

Abstract
The clinical efficacy of opiates for pain control is severely limited by analgesic tolerance and hyperalgesia. Herein we show that chronic morphine upregulates both the sphingolipid ceramide in spinal astrocytes and microglia, but not neurons, and spinal sphingosine-1-phosphate (S1P), the end-product of ceramide metabolism. Coadministering morphine with intrathecal administration of pharmacological inhibitors of ceramide and S1P blocked formation of spinal S1P and development of hyperalgesia and tolerance in rats. Our results show that spinally formed S1P signals at least in part by (1) modulating glial function because inhibiting S1P formation blocked increased formation of glial-related proinflammatory cytokines, in particular tumor necrosis factor-α, interleukin-1βα, and interleukin-6, which are known modulators of neuronal excitability, and (2) peroxynitrite-mediated posttranslational nitration and inactivation of glial-related enzymes (glutamine synthetase and the glutamate transporter) known to play critical roles in glutamate neurotransmission. Inhibitors of the ceramide metabolic pathway may have therapeutic potential as adjuncts to opiates in relieving suffering from chronic pain.
Introduction
Opiate/narcotic analgesics, typified by morphine (Mor), are the most effective treatments for acute and chronic severe pain. However, their clinical utility is often hampered by the development of analgesic tolerance, which requires escalating doses to achieve equivalent pain relief (Foley, 1995). This complex pathophysiological cycle represents a critical barrier to the quality of life of these patients because of the resulting oversedation, reduced physical activity, constipation, respiratory depression, high potential for addiction, and other side-effects (Foley, 1995). Adaptive modifications in cellular responsiveness have been proposed as contributing to tolerance (Taylor and Fleming, 2001). An alternative hypothesis with in vivo evidence in animals (Mao et al., 1995) and in humans (Arnér et al., 1988) is that chronic opioid receptor stimulation triggers the activation of anti-opioid systems that reduce sensory thresholds, thereby resulting in hypersensitivity to tactile and noxious thermal stimulation (Simonnet and Rivat, 2003). As a corollary to this hypothesis, such opioid-induced hypersensitivity paradoxically diminishes the net analgesic effect of the opioid agonist (Ossipov et al., 2003; Simonnet and Rivat, 2003). Although progress has been made in modifying this drug class to improve their formulated delivery, pharmacokinetics, and potential for abuse, little progress has been made in preventing the development of antinociceptive tolerance and hyperalgesia.
Ceramide, a potent proinflammatory and proapoptotic sphingolipid (Hannun and Obeid, 2008), is generated by enzymatic hydrolysis of sphingomyelin by sphingomyelinases (sphingomyelin pathway) and from de novo synthesis by serine palmitoyltransferase and ceramide synthase (de novo pathway) (Delgado et al., 2006). We have reported recently that the development of morphine-induced tolerance is associated with increased formation of ceramide in the spinal cord and inhibition of its biosynthesis blocked the development of antinociceptive tolerance (Bryant et al., 2009; Ndengele et al., 2009). The steady-state availability of ceramide is further regulated by ceramidases that convert ceramide to sphingosine with the latter then converted by sphingosine kinase 1 and/or 2 (Melendez, 2008; Takabe et al., 2008) to S1P, the G-protein-coupled receptor-signaling end product in the ceramide metabolic pathway (Melendez, 2008; Takabe et al., 2008). Once generated, sphingosine-1-phosphate (S1P) diffuses extracellularly acting on G-protein-coupled S1P receptors (S1PR1–S1PR5) found on several cells of the CNS, including neurons and glial cells (Taha et al., 2004; Melendez, 2008; Takabe et al., 2008). The objectives of our studies were to determine whether the involvement of spinal ceramide in opioid antinociceptive tolerance and hyperalgesia occurs via S1P signaling. Through the pharmacological targeting of the de novo and sphingomyelin pathways, we now demonstrate, for the first time, that spinally formed S1P after activation of sphingosine kinase is the key second messenger contributing to morphine-induced hyperalgesia and antinociceptive tolerance at least in part through modulation of glial cell function. Finally and when analyzed collectively with our previous findings (Bryant et al., 2009; Ndengele et al., 2009), we have established a varied platform approach of pharmacological manipulation that can intercept the ceramide metabolic pathway at many levels to preempt opioid-induced hyperalgesia and tolerance. Results from these studies will most certainly lead to novel mechanism-based therapeutic strategies.
Materials and Methods
Experimental animals
Male Sprague Dawley rats (200–230 g) were purchased from Harlan, housed three to four per cage, and maintained in controlled environment (12 h light/dark cycle) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the National Institutes of Health guidelines on laboratory animal welfare and the recommendations by Saint Louis University Institutional Animal Care and Use Committee. Animal use at the University of Magna Graecia (Catanzaro, Italy) likewise complied with Italian regulations for the protection of animals used for experimental and other scientific purpose (D.M. 116192) and with European Economic Community regulations. All experiments were conducted with the experimenters blinded to treatment conditions.
Osmotic pump implantation
Male Sprague Dawley rats were lightly anesthetized with isoflurane and were subcutaneously implanted (in the interscapular region) with primed osmotic minipumps (Alzet 2001; Alza) to deliver saline (Sal) at 1 μl/h or morphine at 75 μg · μl−1 · h−1 over 7 d as described (King et al., 2007; Vera-Portocarrero et al., 2007). The concentrations of morphine sulfate resulted in a daily dose of ∼8–9 mg/kg (depending on the weight of the rat). Minipumps were filled according to the specifications of the manufacturer. The use of the osmotic pump ensures a continuous subcutaneously delivery of morphine avoiding intermittent periods of withdrawal. Rats were tested for analgesia at 2 h after minipump implant to verify that they are analgesic: ∼100% analgesia was achieved. This helps verify that the pumps are working well, which is typically not a problem. The integrity of the pump delivery system was reexamined at the end of each experiment in which the spinal cords are harvested.
Drug administration
The test substances fumonisin B1 (FB1) (Sigma-Aldrich), myriocin (Myr) (Sigma-Aldrich), D609 (tricyclodecan-9-yl-xanthogenate, K) (Calbiochem), DMS (N,N-dimethylsphingosine) (Caymen Chemical), SK-I [2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole] (Calbiochem), or their vehicle (Veh) (0.25% in DMSO) were delivered by intrathecal injection in rats chronically implanted with intrathecal cannulas using the L5–L6 lumbar approach, as described previously (Størkson et al., 1996) and commonly used for drug delivery (Schoeniger-Skinner et al., 2007; Doyle et al., 2010; Ramos et al., 2010). Test substances or vehicle were injected intrathecally once a day for 5 d in a total volume of 10 μl, followed by a 10 μl flush with sterile physiological saline. Injections were made after completion of the behavioral tests.
Behavioral tests
Thermal hyperalgesia.
