

Abstract
Objective
The singular phenomenon of switching from depression to its opposite state of mania or hypomania, and vice versa, distinguishes bipolar disorder (BPD) from all other psychiatric disorders. Despite the fact that it is a core aspect of the clinical presentation of BPD, the neurobiology of the switch process is still poorly understood. In this review we summarize the clinical evidence regarding somatic interventions associated with switching, with a particular focus on the biological underpinnings presumably involved in the switch process.
Data Sources
Literature for this review was obtained through a search of the MEDLINE database (1966–2008).
Study Selection
All English-written, peer-reviewed, published literature, including randomized controlled studies, naturalistic and open-label studies, and case-reports were eligible for inclusion.
Data Synthesis
Converging evidence suggests that certain pharmacological and non-pharmacological interventions with very different mechanisms of action, such as sleep deprivation, exogenous corticosteroids, and dopaminergic agonists, can trigger mood episode switches in patients with BPD. The switch-inducing potential of antidepressants is unclear, although tricyclic antidepressants (TCAs), which confer higher risk of switching than other classes of antidepressants, are a possible exception. Several neurobiological factors appear to be associated with both spontaneous and treatment-emergent mood episode switches; these include abnormalities in catecholamine levels, upregulation of neurotrophic and neuroplastic factors, HPA-axis hyperactivity, and circadian rhythms.
Conclusions
There is a clear need to improve our understanding of the neurobiology of the switch process; research in this field would benefit from the systematic and integrated assessment of variables associated with switching.
Keywords: bipolar disorder (BPD), neurobiology, switch process, treatment-emergent affective switch (TEAS), predictors of switch
1. Introduction
The singular phenomenon of switching from depression to its opposite state of mania or hypomania, and vice versa, distinguishes bipolar disorder (BPD) from all other psychiatric disorders. Although symptoms such as depressed mood, insomnia, paranoid ideation, anxiety, and appetite changes are experienced across many psychiatric disorders, the process of switching from depression to a state of mania or hypomania is a unique and core feature of BPD. Currently, no uniform definition exists to describe the switch phenomenon; herein, we have defined it as a sudden transition from a mood episode to another episode of the opposite polarity. The importance of the switch process as the hallmark of BPD was described in Falret’s original (1854) conceptualization of ‘circular insanity’, which he defined as a form of illness in which “depression and mania must succeed one another for a long time, usually for the whole of the patient’s life, and in a fashion very nearly regular, and with intervals of rationality, which are usually short compared with the length of the episodes”1 (translated into English by Sedler and Desain2, page 1130).
Historically, abrupt changes in mood polarity were described well before the beginning of the psychopharmacological era. Manic features observed after a depressive episode were commonly described as “post-melancholic reactive hyperthymia”, while mania that evolved into depression was referred to as “reactive depression”3. Retrospective data obtained from patients hospitalized between 1920 and 1959 show a rate of 29% for spontaneous switching from depression to hypomania4. However, in modern psychiatry, the term “switch” connotes not only switches in mood polarity as a core feature of BPD, but also treatment-emergent affective switch (TEAS)—often from depression to mania/hypomania. The fact that the term switch is now used synonymously to encompass both types of mood shifts has led psychiatrists to neglect the study of both spontaneous switches (i.e. non-treatment-related) as well as the transition from mania/hypomania to depression.
A possible reason for the dearth of modern studies addressing the neurobiology of spontaneous switches is that, presently, most patients with BPD have complex treatment regimens involving multiple drugs, so that the vast majority of clinical trials done in patients with BPD enroll patients who are already medicated. Conversely, most of the data regarding non-treatment-induced switches (spontaneous switches) derive from studies conducted two to three decades ago when the efficacy of lithium as a prophylactic agent in BPD was still debated, and when monotherapy trials with antidepressants were still being conducted, at least in the United States. As a result, most of the evidence presented in this paper refers to TEAS, unless otherwise specified. The issue of TEAS itself is also one that continues to be the center of considerable controversy. There is genuine uncertainty regarding the potential benefit or harm associated with the use of antidepressants during depressive episodes of BPD. Because the depressed phase of BPD is associated with significant morbidity and increased risk of suicide, this is a significant public health challenge.
Despite the importance of the switch phenomenon, the precise mechanisms underlying the process have yet to be elucidated. Moreover, the neurobiology and long-term clinical consequences of the switch process are still poorly understood. Switching from depression to mania/hypomania can occur spontaneously over the course of the illness, but can also be precipitated by stress, sleep deprivation, or standard treatment for bipolar depression such as electroconvulsive therapy (ECT) and some antidepressants5, 6 (see below), as well as various other agents (e.g., amphetamines and glucocorticoids). In addition, recent evidence suggests that genes that regulate monoaminergic transmission or circadian rhythms might increase individual susceptibility for switching7–9. Another key issue is that most of the research on both spontaneous switches and TEAS has observed the switch from depression into mania. Because the switch from mania to depression occurs in a relatively smaller proportion of patients with BPD than the switch from depression into mania10, data regarding this phenomenon are very sparse, but in this review are noted whenever possible.
Across the spectrum of BPD, there is wide individual variability in how often any given individual diagnosed with BPD will have either spontaneous switching or TEAS. Clinically, this information can be quite important, because having a pattern of switching is associated with several clinical consequences. For instance, evidence suggests that, compared to non-switchers, switchers have a higher genetic loading for mood disorders, spend more time ill during the course of their lifetime, experience significantly more comorbidities, and are at greater risk for developing substance abuse or committing suicide11–14. TEAS in particular is believed to be associated with worsening clinical outcome, including cycle acceleration15, 16. It is also unknown whether individuals switch only when exposed to particular triggers (e.g. antidepressants, glucocorticoids, sleep deprivation), whether switchers have a general propensity to switch in response to any given treatment known to induce switch, or whether some individuals are genetically more likely to experience switching regardless of triggers. Understanding this unique process is crucial to our understanding of the pathophysiology of BPD. Notably, although the switch process may be involved in the phenomenon of rapid cycling and cycle acceleration, the intention of this review is to focus primarily on the switch process and not necessarily on rapid cycling and cycle acceleration or the possible long-term prognostic implications of switching. Switching is an event circumscribed to a period of time that facilitates its study and is more likely to yield information on the molecular underpinnings of the switch process per se; in contrast, rapid cycling and cycle acceleration occur over a longer period of time and are likely to be associated with distinct neurobiological correlates that may or may not be involved in switching.
In this review, we will first discuss the clinical predictors of switching and their significance. We will then discuss the clinical evidence regarding pharmacological interventions associated with switching, with a particular focus on the individual neurotransmitter systems and the possible biological mechanisms involved in this process. Literature for this review was obtained by searching the MEDLINE database (1966–2008) using the following keywords and phrases: switch, bipolar disorder, bipolar depression, antidepressant, SSRIs, tricyclic antidepressants, norepinephrine, serotonin, treatment emergent affective switch, mania, hypomania, HPA-axis, glucocorticoids, amphetamine, dopamine, and sleep deprivation. All English-written, peer-reviewed, published studies, including randomized controlled trials, naturalistic and open-label studies, and case-reports were eligible for inclusion.
2. Clinical predictors of switch and their significance
Few studies have tried to characterize the clinical characteristics of the switch process in BPD or its prognostic significance. To study the clinical and prognostic correlates of the phenomenon of switching in patients with BPD, Maj and colleagues14 prospectively compared a group of patients who experienced a mood switch (defined as a sudden transition from a mood episode to another episode of the opposite polarity with an intervening period of no more than one month) to a comparison group of subjects who did not experience any switches during an observational period of at least three years. The study found that switchers were more likely to have a greater number of hospitalizations previous to their study index episode and to need more time to recover from their index episode. Furthermore, the time to 50% probability of recovery was significantly longer for patients who experienced more than one switch (i.e., those having a polyphasic episode) during their index episode (44 weeks) compared to patients who had only one switch (i.e., those having a biphasic episode) (12 weeks) or to non-switchers (seven weeks). Likewise, patients with more than one switch spent more time in mood episodes during the observational period following the index episode than the other two groups. In this study, switching from depression to mania/hypomania was associated with a poorer prognosis as well as an increased risk of switching during subsequent episodes than switching from mania/hypomania to depression. In addition, switchers were more likely to show psychomotor retardation than non-switchers. However, neither gender, a positive family history of BPD, nor age at recruitment were significant predictors of switching14.
