined by
where θz is the angle between the vector and the z axis of the simulation box. The brackets imply averaging over time and molecules. An order parameter of 1 indicates a bond perfectly aligned with the bilayer normal, an order parameter of zero indicates completely random bond directions, and an order parameter of −1/2 indicates a bond perpendicular to the bilayer normal. The lipid tail order parameters shown in Fig. 4 depict the thinning effect of SAL on the DMPC bilayer in the presence of K+, in agreement with Raman scattering experiments (11). Once again, however, when Na+ counterions are used, the strong condensing effect of Na+ masks the disordering effect of SAL (Fig. 4).
Figure 4.
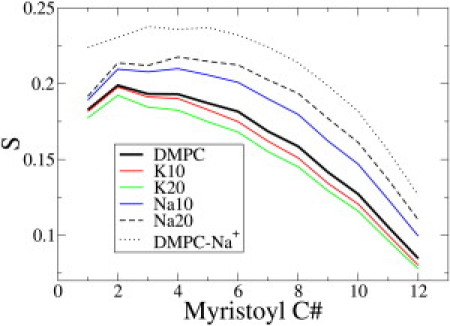
Lipid tail order parameters for the sn-1 acyl chain of DMPC in presence of 50 mM and 100 mM SAL. When K+ is used as a counterion, lipid tails get disordered, in excellent agreement with experiments (8). However, when Na+ is present as a counterion, the bilayer appears more ordered, as seen in prior simulations (13).
Effect of DPAC on DMPC
Three DPAC simulations were implemented with Na+, K+ or TMA+ as positive counterions. The results from the DPAC-K+ simulation are similar to those from the DPAC-TMA+ simulations. We will discuss only the DPAC-Na+ and DPAC-TMA+ simulations for brevity. Being anionic, DPAC binds to the DMPC headgroup near the positively charged choline region irrespective of the choice of counterion. The radial distribution functions shown in Fig. 5 D show that the carboxyl group of DPAC interacts with the positively charged choline group. The insertion of DPAC into the headgroup region thins the bilayer significantly, as apparent from the increase in the projected area per lipid in the DPAC-TMA+ simulation (Fig. 5 B), the decreased bilayer thickness (Fig. 5 A), and the decrease in the lipid tail order parameters (Fig. 5 C). It can be safely concluded that DPAC is responsible for whatever disorder is induced in the bilayer because the effect of TMA+ on the lipid tail order parameters is negligible (see Fig. 6). However, the strong ordering effect of Na+ hides whatever effect DPAC exerts on the order of the membrane. Thus, in the presence of Na+ counterions, it appears that DPAC actually orders the lipid tails and thickens the bilayer (Fig. 4 C).
Figure 5.
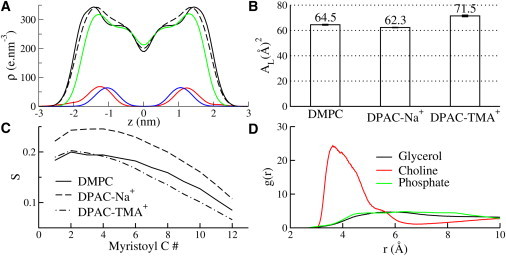
Results from the DPAC-DMPC-Na+ and DPAC-DMPC-TMA+ simulations. (A) Electron density profiles for DMPC and for DPAC. The legends for the curves are as follows: (Dashed black) DMPC without ions or DPAC; (solid black) DMPC in DMPC-DPAC-Na+; (green) DMPC in DMPC-DPAC-TMA+; (red) DPAC in DMPC-DPAC-Na+; and (blue) DPAC in DMPC-DPAC-TMA+. DPAC partitions into the interface in the presence of both Na+ and TMA counterions (red and blue curves). The bilayer is thickened in the presence of Na+ (black) but thinned in the presence of TMA+ (green). (B) Projected area per lipid (AL). A large increase AL in the presence of TMA+ suggests that DPAC thins the bilayer. However, the effect is completely masked in the presence of Na+. (C) Order parameters for the sn-1 acyl tail of DMPC show that DPAC disorders the tail part of the lipids, but Na+ conceals the disordering effect. (D) Radial distribution functions between the carboxyl functional group of DPAC and lipid moieties. The anionic carboxyl group interacts with the positively charged choline group.