Hyperalgesic responses to heat were determined by the Hargreaves' method using a Basile Plantar Test (Ugo Basile) (Hargreaves et al., 1988) with a cutoff latency of 20 s used to prevent tissue damage. Animals were allowed to acclimate within a Plexiglas enclosure on a clear glass plate in a quiet testing room. A mobile infrared generator was positioned to deliver a thermal stimulus directly to an individual hindpaw from beneath the chamber. The withdrawal latency period of injected paws was determined with an electronic clock circuit and thermocouple. Foot-withdrawal latencies were taken on day 0 before implantation of the minipumps (baseline) and subsequently on days 1, 3, and 6 of the experimental period to determine whether morphine-tolerant animals develop increased sensitivity to thermal stimulation. A significant (p < 0.05) reduction in paw-withdrawal latency over time and before implantation of the osmotic minipump is characterized as thermal hyperalgesia. After the Hargreave's test, the development of antinociceptive tolerance was determined in all animals as follows.
Acute nociception.
The tail-flick test, which measures withdrawal latencies of the tail from a noxious radiant heat source, was used to measure thermal nociceptive sensitivity with baseline latencies of 2–3 s and a cutoff time of 10 s to prevent tissue injury (D'Amour, 1941). Tolerance to the antinociceptive effect of morphine was indicated by a significant (p < 0.05) reduction in tail-flick latency after challenge with an acute dose of morphine sulfate (6 mg/kg, i.p.) at 30 min after injection time point, a time demonstrated previously to produce maximal antinociception at this dose. Data obtained were converted to percentage maximal possible antinociceptive effect as follows: (response latency − baseline latency)/(cutoff latency − baseline latency) × 100. At the required time points after the behavioral tests, spinal cord tissues from the lumbar enlargement segment of the spinal cord (L5–L6) were removed and tissues were processed for immunohistochemical, Western blot, and biochemical analysis. All experiments were conducted with the experimenters blinded to treatment conditions.
Immunohistochemical and immunofluorescent detection
For nitrated proteins in the spinal cord, the lumbar sections (L5–L6) were fixed and processed for immunohistochemical staining as described (Wang et al., 2004; Ndengele et al., 2008) using a well characterized monoclonal anti-nitrotyrosine (NT) antibody (1:100 in 10% normal horse serum; Calbiochem). The processed samples were visualized by immunolabeling with secondary antibody, avidin–biotin complex, and diaminobenzidine according to the instructions of the manufacturer (Vector ABC Elite kit; Vector Laboratories).
For ceramide expression in spinal cord, formalin-fixed and OCT-embedded (Sakura Finetek USA) lumbar portions of the spinal cord (L5–L6) were sectioned at 10 μm and stored at −20°C until use. The sections were rinsed in PBS, immersed in 40 ml of DIVA decloaker (BioCare Medical), and subjected to antigen retrieval in a pressure cooker at 100°C for 5 min. The slides were cooled to room temperature (RT), rinsed in PBS, and incubated in blocking buffer [PBS, 5% normal goat serum, 1% bovine serum albumin (BSA), and 0.25% Triton X-100] for 2 h, humidified at RT. The blocked sections were rinsed in PBS and dual labeled for 16 h, humidified at 4°C with a well characterized rabbit polyclonal anti-ceramide antibody (Krishnamurthy et al., 2007) (1:50 or 1:100) and with murine monoclonal anti- neuronal-specific nuclear protein (NeuN) (1:200) (Millipore) for neurons, murine monoclonal anti-glial fibrillary acidic protein (GFAP) (1:100) (Abcam) for astrocytes, or murine monoclonal anti-ionized calcium-binding adaptor molecule 1 (Iba1) (1:100) (Abcam) for microglia in diluted blocking buffer (1:10). The sections were rinsed in PBS, and the proteins were detected with goat anti-rabbit rhodamine and goat anti-mouse Alexa Fluor 488 (1:300; Invitrogen) in diluted blocking buffer (1:10) for 2 h, humidified at RT. After a PBS rinse, the sections were mounted in VectaShield with 4′,6′-diamidino-2-phenylindole (Vector Laboratories), coverslipped, and photographed with an Olympus FV1000 MPE confocal microscope. Sections treated with normal rabbit IgG or normal mouse IgG (Sigma) at equivalent concentrations to primary antibodies were used as controls yielding only nonspecific background fluorescence.
Immunoprecipitation and immunoblotting
Cytosolic fractions and P2 membranes were obtained as described previously (Takagi et al., 2000; Wang et al., 2004; Muscoli et al., 2007) and stored immediately at −80°C. Immunoprecipitation and Western blot analyses were performed as described (Takagi et al., 2000; Wang et al., 2004; Muscoli et al., 2007). Proteins were resolved with 7.5% [glutamate transporter-1 (GLT-1)], 10% [glutamine synthetase (GS) and GFAP], or 4–20% (Iba1) SDS-PAGE before electrophoretic transfer. Membranes were blocked for 1 h at RT in 1% BSA in TBS-T (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, and 0.01%Tween-20) and 0.1% thimerosal, and then probed with mouse monoclonal anti-glutamine synthetase (1:2000; BD Biosciences), polyclonal rabbit anti-GLT-1 (1:1000; Alpha Diagnostic), monoclonal mouse anti-GFAP (1:1000; Dako), or polyclonal goat anti-Iba1 (1:1000; Waco) antibodies. Membranes were washed with TBS-T and visualized with horseradish peroxidase-conjugated secondary antibodies (Thermo Fisher Scientific) for 1 h at RT and enhanced chemiluminescence [ECL (GE Healthcare) or Femto kit (Thermo Fisher Scientific)]. Rat brain lysate (Millipore Corporation) containing most of the proteins expressed by nervous tissue was used as positive control. The blots were stripped and probed with a murine monoclonal anti-β-actin antibody (1:2000; Sigma). The relative density of the protein bands of interest were determined from film using ImageQuant 5.2 software (Molecular Dynamics) and normalized to β-actin bands.
Statistical analysis of the data
Data from time course studies were analyzed by ANOVA, followed by Bonferroni's post hoc test against day 0 or Veh–Mor or by Dunnett's post hoc test against Veh–Mor when noted. Statistically significant differences are defined at a p < 0.05. All statistical analysis was performed using GraphPad Prism (release 5.03; GraphPad Software).