Another retrospective study found that the presence of mixed symptoms during a depressive episode was associated with an increased risk of having a manic switch17. In another study, Zarate and colleagues18 found that a mixed manic presentation was a strong predictor of switch from mania to depression. They investigated clinical and demographic predictors of switch from mania to depression in 28 switchers and 148 non-switchers. In this study, switching from mania to depression was not associated with a longer time to recovery or earlier time to relapse during the 24-month follow-up period. In these two studies the treatment status at the time of switching was not controlled for, as the patients were undergoing uncontrolled treatment with multiple classes of drugs. A recent observational study that investigated the switch from mania to depression noted that a history of previous depressive episodes, substance abuse, greater overall severity on the Clinical Global Impressions Scale for Bipolar Disorder (CGI-BP), and benzodiazepine use all increased the risk of this type of switch. Conversely, the authors also identified factors associated with lower switch rates from mania to depression, including atypical antipsychotic use, lower Young Mania Rating Scale (YMRS) severity, and higher CGI-BP depression scores10.
Several clinical variables have been studied specifically as potential predictors of TEAS, including gender, diagnosis, age, number of previous episodes of mania, previous history of TEAS, and polarity of onset episode. Some studies have found that switchers have a higher number of past manic episodes19, while others found more past manic episodes in non-switchers20, and still others found no differences between switchers and non-switchers21 on this variable. Serretti and colleagues found an association between TEAS and depressive polarity of illness onset20, but this was not replicated in subsequent studies22, 23. Two studies reported that switchers were older at intake20, 24, but the opposite association (i.e., earlier age at intake) was reported in a more recent study22. Also, a positive past history for TEAS was found to predict current TEAS in some22 but not all studies19, 25. A positive history of rapid cycling has also been associated with TEAS20, 22. However, gender, family history, age of onset, and substance abuse have not been found to predict TEAS20, 22, 23.
In addition, a number of studies have focused specifically on clinical predictors of TEAS when antidepressants are administered. Data from the Systematic Treatment Enhancement Program for Bipolar Disorder22 suggest that a past history of multiple antidepressant trials is associated with TEAS22. A history of past TEAS also seems to be associated with the development of chronic dysphoria following antidepressant administration26. Another important question is whether patients with BPD-I and BPD-II differ in their risk for TEAS; while some studies detected an increased likelihood of switch in patients with BPD-I27, others reported no difference21, 28, or increased risk for subjects with BPD-II20. However, a recent meta-analysis that combined results from nine different studies assessing TEAS rates in patients with BPD-I and BPD-II noted that patients with BPD-I had a significantly higher risk of TEAS (14.2 vs 7.1 %, respectively)29.
3. Antidepressants and switch
The evidence regarding the likelihood that antidepressant treatment in individuals with BPD confers increased risk of TEAS has long been controversial and inconclusive, and it is beyond the scope of this review to extensively discuss this controversy (we refer the interested reader to some authoritative reviews on the topic5, 30, 31).
Virtually all antidepressants have been associated with increased risk for TEAS; studies have found that antidepressant-induced TEAS ranges from 10% to 70%21, depending on the methodological heterogeneity of the study design, concomitant treatment, and the type of statistical analyses conducted. Many researchers have recently discussed the methodological flaws associated with many of the studies from which this evidence was drawn5, 30. These include investigating switch potential as a secondary aim or post-hoc analysis, heterogeneity in terms of concomitant treatments administered to patients, and lack of agreement on TEAS-defining criteria. Similarly, different diagnostic criteria such as heterogeneity of YMRS score cut-off for defining a switch, and duration of follow-up need to be considered when interpreting the results (see Table 1 for an overview of switch criteria used in the studies described herein). Ideally, in order for results to be comparable across studies, a single a priori definition of switching should be provided, with fulfillment of DSM-IV criteria for mania or hypomania within a short time frame (e.g., six weeks) from the beginning of antidepressant treatment in patients experiencing a depressive episode. Our present lack of a consensus definition or temporal criteria may dilute the biological underpinnings of this phenomenon, because subjects who develop affective switch within very different time frames from the start of antidepressant treatments are considered equivalent. This methodological issue has been recently emphasized by a task force of the International Society for Bipolar Disorder, which recommended empirical testing in clinical trials of the reliability of different definitions of switch6.
Table 1.
Operational criteria for defining “switch” applied in different pharmacological studies.
| Study | Time from start of AD required to define TEAS | Type of study | Definition of switch and treatment-emergent switch |
|---|---|---|---|
| Lewis & Winokur, 198238 | None required | Retrospective | DSM-III mania while hospitalized or within 6 months of discharge |
| Cohn et al., 198942 | 6 weeks | RCT | Not specified |
| Himmelhoch et al, 199140 | 6 weeks | RCT | Mania, RDC criteria |
| Peet, 199433 | Not specified | Retrospective | Not specified |
| Sachs et al, 199443 | 8 weeks | RCT | DSM-IIIR mania or hypomania |
| Altshuler et al., 1995 | 8 weeks | Retrospective | Mania within 8 weeks of the initiation of AD tx |
| Boerlin et al., 199834 | Within 2 months after a depressive episode | Retrospective | DSM-IV mania/hypomania |
| Bottlender et al., 200137 | None required | Retrospective | Mania/hypomania according to the physician's assessment based on DSM-IV criteria |
| Henry et al., 200121 | 6 weeks | Naturalistic | DSM-IV mania/hypomania or mixed episode within 6 weeks of initiation of AD tx |
| Mundo et al., 20019 | None required | Retrospective | DSM-IV mania/hypomania while being treated with SSRIs for depression |
| Nemeroff et al., 200139 | 10 weeks | RCT | DSM-IV mania |
| Silverstone, 200141 | 8 weeks | RCT | YMRS score ≥ 10, or study discontinuation for manic sx |
| Joffe et al., 200249 | Not specified; switch attributed to an AD based on clinical judgement | Naturalistic | DSM-IV mania or hypomania |
| McIntyre et al, 200248 | 8 weeks | RCT | Not specified |
| Maj et al., 200214 | None required | Naturalistic | One episode of mania or hypomania and one episode of depression (RDC) with an intervening period of <1 month |
| Vieta et al., 200250 | 6 weeks | RCT | YMRS score >11 and fulfilling DSM-IV criteria for mania or hypomania |
| Rousseva et al., 200356 | None required/90 days | Retrospective | Broad definition: self-report of mood elevation at any time after the introduction of an AD; Narrow definition: self-report of mood elevation within 90 days from the beginning of tx |
| Serretti et al., 200320 | None required | Retrospective | DSM-IV mania/hypomania while being treated with SSRIs for depression |
| Tohen et al., 200353 | 8 weeks | RCT | YMRS score <15 at baseline and >15 at any time thereafter |
| Serretti et al., 200457 | 4 weeks | Retrospective | DSM-IV mania/hypomania while being treated with ADs for depression |
| Tamada et al., 200424 | None required | Naturalistic | DSM-IV hospitalized mania or mixed state; YMRS score ≥ 12 and at least 3 days of AD tx within 2 weeks of hospital admission |
| Amsterdam & Shults, 200552 | 8 weeks | RCT | YMRS score >8 at any visit |
| Fonseca et al., 200654 | 12 weeks | Open-label | YMRS >12 and DSM-IV criteria for manic switch; DSM-IV criteria for hypomania for hypomanic switch |
| Post et al., 200625 | 10 weeks | RCT | Either a 2-point increase on the CGI-BP, or a CGI-BP of at least 3, or a YMRS>13 |
| Schaffer et al., 200655 | 12 weeks | RCT | Not specified |
| Carlson et al., 200723 | None required | Retrospective | DSM-IV mania/hypomania while being treated with ADs or within 30 days of stopping tx |
| Nolen et al., 200745 | 10 weeks | RCT | At least "much worse" on the CGI-BP rating of change in mania as baseline and/or YMRS ≥ 14 |
| Sachs et al., 200719 | 16 weeks | RCT | DSM-IV criteria for mania or hypomania or clinically significant mood elevation needing clinical intervention within 16 weeks or before reaching durable recovery (up to 26 weeks) |
| Truman et al., 200722 | 12 weeks | Retrospective | Non DSM-IV report of mania, hypomania, or mixed episode |
| Amsterdam & Shults, 200851 | 12 weeks | Open-label | Two different criteria: YMRS ≥ 8 or YMRS ≥ 12 at any visit |
Abbreviations: AD: antidepressant; CGI-BP: Clinical Global Impressions Scale for Bipolar Disorder; DSM: Diagnostic and Statistical Manual of Mental Disorders; RDC: research diagnostic criteria; RCT: randomized controlled trial; sx: symptoms; tx: treatment; YMRS: Young Mania Rating Scale.