Properties of DMPC in presence of different ions
The area per lipid and the thickness of the DMPC bilayer in the presence of different ions is shown in Table 2. The larger countercations K+ and TMA+ elicited no change in the structural properties of the bilayer because they do not bind to the bilayer interface, in agreement with prior simulations of K+ with zwitterionic lipid bilayers (18). The smaller sodium cation, on the other hand, binds to the lipid glycerol backbone and condenses the bilayer by ∼10%, similar to previously implemented simulations (14,18,40). The order parameters (S) for the sn-1 acyl chain of DMPC are shown in Fig. 6.
Table 2.
Bilayer thickness for the DMPC bilayer in pure water and with ions
| Ions | Pure water | NaCl | KCl | TMACl |
|---|---|---|---|---|
| Thickness (Å) | 33.78 (0.26) | 36.09 (0.53) | 33.89 (0.11) | 33.78 (0.08) |
| Area/lipid (Å2) | 64.50 (0.39) | 58.18 (0.20) | 64.44 (0.50) | 64.72 (0.15) |
Errors calculated from block averages are given in parentheses.
For pure DMPC, the order parameter values and overall appearance of the curves are in agreement with both experiment (41,42) and prior MD simulations (43–45). Na+ ions order the lipid tails significantly, while the larger K+ and TMA+ cations have no detectable influence on the membrane fluidity. The electron density profiles for the cations in Fig. 7 show that the Na+ ions were predominantly positioned at the membrane interface while the TMA+ and K+ ions were located away from the interface. The strongly bound Na+ ions also create a double layer of ions by attracting Cl– ions to the interface. The radial distribution functions between the ions and various lipid headgroup moieties (Fig. S2) show that the Na+ ions preferentially bind to the lipid glycerol region, while the Cl– ions localize near the choline groups.
Figure 7.
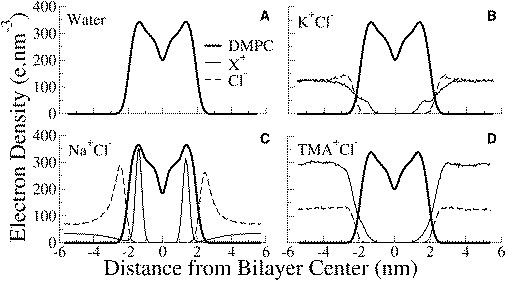
Electron density profiles of components of DMPC lipids and ions in the DMPC simulations. X+ represents the cation in each curve. DMPC is shown as a thick solid line, the cations are shown with a solid line, and a dashed line shows the anion Cl–. Na+ ions bind deep into the headgroup region and also attract Cl– ions from the aqueous phase to form a double layer. TMA+ and K+ ions remain in the aqueous phase.
Discussion
Investigation of the interactions of drugs with membranes is acquiring significance as more evidence accumulates that several drugs, anesthetics and biologically active plant extracts might exert their multifarious clinical effects via modulation of the lipid bilayer component of the cellular plasma membrane. Additionally, such studies are also useful in pharmacology where it is important to quantify the adsorption of drugs into membranes. In this article, we have quantified the interactions of the NSAID salicylate and the terpenoid plant extract perillic acid with a DMPC lipid bilayer. Using a correct choice of counterions in the simulation dispenses with a long-standing conflict between experiments and MD simulations of SAL. The effect on the bilayer induced by the cyclic terpene DPAC, similar to SAL in having a six-membered ring and a carboxyl functional group, was also described in the presence of different cations.
There are almost no experimental studies of DPAC-lipid interactions, but SAL-membrane interactions have been studied extensively because SAL has been in the clinic as an NSAID, and it is possible that some of SAL's biological activity might arise from interactions with the membrane. SAL was shown to disorder membranes isolated from rat kidneys as well as model membranes in the presence of phosphate buffer, which typically contains 150 mM concentration of Na+ (2). Disordering of membranes might lead to inhibition of sulfate transport. A quantity of 100 mM SAL also reduced vesicle membrane viscosity (46) in the presence of HEPES buffer containing 149 mM NaCl and 5 mM KOH and reduced the order of DMPC vesicles at similar concentrations. However, prior MD simulations implemented at similar ion concentrations predicted exactly the opposite, and an ordering of the DMPC bilayer was observed (13). Although the simulations were long enough and were performed carefully, presence of the Na+ counterion skewed the results and masked the disordering effect of SAL on the bilayer. Physiological concentrations of Na+ (150 mM) were present in most experimental investigations of SAL with lipids, suggesting that the Na+ counterion was overestimating membrane condensation in prior simulations (13).