Results
Ceramide, a precursor in the biosynthesis of S1P, colocalizes with glia and not neurons
When compared with rats that received a chronic subcutaneous infusion of saline (Veh–Sal, n = 5) over 7 d, infusion of morphine over the same timeframe (Veh–Mor, n = 5) led to (1) the development of thermal hyperalgesia (Hargreaves et al., 1988) as evidenced by a significant (p < 0.001) reduction in paw-withdrawal latency (seconds) on days 3 and 6 when compared with paw-withdrawal latency from before implantation of the osmotic minipump (baseline, day 0) (Fig. 1A,C) and (2) the development of antinociceptive tolerance over the same timeframe (Fig. 1B,D) (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). The latter was indicated by a significant (p < 0.001) reduction in tail-flick latency 30 min after challenge with an acute dose of morphine (6 mg/kg) given intraperitoneally as evidenced on days 3 and 6 in rats that received chronic morphine infusion over 7 d when compared with rats that received an infusion of saline over the same timeframe (Fig. 1B,D) (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). The development of morphine-induced hyperalgesia and antinociceptive tolerance observed on day 6 was associated with a significant increased formation of ceramide as detected by immunofluorescence, including in superficial layers of the spinal cord from the lumbar enlargement (L5-L6) (Fig. 1F) (for a photomontage including the whole spinal dorsal horn, see supplemental Fig. S2B, available at www.jneurosci.org as supplemental material). These results confirm our previous studies (Ndengele et al., 2009). Interestingly, ceramide was preferentially upregulated and colocalized in astrocytes (Fig. 2D) [using GFAP, a cellular marker for astrocytes (Romero-Sandoval et al., 2008a)] and microglial cells (Fig. 2E) [using Iba1, a cellular marker for microglia (Narita et al., 2006)] but not in neurons (Fig. 2F, using NeuN, neuronal marker). Because of space restriction, only merged images are shown in Figure 2. Individual photos displaying single labeling are shown in supplemental Figs. S3–S5 (available at www.jneurosci.org as supplemental material).
Figure 1.

Morphine-induced hyperalgesia and tolerance is associated with increased ceramide and S1P derived from activation of sphingosine kinase. When compared with rats that received a chronic subcutaneous infusion of saline (Veh–Sal, n = 5, ● or open bar) over 7 d, infusion of morphine over the same timeframe (Veh-Mor, n = 5, ■ or gray bar) led to the development of thermal hyperalgesia as evidenced by a significant reduction in paw-withdrawal latency (seconds) on days 3 and 6 when compared with paw-withdrawal latency from before implantation of the osmotic minipump (day 0) (A, C) and with the development of antinociceptive tolerance (B, D). Results shown in C and D are from behavioral measurements taken on day 6. These events were associated with increased formation of ceramide (red, E, F) and S1P (G, H) in dorsal horn tissues. Coadministration of morphine with DMS (0.3 μm/d for /5 d, n = 5, ● or black bar) or SK-I (0.3 μm/d for 5 d, n = 5, ♦ or hashed bar) blocked the development of hyperalgesia (A), tolerance (B), and S1P (G, H). Dose–response curves for DMS (0.03–0.3 μm/d for 5 d, n = 5, ● or black bar) or SK-I (0.03–0.3 μm/d for 5 d, n = 5, ○ or hashed bar) on day 6 are shown in C for hyperalgesia, D for tolerance, and H for S1P. Micrographs shown in E and F are representative of at least three images of the superficial layers of dorsal horn (L5–L6) harvested on day 6 from three different animals. Results are expressed as mean ± SEM for n = 5 animals and analyzed by ANOVA with Bonferroni's post hoc test: *p < 0.001 for Veh–Mor versus Veh–Sal; §p < 0.01 or §§p < 0.001 versus day 0; and †p < 0.01 or ††p < 0.001 for Drugs–Mor versus Veh–Mor.
Figure 2.

Colocalization of ceramide with activated glial cells but not neurons. Fixed frozen spinal cord sections from Veh–Sal (A–C) and Veh–Mor (D–F) rats were stained for ceramide (red) and for astrocytes (GFAP+, green) (A, D), microglia (Iba1+, green) (B, E), or neurons (NeuN+, green) (C, F). In these merged images, ceramide was low or absent in spinal cords from Veh–Sal animals and substantially increased in spinal cords from Veh–Mor animals. Furthermore, ceramide levels colocalized (yellow) with astrocytes (D) and microglia (E) but not with neurons (F). Negative controls using normal rabbit serum (for ceramide) or normal mouse IgG (for GFAP, Iba1, and NeuN) at the same concentrations as for the immune serum and immune IgG exhibited only low levels of background fluorescence. Micrographs are representative of at least three images of the superficial layers of dorsal horn (L5–L6) from three different animals performed on different days.
The development of morphine-induced hyperalgesia and antinociceptive tolerance observed on days 3 and 6 was associated with a significant (p < 0.001) increased formation in S1P levels in dorsal horn tissues as measured by ELISA (Echelon Biosciences) after extraction of the sphingolipids (Fig. 1G,H). Coadministration of morphine with daily intrathecal injections of myriocin (Myr–Mor; 0.3 μm/d for 5 d, n = 4), an inhibitor of serine palmitoyltransferase (Delgado et al., 2006), FB1 (FB1–Mor; 1 μm/d for 5 d, n = 4), a competitive and reversible inhibitor of ceramide synthase (Delgado et al., 2006), or D609 (D609–Mor; 1 μm/d for 5 d, n = 4), an inhibitor of sphingomyelinase (Delgado et al., 2006), blocked the increased production of S1P (Fig. 3A) and the development of hyperalgesia (Fig. 3B) and antinociceptive tolerance (Fig. 3C) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). We have shown previously that, at these doses, these inhibitors effectively block the formation of ceramide in the spinal cord (Ndengele et al., 2009).
Figure 3.

Therapeutic manipulation with inhibitors of ceramide biosynthesis blocks hyperalgesia and antinociceptive tolerance. Intrathecal delivery of inhibitors of the de novo (Myr, 0.3 μm/d for 5 d; FB1, 1 μm/d for 5 d) and sphingomyelin (D609, 1 μm/d for 5 d) pathways blocked increased S1P levels in dorsal horn tissues (A) and subsequent development of morphine-induced thermal hyperalgesia (B) and antinociceptive tolerance (C). Results are expressed as mean ± SEM for n = 5 animals and analyzed by ANOVA with Dunnett's post hoc test: *p < 0.001 for Veh–Mor versus Veh–Sal and †p < 0.001 for drugs–Mor versus Veh–Mor.
These results suggest that glial-derived ceramide contributes to the development of morphine-induced hyperalgesia and antinociceptive tolerance through the formation of S1P.
Inhibitors of S1P biosynthesis block the development of morphine-induced hyperalgesia and antinociceptive tolerance
That spinally formed S1P is the key second messenger (downstream of ceramide) contributing to morphine-induced hyperalgesia and antinociceptive tolerance was established in the following studies using inhibitors of the enzymatic (sphingosine kinase) bioconversion of ceramide to S1P. As observed on days 3 and 6, coadministration of morphine with daily intrathecal injections of two structurally unrelated and commonly used inhibitors of sphingosine kinase, namely DMS (DMS–Mor; 0.3 μm/d for 5 d, n = 5) or SK-I (SK-I–Mor; 0.3 μm/d for 5 d, n = 5) (French et al., 2003; Lee et al., 2004; Delgado et al., 2006), blocked increased production of S1P (Fig. 1G) (p < 0.001) in dorsal horn tissues and the development of hyperalgesia (Fig. 1A) (p < 0.001) and antinociceptive tolerance (Fig. 1B) (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material) (p < 0.001). Full dose–response curves for DMS (DMS–Mor; 0.03–0.3 μm/d for 5 d, n = 5) or SK-I (SK-I–Mor; 0.03–0.3 μm/d for 5 d, n = 5) are shown at time of maximal effects (day 6) in Figure 1C (for hyperalgesia), Figure 1D and supplemental Figure S1B (available at www.jneurosci.org as supplemental material) (for tolerance), and Figure 1H (for S1P). These results establish the role of the sphingosine kinase to S1P signaling pathway. When given alone daily and over 5 d to rats that received saline infusion (Veh–Sal), these drugs when tested at the highest dose had no effect on withdrawal thresholds (Fig. 1) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), thus suggesting that S1P, like ceramide (Ndengele et al., 2009), is not involved in normal pain processing (Ndengele et al., 2009).