Another critical issue is the uncertainty regarding switch rates in unmedicated patients; for instance, retrospective data obtained from patients hospitalized between 1920 and 1959 found a rate of 29% for spontaneous switching from depression to hypomania4. Without a clear benchmark estimating the rate at which patients are likely to switch spontaneously, it can be difficult to assess the degree to which antidepressants increase that risk. Relatedly, the fact that most patients with BPD receive antidepressants concomitantly with mood stabilizers32 makes switch rates even more difficult to estimate accurately.
Despite these limitations, results from clinical trials may provide important clues to understanding the neurobiology of the switch process by analyzing switch rates for antidepressants that target different neurotransmitter systems (for an excellent and extensive recent review of this topic, see 5). Below, we review what is known about the various classes of antidepressants and their propensity to cause TEAS in individuals with BPD.
3.1 TEAS associated with the use of various classes of antidepressants
Tricyclic antidepressants (TCAs) have consistently been associated with a high risk of TEAS compared to other antidepressants; naturalistic and retrospective studies have reported TEAS incidence rates ranging from 9% to 69%33–38. Because much of this knowledge has been previously and extensively reviewed by others and is already familiar to the reader31, we offer here only a brief discussion of the evidence concerning the mood-elevating potential of TCAs; when possible, we also include data from randomized controlled trials in bipolar depression.
Bunney and colleagues35 reviewed 80 studies involving 3923 patients mostly treated with TCAs for depression and found that the incidence of TEAS into mania or hypomania was 9.5%. A later study by Wehr and Goodwin36 of 26 patients with BPD-I and II found that 18 experienced manic or hypomanic switches while on TCAs after an average of 21 days for those with BPD-I and 35 days for those with BPD-II. Pooled data have similarly shown that mood switches are considerably more frequent with TCAs (11.2%) than with selective serotonin reuptake inhibitors (SSRIs) (3.7%) or placebo (4.2%)33. Bottlender and colleagues37 evaluated the incidence of mania and hypomania in 158 patients with BPD-I treated for depression. They describe switch rates of 34% for patients receiving TCAs. Similar switch rates were reported in a naturalistic study by Boerlin and colleagues34, who found that both TCAs and monoamine oxidase inhibitors (MAOIs) were associated with higher switch rates than the SSRI fluoxetine (32%, 35%, and 12%, respectively). The TCA imipramine has also been associated with TEAS (rates between 6.6 and 17.8%) in four studies39–42. These rates are considerably lower than those obtained from naturalistic and retrospective studies, but the enrollment of patients with milder forms of BPD in clinical trials compared to observational/naturalistic studies might explain this difference.
Evidence from a clinical trial in bipolar depression suggests that use of the TCA desipramine, which is a selective inhibitor of norepinephrine reuptake, was associated with a high frequency of switches into mania or hypomania (30%)43. However, no definitive conclusions can be drawn from this study, as few patients were enrolled (n= 10); furthermore, there have been no studies evaluating desipramine’s propensity to cause TEAS since 1994. One case report noted that reboxetine, another norepinephrine reuptake inhibitor (though not available in the U.S.), induces hypomania44.
Only three randomized clinical trials have evaluated TEAS in monoamine oxidase inhibitors (MAOIs). In the first trial, 3.7% of patients experienced manic/hypomanic symptoms leading to study withdrawal. Also, a YMRS score ≥ 10 was described in 9.3% of all patients taking moclobemide41. In the second study, the MAOI tranylcypromine caused manic or hypomanic switches in 11% of patients40. Finally, Nolen and colleagues45 reported no manic switches in eight patients with BPD openly randomized to tranylcypromine for 10 weeks as an add-on to mood stabilizers. Interestingly, a recent retrospective analysis of STEP-BD data suggests that TEAS is less likely to occur when MAOIs are administered in conjunction with mood stabilizers compared to other classes of antidepressants22.
Bupropion, a norepinephrine and dopamine reuptake inhibitor (NDRI), is associated with low TEAS potential, and its lower mood-elevating potential compared to TCAs has been described since the 1980s46, 47. Five clinical trials have evaluated its switch-inducing potential in patients with bipolar depression, with a frequency of mood episode switches ranging from 0 to 17.9%19, 25, 43, 48, 49. Notably, all the patients enrolled in bupropion trials were concomitantly treated with mood stabilizers, a factor that may have contributed to the low TEAS rates observed. The highest TEAS rates were reported in the trial with the longest temporal operational criteria for defining TEAS19 (see Table 1).
Two clinical trials have evaluated the switch-inducing potential of the selective norepinephrine reuptake inhibitor (SNRI) venlafaxine, which is a double inhibitor of serotonin and norepinephrine reuptake, in patients with bipolar depression (both BPD-I and BPD-II), and reported TEAS rates ranging between 13.3% and 29%25, 50; these rates were higher than TEAS rates reported for the other treatment arms (which used the SSRI paroxetine or sertraline, and the NDRI bupropion), thus suggesting that the perturbation of two monoaminergic systems is more likely to induce TEAS than when a single SSRI is used. The study by Post and colleagues25 also showed that different operational criteria are likely to account for the variability in TEAS rates associated with antidepressant treatment across the different trials. In fact, switch rates varied from 15 to 31% for venlafaxine, from 4 to 14% for bupropion, and from 7 to 16% for sertraline, depending on the criteria used to define the switch (either a two-point increase at any point in the trial on the CGI–BP, or a CGI–BP manic severity score of at least 3 (i.e. at least mildly manic) or a YMRS score above 13 at any visit). In addition, this trial was one of the largest to define the study of TEAS as one of its primary aims. The fact that the vast majority of the subjects enrolled in the studies by Post 25 and Vieta50 had a diagnosis of BPD-I (73% and 67%, respectively) might explain the apparent discrepancy in these findings; for instance, a recent study of patients with BPD-II only showed low TEAS rates for venlafaxine (2.4%) even when it was administered as monotherapy 51.
Post-hoc analyses of data from randomized controlled trials (RCTs) in bipolar depression usually show low switch rates associated with SSRIs. For example, no TEAS was reported in two trials of fluoxetine in bipolar depression42, 52. However, the pooled number of patients who received fluoxetine was low (n=38), and most of the patients came from a study that also detected low TEAS rates for the TCA imipramine42. Another major limitation of the study was the low completion rate: 43% of the patients randomized to fluoxetine dropped out of the study. Moreover, 18 of these patients (20%) concomitantly received lithium. Another study found no difference in mood episode switch between patients randomized to receive either olanzapine (an atypical antipsychotic) plus fluoxetine or placebo52. Five patients participating in this trial (15%) were also receiving lithium or valproate. Another study that compared the efficacy and TEAS rates for olanzapine monotherapy, placebo, or a combination of olanzapine plus fluoxetine found a TEAS rate of 6% in the latter group; however, this switch rate did not differ from the placebo group53. Consistent with these findings, analyses of pooled data from databases of pharmaceutical industry research show that mood switches occur in 3.7% of patients treated with SSRIs33.