DPAC was much more disruptive to the lipid bilayer compared to SAL, as reflected in the greater extent of membrane thinning. The difference probably arises from the higher amphipathicity of DPAC and the presence of an extra isopropenyl moiety at a position 3′ from the carboxyl group, which increases the penetration of DPAC in the hydrophobic region of the bilayer. Like in the case of SAL, the membrane-disruptive effect of DPAC is also masked by Na+ ions. The remainder of this section is devoted to a short comment on the choice of positive counterions in simulations that employ the Berger force field for lipids.
Counterions are required in simulations to keep systems electrostatically neutral for accurate calculation of electrostatic forces using particle-mesh Ewald. In addition, the inclusion of electrolytic ions may be desirable to mimic biological conditions more closely. Like prior studies, our simulations show that Na+ ions condensed the lipid bilayer and interacted with the ester oxygen atoms, causing some structural rearrangements in the headgroup region of the bilayer. The binding of Na+ ions had a strong ordering effect on the zwitterionic lipid bilayers that resulted in a lower area per lipid, higher acyl chain order parameters and an increased bilayer thickness. These results are fully in agreement with the published literature (17,47,48). Whether the condensing effect of the Na+ is a genuine property of the DMPC lipid bilayers or a force-field artifact is debatable. In any case, the use of Na+ as counterions might mask whatever effect the molecule under investigation (in this case, DPAC and SAL) has on the membrane. From a different perspective, Na+ ions can also be viewed as stabilizing the lipid bilayer and thus protecting it from the disruptive effects of bound drugs.
Since it is not straightforward to distinguish the effects on the properties of the lipid bilayer caused by Na+ ions from those caused by DPAC/SAL, we have, as a possible solution, attempted to use two larger cations, K+ and TMA+, as counterions. Both K+ and TMA+ ions do not mask the effects of the external molecule on the bilayer. Based on the two case studies in this article and prior experience with modeling lipid mixtures (49), we recommend that with nonpolarizable ion force fields, larger countercations and larger electrolytic cations should be used in simulations with the Berger force field for lipids. Use of larger counterions becomes particularly important when investigating the effect of small molecules on the lipid bilayer, and perhaps even when modeling small trans-membrane peptides (50,51), whose orientation will change as the hydrophobic thickness of the bilayer increases in the presence of Na+. Without further experiments, it is difficult to decide whether the condensing effect of Na+ on zwitterionic lipids in the Berger force field is realistic, but it does appear that the effect is overestimated. Whether K+ and TMA+ will introduce other artifacts into the simulation is also unknown, but using the larger cation seems to be the safer bet at the moment, at least until improved force fields or more analytical measurements can resolve the matter.
Acknowledgments
The computations were done at the Danish Center for Scientific Computing.
MEMPHYS-Center for Biomembrane Physics is supported by the Danish National Research Foundation.
Supporting Material
References
- 1.Vane J.R. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs. Nat. New Biol. 1971;231:232–235. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 2.Balasubramanian S.V., Straubinger R.M., Morris M.E. Salicylic acid induces changes in the physical properties of model and native kidney membranes. J. Pharm. Sci. 1997;86:199–204. doi: 10.1021/js9602756. [DOI] [PubMed] [Google Scholar]
- 3.Tomisato W., Tanaka K., Mizushima T. Membrane permeabilization by non-steroidal anti-inflammatory drugs. Biochem. Biophys. Res. Commun. 2004;323:1032–1039. doi: 10.1016/j.bbrc.2004.08.205. [DOI] [PubMed] [Google Scholar]
- 4.Jung T.T.K., Rhee C.K., Choi D.C. Ototoxicity of salicylate, nonsteroidal antiinflammatory drugs, and quinine. Otolaryngol. Clin. North Am. 1993;26:791–810. [PubMed] [Google Scholar]
- 5.Lichtenberger L.M., Wang Z.M., Barreto J.C. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat. Med. 1995;1:154–158. doi: 10.1038/nm0295-154. [DOI] [PubMed] [Google Scholar]
- 6.Søgaard R., Werge T.M., Lundbaek J.A. GABAA receptor function is regulated by lipid bilayer elasticity. Biochemistry. 2006;45:13118–13129. doi: 10.1021/bi060734+. [DOI] [PubMed] [Google Scholar]