As reported previously, Myr, FB1, or D609 do not potentiate antinociceptive effects of acute morphine (Ndengele et al., 2009). Likewise, the inhibitory effects of DMS or SK-I were not attributable to acute antinociceptive interactions between these drugs and acute morphine. Thus, intrathecal administration of DMS (0.3 μm, n = 3) or SK-I (0.3 μm, n = 3) 15 min before an acute intraperitoneal injection of morphine known to produce ∼40–50% antinociception at 30 min (3 mg/kg, n = 3) did not potentiate the antinociceptive responses to acute morphine (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). When compared with the vehicle group, DMS or SK-I had no effect on baseline latencies (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Inhibition of S1P with acute intrathecal injection of DMS or SK-I did not reverse tolerance once established (n = 4; data not shown), indicating that ceramide-derived S1P contributes to events leading to the development but not expression of tolerance.
Although pilot studies performed in our laboratories clearly demonstrated that catheterized animals show similar baseline nociceptive thresholds as naive animals, one cannot exclude the possibility that chronic intrathecal catheters may have an effect on the spinal environment that leads to changes in pain processing. To the best of our knowledge, no one in the scientific community has yet to report concerns of this type when the lumbar approach method is used for intrathecal catheterization, as routinely used in many other laboratories. By analogy, one cannot exclude the possibility that temporary or repeated direct lumbar injections also frequently used in many laboratories has effects on the spinal environment that can also lead to changes in pain processing.
S1P contributes to the development of morphine-induced hyperalgesia and antinociceptive tolerance by increasing formation of glia-related cytokines
In support of previous studies and when compared with nontolerant animals (supplemental Figs. S3B, S4B, available at www.jneurosci.org as supplemental material), the development of morphine-induced hyperalgesia and antinociceptive tolerance was associated with an upregulation of astrocyte and microglial cell expression of GFAP and Iba1 immunofluorescence, respectively (supplemental Figs. S3E, S4E, available at www.jneurosci.org as supplemental material) and protein expression by Western blot (Fig. 4A,A1 for GFAP and B,B1 for Iba1) (p < 0.001) and a significant (p < 0.001) increase in tumor necrosis factor-α (TNF-α) (Fig. 4C), interleukin 1β (IL-1β) (Fig. 4D), and interleukin-6 (IL-6) (Fig. 4E). Coadministration of morphine with intrathecal delivery of DMS (0.3 μm/d per 5 d, n = 5) significantly attenuated the morphine-induced increased glial cell expression of these markers (evidenced by decreased GFAP, p < 0.01, and Iba1, p < 0.001, protein expression by Western blot) (Fig. 4A,A1 for GFAP and B,B1 for Iba1) and decreased production of TNF-α, IL-1β, and IL-6 (Fig. 4C–E).
Figure 4.

DMS blocks glial cell activation and increased spinal production of cytokines. When compared with Veh–Sal, the development of tolerance (Veh–Mor) was associated with significant activation of astrocytes and microglial cells as evidenced, respectively, by increased GFAP (A, A1) and Iba1 (B, B1) protein expression and increased formation of TNF-α (C), IL-1β (D), and IL-6 (E) levels in dorsal horn tissues. Coadministration of morphine with DMS (0.3 μm/d for 5 d) prevented glial cell activation (A, A1, B, B1) and increased cytokine formation (C–E). Composite densitometric analyses for gels of five rats are expressed as mean ± SEM of percentage β-actin as shown in A1 and B1. Results for cytokines are expressed as mean ± SEM for n = 5 animals. Data were analyzed by ANOVA with Dunnett's post hoc test: *p < 0.001 for Veh–Mor versus Veh–Sal and †p < 0.01 or ††p < 0.001 for DMS–Mor versus Veh–Mor.
S1P contributes to the development of morphine-induced hyperalgesia and antinociceptive tolerance via nitration of glia-related GLT-1 and GS
We have reported recently that peroxynitrite (ONOO−, PN) is a key player in the development of morphine-induced hyperalgesia and antinociceptive tolerance and have provided data to show that formation of 3-nitrotyrosine (NT) in the superficial layers of the dorsal horn originates from spinal production of PN (Muscoli et al., 2007). Detection of NT in this setting can therefore be reliably used as marker of PN (Muscoli et al., 2007). We now show that, when compared with nontolerant rats (Fig. 5A), the appearance of NT staining in tolerant rats (Fig. 5B) was blocked by coadministration of morphine with DMS (0.3 μm/d for 5 d, n = 5) (Fig. 5C). These findings provide a mechanistic link between chronic administration of morphine and increased formation of spinal PN. Two important glial cell proteins known to play essential roles in glutamate handling and thus glutamatergic neurotransmission, namely GLT-1 and GS, have been found to be nitrated by PN in the spinal cord during the development of morphine-induced hyperalgesia and antinociceptive tolerance (Muscoli et al., 2007). Nitration of these proteins is intimately linked to inactivation of their biological function (Trotti et al., 1996, 1999; Miñana et al., 1997; Gorg et al., 2005), and their inactivation has important ramifications manifested by enhancing glutamatergic neurotransmission, which is key to central sensitization (Salvemini and Neumann, 2010). As can be seen in Figure 5, when compared with nontolerant animals, the development of antinociceptive tolerance and hyperalgesia was associated with nitration of GLT-1 (Fig. 5D,D1) and GS (Fig. 5E,E1), and this was significantly (p < 0.001) attenuated by intrathecal delivery of DMS, thus establishing the link between chronic morphine administration to S1P formation and PN-mediated nitration of GLT-1 and GS (Fig. 5). Total GLT-1 and GS protein levels did not change among the various groups (n = 4) (Fig. 5D,E).
Figure 5.