Slightly higher TEAS rates were reported for the SSRI escitalopram in an open study led by Fonseca and colleagues54, where 15% (3/20) of the patients who received add-on escitalopram to their current mood stabilizer regimen dropped out of the study because of manic/hypomanic symptoms. Recently, Schaffer and colleagues55 reported that one out of 10 patients (10%) developed a manic switch during an open-label trial of citalopram added on to mood stabilizers. Similar TEAS rates were described in two uncontrolled retrospective studies37,34; the former found a switch rate of 12% when SSRIs were administered, despite the inclusion of patients who were currently taking mood stabilizers37. Notably, no significant differences in switch rates were found between patients on SSRI monotherapy and patients who took SSRIs as an add-on to mood stabilizers; however, only eight patients received SSRIs without mood stabilizers37. The TEAS rate of 12% observed in this study during treatment with SSRIs was higher than the one reported from the RCTs described above or from the pooled data from pharmaceutical companies, but it is possible that the less stringent inclusion criteria used in these naturalistic/observational studies (e.g., inclusion of rapid cyclers, patients with comorbidities) might explain this discrepancy. Furthermore, it is interesting to note that both of these two naturalistic studies found a significantly lower rate of TEAS when patients were treated with SSRIs than with TCAs37,34.
Finally, a recent meta-analysis of randomized placebo-controlled trials by Gijsman and colleagues32 concluded that available evidence suggests that antidepressants other than TCAs do not induce significantly more TEAS than placebo (4.7% vs. 3.8%); however, it is important to note that 75% of the subjects were receiving a concurrent mood stabilizer or atypical antipsychotic. The authors also recommended that TCAs not be used as first-line treatment in patients with bipolar depression, because they were associated with higher switch risk (in that study, 10%). Thus, the evidence suggests that TCAs—for which particularly high switch rates have been described—are more likely to trigger TEAS in patients with BPD. In contrast, relatively low switch rates have been reported for SSRIs and MAOIs. The data also indirectly suggest that the concomitant perturbation of more than one monoaminergic system might carry a higher risk of TEAS.
3.2 The role of the serotonergic, catecholaminergic, noradrenergic, and dopaminergic systems in the switch process
As the preceding section emphasized, antidepressants targeting the serotonergic, noradrenergic, and dopaminergic systems have been associated with various degrees of propensity to induce TEAS, providing valuable clues regarding the underlying mechanisms of the switch process.
Data from genetic studies that investigated polymorphisms involved in the homeostasis of the serotonergic system suggest it has a negligible role in the switch process, with one exception. Mundo and colleagues9 found that the short allele polymorphism of the serotonin transporter (5HTTLPR) was overrepresented in patients who developed treatment-emergent hypomania/mania after receiving SSRIs. However, this association was not confirmed in a subsequent study that applied both a broad and a narrow definition of TEAS56; failure to replicate the association between the switch pattern and the short variant of the serotonin transporter might be due to higher age of onset in the second study compared to the first. Another study investigated other potential candidate genes that regulate serotonergic system homeostasis and switch, such as 5HTTLPR, 5-HT2a, and tryptophan hydroxylase, but no association was found57. Tryptophan depletion, a procedure that depletes serotonin, does not generally cause mood changes in lithium-treated euthymic patients with BPD58, while catecholamine depletion evokes a rebound hypomania in patients with BPD (see below).
The role of the noradrenergic and dopaminergic systems in the switch process is not clearly defined or well-studied. Some historical studies tried to investigate the potential role of the noradrenergic and dopaminergic systems in TEAS in BPD by measuring peripheral metabolites of monoaminergic systems activity. Most of these case reports or case series were carefully conducted with inpatients studied across sequential episodes of switches. With the exception of three studies35, 59, 60, all other reports described here are single case studies of patients with rapid- or ultra-rapid cycling. The data summarized here generally refer to drug-free patients, with few exceptions35, 61. Higher urinary cyclic adenosine 3’5’monophosphate (cAMP)59, 62, urinary norepinephrine35, 63, 64, and dopamine35, 63 have all been associated with mania and, more relevant to the present discussion, the switch to mania. Increased urinary 3-methoxy-4-hydroxyphenylglycol (MHPG) has also been described in this context60, 65. Increased post-synaptic receptor sensitivity interacting with high levels of catecholamines has also been hypothesized to trigger manic switches in some patients with BPD60, 66.
Several genetic polymorphisms in the catecholaminergic system (D4 receptor, D2 receptor, Catechol-O-methyl transferase [COMT], MAO-A) have been proposed as putative risk factors for TEAS in BPD, but no polymorphism was specifically found to be associated with the switch process57. Interestingly, this study analyzed genetic polymorphisms that had previously been associated with antidepressant response67, thus suggesting that the process associated with spontaneous switching might have a very different mechanism from that associated with antidepressant response.
Evidence from rodent studies further supports a putative role for the catecholaminergic system in the switch process. Drugs that deplete norepinephrine in the CNS (reserpine-like drugs) produce depression-like symptoms (e.g., locomotor hypoactivity) in animal models, whereas drugs that increase norepinephrine levels, such as MAOIs and TCAs, are associated with antidepressant-like effects68. One hypothesis is that these antidepressant-like effects may occur through delayed postsynaptic receptor desensitization, leading to increased receptor responsivity69. This is theorized to be a critical physiological protective mechanism against acute and chronic receptor overstimulation that, in turn, might be associated with an increased risk for switching in BPD. Also, receptor supersensitivity, altered internalization of cell surface receptors, and changes in critical mRNA expression might result in altered monoaminergic activity in the prefrontal areas, leading to manic-like behavioral changes 70.
4. The glutamatergic system and switch
Abundant evidence now implicates glutamatergic system dysfunction in the pathophysiology and treatment of unipolar depression and BPD (reviewed in 71). For instance, animal models of BPD suggest that the glutamatergic system plays a major role in manic-like behaviors. Du and colleagues72 found that inhibition of glutamate receptor type 1/2 (GluR1/2) subunit of the alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor significantly attenuates amphetamine-induced hyperactivity in rodents. In addition, disruption of GluR6, a subunit of the kainate receptor (kainate receptors, along with AMPA receptors, are glutamatergic non-N-methyl D-aspartate (NMDA) ionotropic receptors), produces a complex set of symptoms in mice that resemble the behavioral symptoms of mania, including increased risk-taking behaviors and aggressiveness, hyperactivity, and less despair-type manifestations73. Whether and how these findings might be related to the switch process will need to be addressed in future studies.
Although the study of glutamatergic drugs in the treatment of mood disorders is still in its infancy, preliminary evidence from small trials and case reports suggests that drugs that modulate the glutamatergic system have low risk of inducing TEAS. For example, lamotrigine—an FDA-approved mood stabilizer that inhibits glutamate release through sodium and calcium channel blockage74—is not associated with significant risk of switch in patients with bipolar depression75. In another study of 14 patients with bipolar depression, riluzole, another inhibitor of glutamate release, was not associated with increased risk of switching; in that eight-week study, patients received riluzole as an add-on to lithium76.
The switch-inducing potential of glutamatergic drugs that act by blocking NMDA receptors (i.e., ketamine, memantine) is essentially unknown, as the clinical evidence for their use in BPD is small77. Studies in healthy volunteers found that individuals who received intravenously-administered ketamine showed significantly more euphoria than those who received amphetamine or placebo78, possibly indicating some switch-inducing potential. However, it is unclear whether ketamine or memantine elicit core manic symptoms in BPD patients, and not only euphoria. Clinical trials conducted with these agents have not noted any increased risk of switch associated with their use71. While no conclusions can yet be drawn about the propensity of these agents to induce mood switches in BPD, this is nevertheless an important new avenue of research that will undoubtedly further our understanding of the molecular underpinnings of the switch process.