- 7.Lichtenberger L.M., Zhou Y., Raphael R.M. NSAID injury to the gastrointestinal tract: evidence that NSAIDs interact with phospholipids to weaken the hydrophobic surface barrier and induce the formation of unstable pores in membranes. J. Pharm. Pharmacol. 2006;58:1421–1428. doi: 10.1211/jpp.58.10.0001. [DOI] [PubMed] [Google Scholar]
- 8.Zhou Y., Raphael R.M. Effect of salicylate on the elasticity, bending stiffness, and strength of SOPC membranes. Biophys. J. 2005;89:1789–1801. doi: 10.1529/biophysj.104.054510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhi M., Ratnanather J.T., Brownell W.E. Hypotonic swelling of salicylate-treated cochlear outer hair cells. Hear. Res. 2007;228:95–104. doi: 10.1016/j.heares.2007.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Greeson J.N., Raphael R.M. Amphipath-induced nanoscale changes in outer hair cell plasma membrane curvature. Biophys. J. 2009;96:510–520. doi: 10.1016/j.bpj.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox C.B., Horton R.A., Harris J.M. Detection of drug-membrane interactions in individual phospholipid vesicles by confocal Raman microscopy. Anal. Chem. 2006;78:4918–4924. doi: 10.1021/ac0605290. [DOI] [PubMed] [Google Scholar]
- 12.Kyrikou I., Hadjikakou S.K., Mavromoustakos T. Effects of non-steroid anti-inflammatory drugs in membrane bilayers. Chem. Phys. Lipids. 2004;132:157–169. doi: 10.1016/j.chemphyslip.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 13.Song Y., Guallar V., Baker N.A. Molecular dynamics simulations of salicylate effects on the micro- and mesoscopic properties of a dipalmitoylphosphatidylcholine bilayer. Biochemistry. 2005;44:13425–13438. doi: 10.1021/bi0506829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Böckmann R.A., Hac A., Grubmüller H. Effect of sodium chloride on a lipid bilayer. Biophys. J. 2003;85:1647–1655. doi: 10.1016/S0006-3495(03)74594-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petrache H.I., Tristram-Nagle S., Parsegian V.A. Swelling of phospholipids by monovalent salt. J. Lipid Res. 2006;47:302–309. doi: 10.1194/jlr.M500401-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mukhopadhyay P., Monticelli L., Tieleman D.P. Molecular dynamics simulation of a palmitoyl-oleoyl phosphatidylserine bilayer with Na+ counterions and NaCl. Biophys. J. 2004;86:1601–1609. doi: 10.1016/S0006-3495(04)74227-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gurtovenko A.A. Asymmetry of lipid bilayers induced by monovalent salt: atomistic molecular-dynamics study. J. Chem. Phys. 2005;122:244902. doi: 10.1063/1.1942489. [DOI] [PubMed] [Google Scholar]
- 18.Gurtovenko A.A., Vattulainen I. Effect of NaCl and KCl on phosphatidylcholine and phosphatidylethanolamine lipid membranes: insight from atomic-scale simulations for understanding salt-induced effects in the plasma membrane. J. Phys. Chem. B. 2008;112:1953–1962. doi: 10.1021/jp0750708. [DOI] [PubMed] [Google Scholar]
- 19.Lee S.-J., Song Y., Baker N.A. Molecular dynamics simulations of asymmetric NaCl and KCl solutions separated by phosphatidylcholine bilayers: potential drops and structural changes induced by strong Na+-lipid interactions and finite size effects. Biophys. J. 2008;94:3565–3576. doi: 10.1529/biophysj.107.116335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao W., Róg T., Karttunen M. Atomic-scale structure and electrostatics of anionic palmitoyloleoylphosphatidylglycerol lipid bilayers with Na+ counterions. Biophys. J. 2007;92:1114–1124. doi: 10.1529/biophysj.106.086272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crowell P.L., Gould M.N. Chemoprevention and therapy of cancer by d-limonene. Crit. Rev. Oncog. 1994;5:1–22. doi: 10.1615/critrevoncog.v5.i1.10. [DOI] [PubMed] [Google Scholar]
- 22.Marostica M.R., Pastore G.M. Limonene and its oxyfunctionalized compounds: biotransformation by microorganisms and their role as functional bioactive compounds. Food Sci. Biotechnol. 2009;18:833–841. [Google Scholar]
- 23.Berger O., Edholm O., Jähnig F. Molecular dynamics simulations of a fluid bilayer of dipalmitoylphosphatidylcholine at full hydration, constant pressure, and constant temperature. Biophys. J. 1997;72:2002–2013. doi: 10.1016/S0006-3495(97)78845-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berendsen H.J.C., Postma J.P.M., Hermans J. Interaction models for water in relation to protein hydration. In: Pullman B., editor. Intermolecular Forces (Jerusalem Symposia) Springer; New York: 1981. pp. 331–342. [Google Scholar]