DMS blocks posttranslation protein nitration in spinal cord. When compared with Veh–Sal (A), the development of tolerance (Veh–Mor) was associated with significant protein nitration as detected by immunohistochemistry (B, see arrows), in particular significant nitration of the glutamate transporter (GLT-1; D, D1) and GS (E, E1). These events were attenuated by coadministration of morphine with DMS (0.3 μm/d for 5 d; C–E). Gels shown in D and E are representative of gels from n = 5 rats. Composite densitometric analyses for gels of nitrated proteins of five rats are expressed as mean ± SEM of percentage β-actin as shown in D1 and E1 and analyzed by ANOVA with Dunnett's post hoc test: *p < 0.001 for Veh–Mor versus Veh–Sal and †p < 0.001 for DMS–Mor versus Veh–Mor. Micrographs are representative of at least five from different animals performed on different days and are taken from the superficial layers of the dorsal horn (L5–L6), the anatomical site that stains for NT during tolerance (Muscoli et al., 2007).
Discussion
A devastating health problem in our country is the inadequate treatment of pain. One-third of all Americans suffer from some form of chronic pain, and one-third of these have pain that is resistant to current medical therapy. The economic impact of pain is equally large at approximately $100 billion annually (Renfrey et al., 2003). Severe pain syndromes reduce quality of life in patients, partly because reduced analgesic effectiveness with chronic opiate therapy (i.e., hyperalgesia and tolerance) leads to escalating doses and distressing side-effects. Accordingly, there is major interest in new approaches to maintain opiate efficacy during repetitive dosing for chronic pain without engendering tolerance or unacceptable side-effects. Our results have established a key role for ceramide-derived S1P in the development of morphine-induced hyperalgesia and antinociceptive tolerance, providing a novel mechanistic rationale for development of inhibitors of ceramide and S1P as adjunct to opiates in pain management. Whereas in this paper we focused on the role of S1P on glia, we are not excluding the likely possibility that S1P may, in addition, contribute to the development of morphine-induced hyperalgesia and antinociceptive tolerance by affecting neurons. In support, S1P can for example augment the excitability of sensory neurons (Zhang et al., 2002, 2006). The schematic shown in Figure 6 summarizes our findings that raise the possibility that chronic administration of morphine activates the ceramide metabolic pathway in spinal glial cells (astrocytes and microglial cells), resulting in increased production in S1P by sphingosine kinases. The relative contribution of sphingosine kinase 1 versus 2 remains to be addressed in future studies. After its extracellular release, S1P would then bind to its receptors on glial cells initiating a series of signaling pathways, culminating in enhanced production of the well known proinflammatory and neuroexcitatory cytokines IL-1β, TNF-α, and IL-6 (Romero-Sandoval et al., 2008b; Latremoliere and Woolf, 2009; Milligan and Watkins, 2009; Watkins et al., 2009). As shown, inhibition of S1P blocks the formation of these cytokines, suggesting that the attenuation of morphine-induced hyperalgesia and tolerance by inhibitors of S1P formation (such as sphingosine kinase inhibitors) is secondary to suppression of morphine-induced spinal cytokine formation. This presumptive proinflammatory role of S1P and thus the anti-inflammatory action of sphingosine kinase inhibitors is consistent with other findings. Indeed, inhibition of S1P with sphingosine kinase inhibitors is anti-inflammatory in animal models of disease, including arthritis (Kitano et al., 2006; Lai et al., 2008), acute inflammation (Pushparaj et al., 2008), asthma (Jolly et al., 2002; Nishiuma et al., 2008), and colitis (Maines et al., 2008; Snider et al., 2009). A more recent study revealed that increased formation of TNF-α and nitric oxide in microglial cells activated with Escherichia coli lipopolysaccharide is dependent on the production of S1P; blocking the activity of sphingosine kinase with DMS blocked TNF-α and nitric oxide release (Nayak et al., 2010). Conversely, exogenous administration of S1P stimulated the production of TNF-α and nitric oxide from microglial cells (Nayak et al., 2010). These results support a role for the ceramide to S1P signaling pathway in the regulation of cytokines and other pronociceptive mediators such as nitric oxide from glial cells. The intracellular transduction mechanisms whereby S1P modulates cytokine formation from glia are not known with certainty, but a few possibilities are proposed based on existing literature in diverse fields. S1P has been shown to activate redox-sensitive transcription factors [i.e., nuclear factor-κB (NF-κB)] and several mitogen-activated protein kinase (MAPKs), including p38 and extracellular signal-regulated kinase 1/2 (ERK1/2), known to regulate the production of proinflammatory mediators from glial cells (Watkins et al., 2001; Tanga et al., 2006). Furthermore, NF-κB, p38 kinase, and ERK have been implicated in the development of morphine-induced hyperalgesia and antinociceptive tolerance (Cui et al., 2006; Ndengele et al., 2009; Wang et al., 2009). Collectively, these data allow us to put forward the suggestion that glial S1P contributes to the development of morphine-induced hyperalgesia and antinociceptive tolerance by activating glial cells to release proinflammatory cytokines in an NF-κB and/or MAPK (p38 kinase, ERK)-dependent manner.
Figure 6.
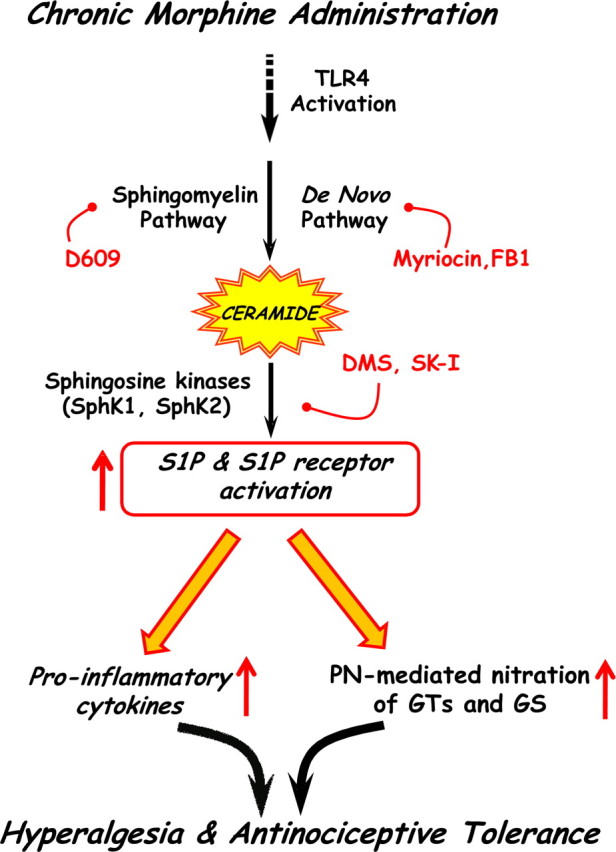
Proposed working hypothesis. Chronic administration of morphine activates the ceramide metabolic pathway, resulting in increased formation of S1P in glial cells during activation of sphingosine kinase (SphK) 1 and/or 2. After its extracellular release, S1P binds S1P receptors (which remain to be identified) on glial cells, initiating a series of events culminating in enhanced production of proinflammatory cytokines and PN-mediated nitration of glutamate transporters (GTs) and GS. Activation of glial TLR4 may provide a link between chronic administration of morphine and activation of the ceramide metabolic pathway in the development of morphine-induced hyperalgesia and antinociceptive tolerance.