5. Dopaminergic agonists (psychostimulants) and switch
Selective dopaminergic drugs, such as psychostimulants, have long been associated with high rates of TEAS, and have been empirically tested in preclinical studies. Murphy and colleagues79 studied the effects of L-dopa, and L-dopa + peripheral decarboxylase inhibitor alpha-methyl dopa hydrazine (MK-485) in a double-blind, randomized, placebo-controlled study in bipolar depression. Six out of seven subjects treated with L-dopa developed hypomanic symptoms after an average of 7.8 days. Interestingly, the symptoms decreased within 24–48 hours of discontinuing L-dopa. These results suggest that, at least for some patients, the switch into mania or hypomania is associated with increased functional brain norepinephrine and dopamine.
Similarly, amphetamines that promote dopamine release and inhibit its reuptake have been shown to either precipitate hypomania in patients with BPD or induce a “hypomanic-like” state in healthy subjects80, 81. Consistent with these findings, a chart review of depressed, medically ill patients found several cases of hypomania one to five days after d-amphetamine was initiated at doses as low as 5–10 mg/day82. Another study found a significant increase in subjective measures of thought processing speed and irritability in healthy volunteers who received 25 mg oral dextro-amphetamine, two symptoms often associated with mania80. However, whether amphetamine can trigger other core manic symptoms (e.g., grandiosity, aggressive behaviors, pressured speech) has yet to be demonstrated. Amphetamine has been shown to trigger euphoria in healthy volunteers, mostly due to increased dopamine levels in the anteroventral striatum83. Polymorphisms in the dopamine (DAT1) and norepinephrine (SLC6A2) transporters are known to modulate the mood-elevating effects of amphetamine84, 85.
Pharmacological evidence supports the notion that manipulating the dopaminergic system can mimic the symptoms of BPD. Investigators have used a catecholamine depletion strategy employing the tyrosine hydroxylase inhibitor alpha-methyl-p-tyrosine (AMPT) in lithium-treated, euthymic patients with BPD to study the pathophysiology of the disorder86. Intriguingly, AMPT was not associated with any mood-lowering effects, but was associated with “rebound” hypomanic symptoms. Although preliminary, these results are compatible with the theory of a dysregulated signaling system wherein the compensatory adaptation to catecholamine depletion results in an “overshoot” due to impaired homeostatic mechanisms. Most recently, McTavish and colleagues87 found that a tyrosine-free mixture lowered both subjective and objective measures of the psychostimulant effects of methamphetamine or amphetamine, as well as manic symptom scores. These preliminary findings suggest that decreased tyrosine availability to the brain attenuates pathological increases in dopaminergic neurotransmission following methamphetamine administration and, putatively, in mania.
Evidence from animal models shows that decreased dopaminergic activity and receptor binding in the mesolimbic cortex and nucleus accumbens is associated with depression-like states that can be reversed by diverse antidepressants that potentiate dopaminergic activity88–91. In contrast, stimulants with dopaminergic properties (such as amphetamine and cocaine), lead to both manic-like effects and increased sensitization in diverse animal models of BPD92. Intriguingly, quinpirole, a D2/D3 agonist, induces a biphasic motor activity response, characterized by initial inhibition followed by hyperactivity, which resembles the switch process in BPD93, 94.
Furthermore, psychostimulants exert opposite effects than mood stabilizers on major intracellular signaling cascades, which might also be relevant for the switch process. For example, increased striatal dopaminergic activity—either in dopaminergic transporter knock-out mice or following amphetamine administration—is mediated by the activation of glycogen-synthase kinase 3 (GSK-3) α and β, whose inhibition is pivotal for the therapeutic actions of lithium and valproate95. Psychostimulants also activate protein kinase C (PKC), a family of enzymes that have been associated with the pathophysiology of BPD (reviewed in 96). Recent evidence shows that the integrity of the PKC pathway is critical for amphetamine-induced behavioral responses97 and that PKC inhibition has robust antimanic effects in patients with BPD98, 99.
These data are intriguing, as they show converging evidence from clinical and preclinical models regarding the major involvement of the dopaminergic system in mania and mood stabilization. However, whether activation of GSK-3 and PKC pathways is necessary for producing mood switching in patients with BPD is a topic that requires further investigation.
6. The Hypothalamic-Pituitary-Adrenal Axis (HPA) and switch
Since the early 1950s, the administration of HPA exogenous hormones has been reported to produce psychiatric symptoms in some patients with no pre-existing psychiatric disorders. In particular, adrenocorticotropic hormone (ACTH) and cortisone have been associated with mood elevation100, 101. A review of the literature prior to 1983 reported that the incidence of psychiatric symptoms in patients receiving corticosteroids ranged from 5.7 to 27.6% in uncontrolled studies, and 6.3 to 32% in controlled studies102. All of these cases were medically ill patients whose onset of psychiatric symptoms occurred within one day to several weeks of initiating treatment with glucocorticoids, and most of the patients developed mania/psychosis103. These psychiatric symptoms were clearly induced in a dose-response fashion, with a higher proportion of manic symptoms occurring in patients who received higher doses (>80 mg/day)104. Recent studies have also confirmed this association between corticosteroid administration and psychiatric symptoms. For example, glucocorticoids elevate mood in patients with multiple sclerosis105, ophthalmologic diseases106, asthma103, 107, 108, and also in healthy volunteers109. Notably, higher rates of BPD-like symptoms were usually associated with a positive personal or family history of psychiatric disorders107, 109.
Patients suffering from BPD are particularly susceptible to developing hypomanic/manic symptoms after receiving steroids. A recent study reviewing clinical charts from patients referred for a psychiatric consultation found nine patients with BPD whose psychiatric symptoms were precipitated by the use of corticosteroids (prednisone, betamethasone, methylprednisolone); seven of the nine (77%) rapidly developed manic/hypomanic symptoms110. Patients with BPD using a beclomethasone inhaler111, 112, as well as those using the androgen hormone dehydroepiandrosterone (DHEA)113, also developed mania. In addition, the single administration of triamcinolone in a celiac plexus block produced manic episodes in two patients with BPD114, confirming that susceptible patients can develop manic symptoms after the administration of even a single dose of glucocorticoids, and within a short period of time114. This relationship between the administration of glucocorticoids and the switch process is more striking when one considers that the administration of prednisone 40–60 mg on alternate days (in an on–off fashion) induced rapid-cycling symptoms in three patients115. These patients developed manic symptoms on the days they received prednisone; the opposite—a relapse into depression—occurred on the days they did not receive the drug.
In addition, hyperactivity of the HPA axis is one of the most replicated biological finding in major depression. Although the evidence for HPA dysfunction in BPD is not as well-validated, several authors have reported abnormalities in urinary and cerebrospinal fluid (CSF) cortisol levels and decreased dexamethasone test suppression in patients with BPD (see 116 for a review). In contrast, this finding is not observed in pure mania117, 118. However, this association does not necessarily implicate a causal relationship, as HPA axis hyperactivity might be an epiphenomenon of mounting mood elevation.
Converging evidence from small studies with rapid cyclers or ultra-rapid cyclers in patients with BPD suggest that HPA hyperactivity is critical for the switch from mania to depression in most of these patients63, 119–121; however, the role of the HPA axis in switching from depression to mania is more controversial. Notably, transgenic mice overexpressing glucocorticoid receptors in the forebrain displayed enhanced depressive-like behaviors and increased sensitization to cocaine and antidepressants122. They also had a wider range of reactivity to stimuli that trigger both negative and positive emotional responses, which might be relevant for the neurobiology of the switch process in BPD. Findings from other rodent studies, albeit not always consistent, further support the role of glucocorticoid receptors in affective-like behaviors (reviewed in 96).