- 25.Witzke S., Duelund L., Khandelia H. Inclusion of terpenoid plant extracts in lipid bilayers investigated by molecular dynamics simulations. J. Phys. Chem. B. 2010 doi: 10.1021/jp108675b. 2010 Nov 11. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 26.Frisch M.J., Trucks G.W., Wallingford C.T. University of Groningen; Groningen, The Netherlands: 2004. Gaussian 03, Rev. D.01. [Google Scholar]
- 27.Siwko M.E., Marrink S.J., Mark A.E. Does isoprene protect plant membranes from thermal shock? A molecular dynamics study. Biochim. Biophys. Acta. Biomembr. 2007;1768:198–206. doi: 10.1016/j.bbamem.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 28.Hess B., Kutzner C., Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 29.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 30.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 31.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 32.Leach A.R. Pearson Prentice Hall; Harlow, England: 2001. Molecular Modeling: Principles and Applications. [Google Scholar]
- 33.Hess B., Bekker H., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 34.Hess B. P-LINCS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008;4:116–122. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 35.Miyamoto S., Kollman P.A. SETTLE: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992;13:952–962. [Google Scholar]
- 36.Darden T., York D., Pedersen L. Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 37.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 38.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 39.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 40.Pandit S.A., Berkowitz M.L. Molecular dynamics simulation of dipalmitoylphosphatidylserine bilayer with Na+ counterions. Biophys. J. 2002;82:1818–1827. doi: 10.1016/S0006-3495(02)75532-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Douliez J.P., Léonard A., Dufourc E.J. Restatement of order parameters in biomembranes: calculation of C-C bond order parameters from C-D quadrupolar splittings. Biophys. J. 1995;68:1727–1739. doi: 10.1016/S0006-3495(95)80350-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrache H.I., Dodd S.W., Brown M.F. Area per lipid and acyl length distributions in fluid phosphatidylcholines determined by 2H NMR spectroscopy. Biophys. J. 2000;79:3172–3192. doi: 10.1016/S0006-3495(00)76551-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosso L., Gould I.R. Structure and dynamics of phospholipid bilayers using recently developed general all-atom force fields. J. Comput. Chem. 2008;29:24–37. doi: 10.1002/jcc.20675. [DOI] [PubMed] [Google Scholar]
- 44.Poger D., Mark A.E. On the validation of molecular dynamics simulations of saturated and cis-monounsaturated phosphatidylcholine lipid bilayers: a comparison with experiment. J. Chem. Theory Comput. 2010;6:325–336. doi: 10.1021/ct900487a. [DOI] [PubMed] [Google Scholar]
- 45.Damodaran K.V., Merz K.M., Jr. A comparison of DMPC- and DLPE-based lipid bilayers. Biophys. J. 1994;66:1076–1087. doi: 10.1016/S0006-3495(94)80889-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abramson S.B., Cherksey B., Weissmann G. Nonsteroidal antiinflammatory drugs exert differential effects on neutrophil function and plasma membrane viscosity. Studies in human neutrophils and liposomes. Inflammation. 1990;14:11–30. doi: 10.1007/BF00914026. [DOI] [PubMed] [Google Scholar]
- 47.Pandit S.A., Bostick D., Berkowitz M.L. Molecular dynamics simulation of a dipalmitoylphosphatidylcholine bilayer with NaCl. Biophys. J. 2003;84:3743–3750. doi: 10.1016/S0006-3495(03)75102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cordomí A., Edholm O., Perez J.J. Effect of force field parameters on sodium and potassium ion binding to dipalmitoyl phosphatidylcholine bilayers. J. Chem. Theory Comput. 2009;5:2125–2134. doi: 10.1021/ct9000763. [DOI] [PubMed] [Google Scholar]
- 49.Khandelia H., Mouritsen O.G. Lipid gymnastics: evidence of complete acyl chain reversal in oxidized phospholipids from molecular simulations. Biophys. J. 2009;96:2734–2743. doi: 10.1016/j.bpj.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kandasamy S.K., Larson R.G. Effect of salt on the interactions of antimicrobial peptides with zwitterionic lipid bilayers. Biochim. Biophys. Acta. 2006;1758:1274–1284. doi: 10.1016/j.bbamem.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 51.Kandasamy S.K., Larson R.G. Molecular dynamics simulations of model trans-membrane peptides in lipid bilayers: a systematic investigation of hydrophobic mismatch. Biophys. J. 2006;90:2326–2343. doi: 10.1529/biophysj.105.073395. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.