Another signaling pathway engaged by S1P, which was unraveled in our studies, occurs through the formation and rapid reactions of PN. S1P can increase the formation of superoxide and nitric oxide by activating, respectively, the NADPH oxidase and nitric oxide synthase (Keller et al., 2006; Nayak et al., 2010). Both superoxide and nitric oxide are precursors in the biosynthesis of PN (Beckman et al., 1990), a potent proinflammatory (Salvemini et al., 1998) and pronociceptive nitroxidative species (Salvemini and Neumann, 2010) also implicated in the development of opiate-induced hyperalgesia and antinociceptive tolerance (Salvemini and Neumann, 2009). We therefore hypothesized and showed herein that S1P may contribute to the development of morphine-induced hyperalgesia and antinociceptive tolerance by favoring the production of PN, which in turn nitrates key glial cell proteins known to be involved in maintaining optimal spinal glutamatergic signaling. Indeed, inhibition of S1P with DMS blocked the formation of NT, a marker for PN in spinal cord (Muscoli et al., 2007; Ndengele et al., 2009). It is well established that glutamate neurotransmission, in particular, mediated via NMDA receptors is key in the development of opioid tolerance (Trujillo and Akil, 1991). The homeostasis of extracellular glutamate is tightly regulated by sodium-dependent high-affinity glutamate transporters in the plasma membranes of both neurons and glia, although the bulk (>90%) of functional glutamate uptake is mediated by the glial transporters glutamate/aspartate transporter (GLAST) and GLT-1 (Danbolt, 2001). If GLAST/GLT-1 function is impacted on (i.e., reduced or eliminated), glutamate can increase in the CSF contributing to rapid alterations in synaptic transmission (Nakagawa et al., 2001). In contradistinction to the central role of glutamate transporters in regulating the homeostasis of extracellular glutamate, the glial cell enzyme GS plays a pivotal role in its intracellular metabolic fate (Suárez et al., 2002). In the CNS, GS is located mainly in astrocytes and one of the primary roles of these cells is to protect neurons against excitotoxicity by taking up excess ammonia and glutamate and converting them into glutamine (Suárez et al., 2002). Glutamine is then transported into neurons, in which it serves as a precursor for the formation of glutamate and GABA (Waniewski and Martin, 1986). Enzymatic inactivation of GS facilitates neuronal excitation (Suárez et al., 2002; Muscoli et al., 2005). Furthermore, through feedback regulation, a decrease in the activity of glutamine synthetase can reduce the activity of glutamate transporter (Suárez et al., 2002), underscoring the reciprocal interaction between these two pathways. Inhibition of GS activity blocks central sensitization associated with inflammatory hyperalgesia (Chiang et al., 2007). Our results demonstrate that nitration of GLT-1 and GS was blocked by DMS. These data suggest that inhibition of hyperalgesia and antinociceptive tolerance by inhibitors of S1P is secondary, at least in part, to inhibition of PN generation and subsequent posttranslational nitration and inactivation of GLT-1 and GS. By preventing nitration of these glial cell proteins, DMS would reduce the level of glutamate to basal, thus restoring optimal glutamatergic neurotransmission.
The mechanistic connections between chronic administration of morphine, activation of the ceramide metabolic pathway, and the development of hyperalgesia and antinociceptive tolerance remains unknown. A growing body of data has emerged recently that implicates activation of Toll-like receptor 4 (TLR4) on glial cells in the development of opiate-induced hyperalgesia and antinociceptive tolerance as well as neuropathic pain (Tanga et al., 2005; Watkins et al., 2009). Interestingly, activation of TLR4 by lipopolysaccharide (a well known exogenous ligand for TLR4) on monocytes and macrophages activates enzymes in the de novo and sphingomyelinase pathways, leading to increased production of ceramide that, in turn, activates NF-κB and MAPKs to increase the production of nitric oxide and superoxide, as well as TNF-α and IL-1β events that have been linked to the development of sepsis and septic shock (Delogu et al., 1999; Claus et al., 2005; Cuzzocrea et al., 2009). Therefore, when analyzed collectively, it is reasonable to hypothesize that activation of the TLR4-derived signaling pathway by morphine, a recently documented TLR4 agonist (Watkins et al., 2009), is one potential pathway that links chronic morphine administration to activation of the ceramide metabolic pathway and hence the development of hyperalgesia and antinociceptive tolerance. We are currently testing this hypothesis.
Considering the appreciable molecular, biochemical, and pharmacological similarities between opiate-mediated hypersensitivity and hypersensitivity associated with chronic neuropathic pain from diabetes mellitus and other sensory neuropathies (Mao et al., 1995; Watkins et al., 2005), ceramide and S1P may be viable therapeutic targets in both conditions.
Footnotes
This work was supported by National Institutes of Health Grants R01 DA024074 and R21 DA023056 (D.S.). We thank Dr. Lina M. Obeid (Medical University of South Carolina, Charleston, SC) for critical input to this manuscript.
The authors declare no conflicts of interest.