7. Sleep deprivation and switch
Sleep deprivation has historically been proposed as a final common pathway prior to the onset of mania, and it can be triggered by diverse environmental, psychological, interpersonal, or pharmacological factors associated with the onset of mania123. Studies have consistently shown that sleep deprivation produces an acute antidepressant response in as many as 80% of subjects with bipolar depression and 60% of patients with unipolar depression124. Spontaneous switch rates after sleep deprivation vary from 10%125 to 30%123, 126 across studies, and this wide range is likely due to sample heterogeneity and the different treatment status of the patients. The fact that sleep deprivation acts quickly makes it an ideal tool to study the molecular basis of the switch process. However, it remains unclear why sleep deprivation causes temporary recovery in some patients, but triggers manic switches in others, and whether these two phenomena share the same neurobiological mechanism.
Sleep deprivation produces several behaviors in rats that suggest it may be a useful model for mania, including insomnia, hyperactivity, irritability127, 128, aggressive behavior129, novelty seeking preference130, and hypersexuality131. Moreover, rats exposed to serial sleep deprivation display behavioral sensitization, with worse manic-like symptoms emerging over repetition of the procedure, which parallels clinical findings of increased severity of illness over cumulative relapses in patients with BPD132. Sleep deprivation induces few effects at adrenergic or serotonergic receptors133, but directly regulates brain dopaminergic receptor sensitivity134. Increased plasma norepinephrine and norepinephrine metabolites have also been found in responders to sleep deprivation135,136. Decreased MHPG levels have also been found in the CSF of sleep deprivation responders compared to nonresponders137, 138. More recent studies have demonstrated that the expression of selected critical genes varies dramatically during sleep and waking139, which likely plays a major role in regulating long-term neuroplastic events related to the antidepressant effects of sleep deprivation. A number of mRNA differential display, microarray, and biochemical studies have also shown that short-term sleep deprivation is associated with both increased cyclic AMP response element binding protein levels (pCREB, the active form of this transcription factor) levels and increased the expression of brain-derived neurotrophic factor (BDNF) (and its receptor tyrosine kinase B, TrkB) expression (reviewed in 140).
In an extension of these gene expression studies, Cirelli and Tononi139 hypothesized that the level of activity of the neuromodulatory noradrenergic and serotonergic systems is a key factor in the induction of plasticity genes. Both of these systems project diffusely in the brain, where they regulate gene expression, and are quiescent only during REM sleep. To delineate the putative roles of the noradrenergic and serotonergic projections in regulating the expression of plasticity genes, a series of lesioning studies was undertaken. These studies showed that the expression of these molecules was regulated by the noradrenergic system, and that lesions in the locus coeruleus (LC) abolished the upregulation of their expression. In contrast, lesions of the serotonergic system had no effect on the level of expression of these genes (reviewed in 141), thus implying a negligible role for the serotonergic system in the neurobiology of response to sleep deprivation.
It has been suggested that sleep deprivation may bring about its rapid antidepressant effects by activating the LC noradrenergic system at a time when it would normally be quiescent (i.e., during periods of REM sleep at night). This would then allow the interaction of released norepinephrine with a primed, sensitized postsynaptic milieu in critical circuits, resulting in the rapid and robust expression of plasticity genes such as CREB, BDNF, and TrkB and, consequently, a rapid antidepressant response, as well as a switch into mania/hypomania141. Notably, an early case report by Gillin and colleagues142 documented nocturnal EEG recordings in a rapid-cycling patient who experienced four manic switches while asleep, and showed that on every occasion the last sleep stage recorded was REM; a possible role of increased LC firing rate during REM was hypothesized as one of the pathophysiological mechanisms underlying the switch process.
Supporting the importance of neuroplasticity in manic-like behaviors, studies have shown that BDNF gene mutations are associated with increased spontaneous locomotion and aggression in response to acute amphetamine and chronic cocaine in rodents, symptoms that often characterize manic episodes 143.
8. Other neurobiological factors implicated in the switch process: role of circadian rhythms
Observational studies conducted as early as the 1970s hypothesized that a disruption in circadian rhythms in BPD was a core feature of this illness. For example, an early report of patients hospitalized at the National Institute of Mental Health (NIMH) showed that switches into mania were more likely to happen in the morning than at night, suggesting a possible role for circadian factors in this process144. Marked alterations in body temperature, sleep patterns, cortisol secretion, thyroid-stimulating hormone (TSH) secretion, and motor activity have been described during episodes of BPD (reviewed in 7). Increased motor activity and decreased REM sleep, in particular, were found to strongly predict an imminent manic switch35, 59, 64. According to some researchers123, sleep loss might be the final common pathway triggering switches into mania. According to this model, the interaction between sleep reduction and a sleep-sensitive circadian phase interval could promote switches from depression. However, sleep disruption might also be the sign that the manic process is already mounting, rather than the specific trigger.
Both genetic and environmental factors might act as susceptibility factors through circadian rhythm regulation, increasing the desynchronization between the central pacemaker (i.e., the suprachiasmatic nucleus) and other internal oscillators. Increased external desynchronization between the timing of body rhythms and the light-dark cycle has been also hypothesized as a predisposing factor for mood episodes145. Interestingly, different mood stabilizers modulate the circadian clock, controlling the expression of genes involved in circadian rhythm regulation. For example, the mood stabilizer lithium inhibits GSK-3, and through this mechanism increases circadian period length7, 146. Studies conducted in Drosophila have shown that the orthologue of GSK-3, a protein called SHAGGY, is an important regulator of circadian cycles7. However, studies that have investigated GSK-3 polymorphisms as a putative susceptibility gene for BPD have produced conflicting results147–149.
The CLOCK gene is another major determinant of circadian cycles and might be involved in the switch to mania in patients with BPD; indeed, such evidence has arisen in animal models of BPD150. Disruption of the CLOCK gene produces manic-like behaviors in mice, such as hyperactivity, increased reward-value for cocaine and sucrose, and medial forebrain bundle stimulation150. In humans, CLOCK gene polymorphisms were shown to be associated with illness recurrence but not with diurnal variation in individuals with BPD8. Thus, it appears that polymorphisms in genes that regulate the circadian clock (e.g., CLOCK), along with sleep disruption and consequent increase in neuroplastic factor expression (pCREB, TrkB, BDNF) might have a substantial impact on mood destabilization leading to manic switch.
9. Conclusions and future perspectives
Despite the fact that the switch phenomenon is a core aspect of the clinical presentation of BPD, as well as fundamentally relevant to its therapeutics, it is still poorly understood. The studies conducted on this issue are unfortunately associated with several methodological limitations, and are often retrospective in nature, or the result of secondary analyses. For example, different definitions of TEAS have been used throughout these studies and may produce dramatically different results in terms of both clinical and biological findings. In order for a systematic study of this topic to be successful, the criteria and threshold of rating scales used will need to be uniform across studies. Agreement is also needed regarding how long after the beginning of drug treatment a manic episode should be considered as TEAS. Another major limitation in our understanding of the switch process is the lack of appropriate animal models for manic behaviors; preliminary evidence linking glutamate receptor abnormalities with manic-like behaviors in rodents73 are encouraging in this sense and might provide new evidence about the role of the glutamatergic system in the switch process.
For these reasons, results from clinical trials that have investigated the switch potential of different classes of antidepressants are difficult to interpret and subject to controversy among researchers. Even considering these caveats, it appears that drugs that “perturbate” more than one monoaminergic system, such as TCAs and, possibly, venlafaxine, confer a higher risk for TEAS than SSRIs or other second-generation antidepressants. A putative role for the monoaminergic system in the switch process has been suggested by clinical35, 64 and preclinical studies140, but needs further systematic investigation. Increased catecholamine levels lead to upregulation of factors involved in neuroplasticity cascades and to increased post-synaptic receptor sensitivity, which might ultimately increase the liability to switch (see Figure 1).
Figure 1. Neurobiology of the switch process: a comprehensive overview of the current evidence.