References
- Arnér S, Rawal N, Gustafsson LL. Clinical experience of long-term treatment with epidural and intrathecal opioids: a nationwide survey. Acta Anaesthesiol Scand. 1988;32:253–259. doi: 10.1111/j.1399-6576.1988.tb02725.x. [DOI] [PubMed] [Google Scholar]
- Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant L, Doyle T, Chen Z, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Mazzon E, Petrusca DN, Petrache I, Salvemini D. Spinal ceramide and neuronal apoptosis in morphine antinociceptive tolerance. Neurosci Lett. 2009;463:49–53. doi: 10.1016/j.neulet.2009.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang CY, Wang J, Xie YF, Zhang S, Hu JW, Dostrovsky JO, Sessle BJ. Astroglial glutamate-glutamine shuttle is involved in central sensitization of nociceptive neurons in rat medullary dorsal horn. J Neurosci. 2007;27:9068–9076. doi: 10.1523/JNEUROSCI.2260-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus RA, Bunck AC, Bockmeyer CL, Brunkhorst FM, Lösche W, Kinscherf R, Deigner HP. Role of increased sphingomyelinase activity in apoptosis and organ failure of patients with severe sepsis. FASEB J. 2005;19:1719–1721. doi: 10.1096/fj.04-2842fje. [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Genovese T, Mazzon E, Esposito E, Crisafulli C, Di Paola R, Bramanti P, Salvemini D. Fumonisin b1 reduces the development of multiple organ failure induced by zymosan in mice. Shock. 2009;31:170–177. doi: 10.1097/SHK.0b013e31817fbd4a. [DOI] [PubMed] [Google Scholar]
- D'Amour F. A method for determining loss of pain sensation. J Pharmacol Exp Ther. 1941;72:74–79. [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Delgado A, Casas J, Llebaria A, Abad JL, Fabrias G. Inhibitors of sphingolipid metabolism enzymes. Biochim Biophys Acta. 2006;1758:1957–1977. doi: 10.1016/j.bbamem.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Delogu G, Famularo G, Amati F, Signore L, Antonucci A, Trinchieri V, Di Marzio L, Cifone MG. Ceramide concentrations in septic patients: a possible marker of multiple organ dysfunction syndrome. Crit Care Med. 1999;27:2413–2417. doi: 10.1097/00003246-199911000-00015. [DOI] [PubMed] [Google Scholar]
- Doyle T, Bryant L, Muscoli C, Cuzzocrea S, Esposito E, Chen Z, Salvemini D. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett. 2010;483:85–89. doi: 10.1016/j.neulet.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley KM. Misconceptions and controversies regarding the use of opioids in cancer pain. Anticancer Drugs. 1995;6(Suppl 3):4–13. doi: 10.1097/00001813-199504003-00002. [DOI] [PubMed] [Google Scholar]
- French KJ, Schrecengost RS, Lee BD, Zhuang Y, Smith SN, Eberly JL, Yun JK, Smith CD. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- Görg B, Wettstein M, Metzger S, Schliess F, Häussinger D. Lipopolysaccharide-induced tyrosine nitration and inactivation of hepatic glutamine synthetase in the rat. Hepatology. 2005;41:1065–1073. doi: 10.1002/hep.20662. [DOI] [PubMed] [Google Scholar]
- Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Jolly PS, Rosenfeldt HM, Milstien S, Spiegel S. The roles of sphingosine-1-phosphate in asthma. Mol Immunol. 2002;38:1239–1245. doi: 10.1016/s0161-5890(02)00070-6. [DOI] [PubMed] [Google Scholar]
- Keller M, Lidington D, Vogel L, Peter BF, Sohn HY, Pagano PJ, Pitson S, Spiegel S, Pohl U, Bolz SS. Sphingosine kinase functionally links elevated transmural pressure and increased reactive oxygen species formation in resistance arteries. FASEB J. 2006;20:702–704. doi: 10.1096/fj.05-4075fje. [DOI] [PubMed] [Google Scholar]
- King T, Vardanyan A, Majuta L, Melemedjian O, Nagle R, Cress AE, Vanderah TW, Lai J, Porreca F. Morphine treatment accelerates sarcoma-induced bone pain, bone loss, and spontaneous fracture in a murine model of bone cancer. Pain. 2007;132:154–168. doi: 10.1016/j.pain.2007.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitano M, Hla T, Sekiguchi M, Kawahito Y, Yoshimura R, Miyazawa K, Iwasaki T, Sano H, Saba JD, Tam YY. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 2006;54:742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- Krishnamurthy K, Dasgupta S, Bieberich E. Development and characterization of a novel anti-ceramide antibody. J Lipid Res. 2007;48:968–975. doi: 10.1194/jlr.D600043-JLR200. [DOI] [PubMed] [Google Scholar]
- Lai WQ, Irwan AW, Goh HH, Howe HS, Yu DT, Valle-Oñate R, McInnes IB, Melendez AJ, Leung BP. Anti-inflammatory effects of sphingosine kinase modulation in inflammatory arthritis. J Immunol. 2008;181:8010–8017. doi: 10.4049/jimmunol.181.11.8010. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Xu DZ, Feketeova E, Kannan KB, Yun JK, Deitch EA, Fekete Z, Livingston DH, Hauser CJ. Attenuation of shock-induced acute lung injury by sphingosine kinase inhibition. J Trauma. 2004;57:955–960. doi: 10.1097/01.ta.0000149495.44582.76. [DOI] [PubMed] [Google Scholar]
- Maines LW, Fitzpatrick LR, French KJ, Zhuang Y, Xia Z, Keller SN, Upson JJ, Smith CD. Suppression of ulcerative colitis in mice by orally available inhibitors of sphingosine kinase. Dig Dis Sci. 2008;53:997–1012. doi: 10.1007/s10620-007-0133-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Mechanisms of hyperalgesia and morphine tolerance: a current view of their possible interactions. Pain. 1995;62:259–274. doi: 10.1016/0304-3959(95)00073-2. [DOI] [PubMed] [Google Scholar]
- Melendez AJ. Sphingosine kinase signalling in immune cells: potential as novel therapeutic targets. Biochim Biophys Acta. 2008;1784:66–75. doi: 10.1016/j.bbapap.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miñana MD, Kosenko E, Marcaida G, Hermenegildo C, Montoliu C, Grisolía S, Felipo V. Modulation of glutamine synthesis in cultured astrocytes by nitric oxide. Cell Mol Neurobiol. 1997;17:433–445. doi: 10.1023/A:1026339428059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscoli C, Visalli V, Colica C, Nisticò R, Palma E, Costa N, Rotiroti D, Nisticò G, Mollace V. The effect of inflammatory stimuli on NMDA-related activation of glutamine synthase in human cultured astroglial cells. Neurosci Lett. 2005;373:184–188. doi: 10.1016/j.neulet.2004.09.079. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Ozawa T, Shige K, Yamamoto R, Minami M, Satoh M. Inhibition of morphine tolerance and dependence by MS-153, a glutamate transporter activator. Eur J Pharmacol. 2001;419:39–45. doi: 10.1016/s0014-2999(01)00965-7. [DOI] [PubMed] [Google Scholar]
- Narita M, Yoshida T, Nakajima M, Narita M, Miyatake M, Takagi T, Yajima Y, Suzuki T. Direct evidence for spinal cord microglia in the development of a neuropathic pain-like state in mice. J Neurochem. 2006;97:1337–1348. doi: 10.1111/j.1471-4159.2006.03808.x. [DOI] [PubMed] [Google Scholar]
- Nayak D, Huo Y, Kwang WX, Pushparaj PN, Kumar SD, Ling EA, Dheen ST. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience. 2010;166:132–144. doi: 10.1016/j.neuroscience.2009.12.020. [DOI] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Esposito E, Mazzon E, Di Paola R, Matuschak GM, Salvemini D. Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J. 2008;22:3154–3164. doi: 10.1096/fj.08-108159. [DOI] [PubMed] [Google Scholar]
- Ndengele MM, Cuzzocrea S, Masini E, Vinci MC, Esposito E, Muscoli C, Petrusca DN, Mollace V, Mazzon E, Li D, Petrache I, Matuschak GM, Salvemini D. Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J Pharmacol Exp Ther. 2009;329:64–75. doi: 10.1124/jpet.108.146290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiuma T, Nishimura Y, Okada T, Kuramoto E, Kotani Y, Jahangeer S, Nakamura S. Inhalation of sphingosine kinase inhibitor attenuates airway inflammation in asthmatic mouse model. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1085–L1093. doi: 10.1152/ajplung.00445.2007. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, Vanderah TW, Porreca F. Induction of pain facilitation by sustained opioid exposure: relationship to opioid antinociceptive tolerance. Life Sci. 2003;73:783–800. doi: 10.1016/s0024-3205(03)00410-7. [DOI] [PubMed] [Google Scholar]
- Pushparaj PN, H'Ng SC, Melendez AJ. Refining siRNA in vivo transfection: silencing SPHK1 reveals its key role in C5a-induced inflammation in vivo. Int J Biochem Cell Biol. 2008;40:1817–1825. doi: 10.1016/j.biocel.2008.01.015. [DOI] [PubMed] [Google Scholar]
- Ramos KM, Lewis MT, Morgan KN, Crysdale NY, Kroll JL, Taylor FR, Harrison JA, Sloane EM, Maier SF, Watkins LR. Spinal upregulation of glutamate transporter GLT-1 by ceftriaxone: therapeutic efficacy in a range of experimental nervous system disorders. Neuroscience. 2010;169:1888–1900. doi: 10.1016/j.neuroscience.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renfrey S, Downton C, Featherstone J. The painful reality. Nat Rev Drug Discov. 2003;2:175–176. doi: 10.1038/nrd1038. [DOI] [PubMed] [Google Scholar]
- Romero-Sandoval A, Chai N, Nutile-McMenemy N, Deleo JA. A comparison of spinal Iba1 and GFAP expression in rodent models of acute and chronic pain. Brain Res. 2008a;1219:116–126. doi: 10.1016/j.brainres.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Sandoval EA, Horvath RJ, DeLeo JA. Neuroimmune interactions and pain: focus on glial-modulating targets. Curr Opin Investig Drugs. 2008b;9:726–734. [PMC free article] [PubMed] [Google Scholar]
- Salvemini D, Neumann W. Targeting peroxynitrite driven nitroxidative stress with synzymes: a novel therapeutic approach in chronic pain management. Life Sci. 2010;86:604–614. doi: 10.1016/j.lfs.2009.06.011. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Neumann WL. Peroxynitrite: a strategic linchpin of opioid analgesic tolerance. Trends Pharmacol Sci. 2009;30:194–202. doi: 10.1016/j.tips.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Salvemini D, Jensen MP, Riley DP, Misko TP. Therapeutic manipulations of peroxynitrite. Drug News Perspect. 1998;11:204–214. [PubMed] [Google Scholar]
- Schoeniger-Skinner DK, Ledeboer A, Frank MG, Milligan ED, Poole S, Martin D, Maier SF, Watkins LR. Interleukin-6 mediates low-threshold mechanical allodynia induced by intrathecal HIV-1 envelope glycoprotein gp120. Brain Behav Immun. 2007;21:660–667. doi: 10.1016/j.bbi.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonnet G, Rivat C. Opioid-induced hyperalgesia: abnormal or normal pain? Neuroreport. 2003;14:1–7. doi: 10.1097/00001756-200301200-00001. [DOI] [PubMed] [Google Scholar]
- Snider AJ, Kawamori T, Bradshaw SG, Orr KA, Gilkeson GS, Hannun YA, Obeid LM. A role for sphingosine kinase 1 in dextran sulfate sodium-induced colitis. FASEB J. 2009;23:143–152. doi: 10.1096/fj.08-118109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Størkson RV, Kjørsvik A, Tjølsen A, Hole K. Lumbar catheterization of the spinal subarachnoid space in the rat. J Neurosci Methods. 1996;65:167–172. doi: 10.1016/0165-0270(95)00164-6. [DOI] [PubMed] [Google Scholar]
- Suárez I, Bodega G, Fernández B. Glutamine synthetase in brain: effect of ammonia. Neurochem Int. 2002;41:123–142. doi: 10.1016/s0197-0186(02)00033-5. [DOI] [PubMed] [Google Scholar]
- Taha TA, Argraves KM, Obeid LM. Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta. 2004;1682:48–55. doi: 10.1016/j.bbalip.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60:181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi N, Logan R, Teves L, Wallace MC, Gurd JW. Altered interaction between PSD-95 and the NMDA receptor following transient global ischemia. J Neurochem. 2000;74:169–178. doi: 10.1046/j.1471-4159.2000.0740169.x. [DOI] [PubMed] [Google Scholar]
- Tanga FY, Nutile-McMenemy N, DeLeo JA. The CNS role of Toll-like receptor 4 in innate neuroimmunity and painful neuropathy. Proc Natl Acad Sci U S A. 2005;102:5856–5861. doi: 10.1073/pnas.0501634102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanga FY, Raghavendra V, Nutile-McMenemy N, Marks A, Deleo JA. Role of astrocytic S100beta in behavioral hypersensitivity in rodent models of neuropathic pain. Neuroscience. 2006;140:1003–1010. doi: 10.1016/j.neuroscience.2006.02.070. [DOI] [PubMed] [Google Scholar]
- Taylor DA, Fleming WW. Unifying perspectives of the mechanisms underlying the development of tolerance and physical dependence to opioids. J Pharmacol Exp Ther. 2001;297:11–18. [PubMed] [Google Scholar]
- Trotti D, Rossi D, Gjesdal O, Levy LM, Racagni G, Danbolt NC, Volterra A. Peroxynitrite inhibits glutamate transporter subtypes. J Biol Chem. 1996;271:5976–5979. doi: 10.1074/jbc.271.11.5976. [DOI] [PubMed] [Google Scholar]
- Trotti D, Rolfs A, Danbolt NC, Brown RH, Jr, Hediger MA. SOD1 mutants linked to amyotrophic lateral sclerosis selectively inactivate a glial glutamate transporter. Nat Neurosci. 1999;2:848. doi: 10.1038/12227. [DOI] [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, Porreca F. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45. doi: 10.1016/j.pain.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Ma W, Chabot JG, Quirion R. Cell-type specific activation of p38 and ERK mediates calcitonin gene-related peptide involvement in tolerance to morphine-induced analgesia. FASEB J. 2009;23:2576–2586. doi: 10.1096/fj.08-128348. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Porreca F, Cuzzocrea S, Galen K, Lightfoot R, Masini E, Muscoli C, Mollace V, Ndengele M, Ischiropoulos H, Salvemini D. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Exogenous glutamate is metabolized to glutamine and exported by rat primary astrocyte cultures. J Neurochem. 1986;47:304–313. doi: 10.1111/j.1471-4159.1986.tb02863.x. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Milligan ED, Maier SF. Glial activation: a driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Johnston IN, Maier SF. Glia: novel counter-regulators of opioid analgesia. Trends Neurosci. 2005;28:661–669. doi: 10.1016/j.tins.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The “toll” of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Vasko MR, Nicol GD. Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J Physiol. 2002;544:385–402. doi: 10.1113/jphysiol.2002.024265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YH, Fehrenbacher JC, Vasko MR, Nicol GD. Sphingosine-1-phosphate via activation of a G-protein-coupled receptor(s) enhances the excitability of rat sensory neurons. J Neurophysiol. 2006;96:1042–1052. doi: 10.1152/jn.00120.2006. [DOI] [PubMed] [Google Scholar]