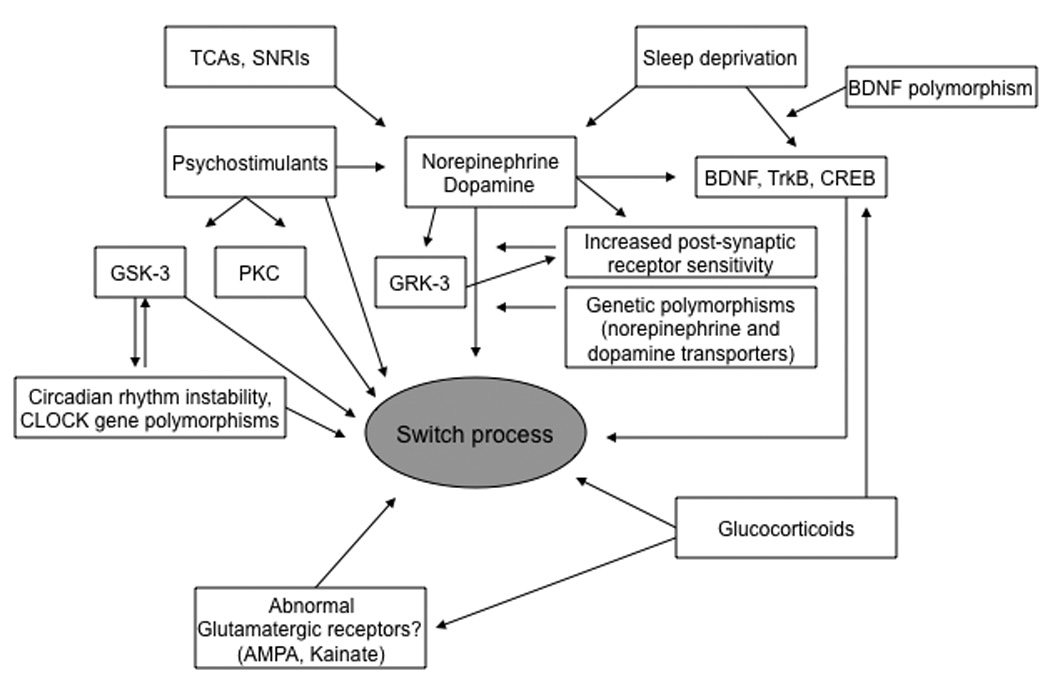
Several factors have been associated with the switch process in BPD, but little is known about how these neurobiological variables are interconnected. Psychostimulants, TCAs, SNRIs and sleep deprivation, three interventions that trigger manic switches in a significant proportion of individuals with BPD, are all known to increase catecholamine levels. Increased catecholamine levels lead to upregulation of factors involved in neuroplasticity cascades and to increased post-synaptic receptor sensitivity, which might ultimately increase the liability to switch. Psychostimulants also act by activating GSK-3 and PKC, two major proteins whose inhibition is important in the mechanism of action of mood stabilizers.
Other major determinants of this complex phenomenon include glucocorticoids, which increase cellular vulnerability to different physiological stressors (e.g., glutamatergic-mediated excitoxicity), abnormal glutamatergic transmission, and circadian rhythm instability. Some genetic polymorphisms that regulate catecholaminergic transmission (norepinephrine and dopamine transporters), neuroplasticity (BDNF), circadian period length (GSK-3), and GRK-3 may also be important mediators of the switch phenomenon.
Abbreviations: AMPA: alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid; BDNF: brain-derived neurotrophic factor; CREB: cyclic AMP response element binding protein; GRK-3: G protein receptor kinase 3; GSK-3: glycogen synthase kinase-3; PKC: protein kinase C; SNRIs: serotonin and norepinephrine reuptake inhibitors; TCAs: tricyclic antidepressants; TrkB: tyrosine receptor kinase B.
Other pharmacological and somatic interventions reviewed here include exogenous corticosteroids, dopaminergic agonists, and sleep deprivation. These interventions are particularly interesting because, in contrast to antidepressants, when they do induce switch, it generally occurs within a short time frame and is seen even in healthy volunteers. Also potentially relevant to the switch process are data from preclinical studies linking the HPA-axis, the dopaminergic system, and sleep deprivation to intracellular signaling pathways that have been extensively investigated in BPD, as well as in the mechanisms of action of mood stabilizers; these include BDNF, GSK-3, and PKC cascades. Other factors that have been linked to this complex phenomenon include abnormal glutamatergic transmission and circadian rhythm instability.
To date, the most convincing evidence suggests that BDNF may play a major role in the switch process, as suggested by preclinical models of the antidepressant effects of sleep deprivation. Human genetic studies further suggest that BDNF plays a key role in BPD and, perhaps, switch 151. For instance, a Valine(66) Methionine variant associated with increased BDNF stimulated release in vitro152 was found to be excessively transmitted in patients with BPD153 and was associated with earlier age of onset154. These preliminary data raise the intriguing possibility that individuals with BPD with the val/val BDNF genotype may be at greater risk for spontaneous, and antidepressant- or sleep deprivation-induced switches into mania. However, future studies are clearly needed to investigate this possibility. Furthermore, BDNF has been already implicated in other facets of BPD, including rapid cycling155, response to lithium156 and suicidality157. Figure 1 highlights the factors and pathways that may be putative determinants of the switch process and warrant further study.
In summary, there is a clear need to refine our understanding of the neurobiology of the switch process. Research with patients who experience mood switching during the course of clinical trials in BPD is not likely to be very informative in terms of understanding the neurobiology involved in this process, given the relatively rare occurrence of switch, the time until a switch occurs, and the multiple confounding factors associated with such investigations. In order to better understand the neurobiology of the switch process, it might be more illuminating to investigate interventions that more consistently produce switch, typically within a short period of time, such as sleep deprivation. In addition, the phenomenon could be studied in distinct groups, such as healthy subjects receiving switch-inducing interventions and individuals with BPD not receiving concomitant medications. Furthermore, a large sample size would allow investigating whether healthy subjects or individuals with BPD with certain “risk polymorphisms” (e.g., homozygous subjects for the BDNF Val66 allele or with certain CLOCK genetic variants) have a higher risk of switch compared to those without this vulnerability. Such group comparisons would also permit the systematic evaluation of neurobiological factors associated with switching (e.g., plasma catecholamines and hormones, sleep parameters, brain imaging data). Finally, preclinical studies conducted in appropriate animal models might provide important hints about the molecular and cellular mechanisms of this understudied but key phenomenon.
Acknowledgments
This study was supported by the Intramural Research Program of the National Institute of Mental Health (Bethesda, Maryland) and a NARSAD Award (CAZ).
References
- 1.Falret J. Mémoire sur la folie circulaire, forme de maladie mentale caractérisée par la reproduction sucessive et réguliäre de l'état maniqaue, de l'état mélancolique, et d'un intervalle lucide plus or moins prolongé. Bull Acad Natl Med. 1854;19:382–415. [Google Scholar]
- 2.Sedler MJ. Falret's discovery: the origin of the concept of bipolar affective illness. Translated by M. J. Sedler and Eric C. Dessain. Am J Psychiatry. 1983;140(9):1127–1133. doi: 10.1176/ajp.140.9.1127. [DOI] [PubMed] [Google Scholar]
- 3.Angst J, Sellaro R. Historical perspectives and natural history of bipolar disorder. Biol Psychiatry. 2000;48(6):445–457. doi: 10.1016/s0006-3223(00)00909-4. [DOI] [PubMed] [Google Scholar]
- 4.Angst J. Switch from depression to mania, or from mania to depression: role of psychotropic drugs. Psychopharmacol Bull. 1987;23(1):66–67. [PubMed] [Google Scholar]
- 5.Licht RW, Gijsman H, Nolen WA, Angst J. Are antidepressants safe in the treatment of bipolar depression? A critical evaluation of their potential risk to induce switch into mania or cycle acceleration. Acta Psychiatr Scand. 2008;118(5):337–346. doi: 10.1111/j.1600-0447.2008.01237.x. [DOI] [PubMed] [Google Scholar]
- 6.Grunze HC. Switching, induction of rapid cycling, and increased suicidality with antidepressants in bipolar patients: fact or overinterpretation? CNS Spectr. 2008;13(9):790–795. doi: 10.1017/s1092852900013912. [DOI] [PubMed] [Google Scholar]
- 7.Hasler G, Drevets WC, Gould TD, Gottesman II, Manji HK. Toward constructing an endophenotype strategy for bipolar disorders. Biol Psychiatry. 2006;60(2):93–105. doi: 10.1016/j.biopsych.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 8.Benedetti F, Serretti A, Colombo C, et al. Influence of CLOCK gene polymorphism on circadian mood fluctuation and illness recurrence in bipolar depression. Am J Med Genet B Neuropsychiatr Genet. 2003;123B(1):23–26. doi: 10.1002/ajmg.b.20038. [DOI] [PubMed] [Google Scholar]
- 9.Mundo E, Walker M, Cate T, Macciardi F, Kennedy JL. The role of serotonin transporter protein gene in antidepressant-induced mania in bipolar disorder: preliminary findings. Arch Gen Psychiatry. 2001;58(6):539–544. doi: 10.1001/archpsyc.58.6.539. [DOI] [PubMed] [Google Scholar]
- 10.Vieta E, Angst J, Reed C, Bertsch J, Haro JM the EMBLEM advisory board. Predictors of switching from mania to depression in a large observational study across Europe (EMBLEM) J Affect Disord. 2009 March 6; doi: 10.1016/j.jad.2009.02.007. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 11.MacKinnon DF, Zandi PP, Gershon ES, Nurnberger JI, Jr, DePaulo JR., Jr Association of rapid mood switching with panic disorder and familial panic risk in familial bipolar disorder. Am J Psychiatry. 2003;160(9):1696–1698. doi: 10.1176/appi.ajp.160.9.1696. [DOI] [PubMed] [Google Scholar]
- 12.MacKinnon DF, Zandi PP, Gershon E, Nurnberger JI, Jr, Reich T, DePaulo JR. Rapid switching of mood in families with multiple cases of bipolar disorder. Arch Gen Psychiatry. 2003;60(9):921–928. doi: 10.1001/archpsyc.60.9.921. [DOI] [PubMed] [Google Scholar]
- 13.MacKinnon DF, Potash JB, McMahon FJ, Simpson SG, Depaulo JR, Jr, Zandi PP. Rapid mood switching and suicidality in familial bipolar disorder. Bipolar Disord. 2005;7(5):441–448. doi: 10.1111/j.1399-5618.2005.00236.x. [DOI] [PubMed] [Google Scholar]
- 14.Maj M, Pirozzi R, Magliano L, Bartoli L. The prognostic significance of "switching" in patients with bipolar disorder: a 10-year prospective follow-up study. Am J Psychiatry. 2002;159(10):1711–1717. doi: 10.1176/appi.ajp.159.10.1711. [DOI] [PubMed] [Google Scholar]
- 15.Altshuler LL, Post RM, Leverich GS, Mikalauskas K, Rosoff A, Ackerman L. Antidepressant-induced mania and cycle acceleration: a controversy revisited. Am J Psychiatry. 1995;152(8):1130–1138. doi: 10.1176/ajp.152.8.1130. [DOI] [PubMed] [Google Scholar]
- 16.Post RM. Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci Biobehav Rev. 2007;31(6):858–873. doi: 10.1016/j.neubiorev.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Bottlender R, Sato T, Kleindienst N, Strauss A, Moller HJ. Mixed depressive features predict maniform switch during treatment of depression in bipolar I disorder. J Affect Disord. 2004;78(2):149–152. doi: 10.1016/s0165-0327(02)00265-3. [DOI] [PubMed] [Google Scholar]
- 18.Zarate CA, Jr, Tohen M, Fletcher K. Cycling into depression from a first episode of mania: a case-comparison study. Am J Psychiatry. 2001;158(9):1524–1526. doi: 10.1176/appi.ajp.158.9.1524. [DOI] [PubMed] [Google Scholar]
- 19.Sachs GS, Nierenberg AA, Calabrese JR, et al. Effectiveness of adjunctive antidepressant treatment for bipolar depression. N Engl J Med. 2007;356(17):1711–1722. doi: 10.1056/NEJMoa064135. [DOI] [PubMed] [Google Scholar]
- 20.Serretti A, Artioli P, Zanardi R, Rossini D. Clinical features of antidepressant associated manic and hypomanic switches in bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27(5):751–757. doi: 10.1016/S0278-5846(03)00104-0. [DOI] [PubMed] [Google Scholar]
- 21.Henry C, Sorbara F, Lacoste J, Gindre C, Leboyer M. Antidepressant-induced mania in bipolar patients: identification of risk factors. J Clin Psychiatry. 2001;62(4):249–255. doi: 10.4088/jcp.v62n0406. [DOI] [PubMed] [Google Scholar]
- 22.Truman CJ, Goldberg JF, Ghaemi SN, et al. Self-reported history of manic/hypomanic switch associated with antidepressant use: data from the Systematic Treatment Enhancement Program for Bipolar Disorder (STEP-BD) J Clin Psychiatry. 2007;68(10):1472–1479. doi: 10.4088/jcp.v68n1002. [DOI] [PubMed] [Google Scholar]
- 23.Carlson GA, Finch SJ, Fochtmann LJ, et al. Antidepressant-associated switches from depression to mania in severe bipolar disorder. Bipolar Disord. 2007;9(8):851–859. doi: 10.1111/j.1399-5618.2007.00424.x. [DOI] [PubMed] [Google Scholar]
- 24.Tamada RS, Issler CK, Amaral JA, Sachs GS, Lafer B. Treatment emergent affective switch: a controlled study. Bipolar Disord. 2004;6(4):333–337. doi: 10.1111/j.1399-5618.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 25.Post RM, Altshuler LL, Leverich GS, et al. Mood switch in bipolar depression: comparison of adjunctive venlafaxine, bupropion and sertraline. Br J Psychiatry. 2006;189:124–131. doi: 10.1192/bjp.bp.105.013045. [DOI] [PubMed] [Google Scholar]
- 26.El-Mallakh RS, Ghaemi SN, Sagduyu K, et al. Antidepressant-associated chronic irritable dysphoria (ACID) in STEP-BD patients. J Affect Disord. 2008;111:372–377. doi: 10.1016/j.jad.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 27.Altshuler LL, Suppes T, Black DO, et al. Lower switch rate in depressed patients with bipolar II than bipolar I disorder treated adjunctively with second-generation antidepressants. Am J Psychiatry. 2006;163(2):313–315. doi: 10.1176/appi.ajp.163.2.313. [DOI] [PubMed] [Google Scholar]
- 28.Bauer M, Rasgon N, Grof P, et al. Do antidepressants influence mood patterns? A naturalistic study in bipolar disorder. Eur Psychiatry. 2006;21(4):262–269. doi: 10.1016/j.eurpsy.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 29.Bond DJ, Noronha MM, Kauer-Sant'Anna M, Lam RW, Yatham LN. Antidepressant-associated mood elevations in bipolar II disorder compared with bipolar I disorder and major depressive disorder: a systematic review and meta-analysis. J Clin Psychiatry. 2008;69(10):1589–1601. doi: 10.4088/jcp.v69n1009. [DOI] [PubMed] [Google Scholar]
- 30.Ghaemi SN, Goodwin FK. Antidepressants for bipolar depression. Am J Psychiatry. 2005;162(8):1545–1546. doi: 10.1176/appi.ajp.162.8.1545-a. author reply 1547–1548. [DOI] [PubMed] [Google Scholar]
- 31.Salvi VFA, Swartz HA, Maina G, Frank E. The Use of Antidepressants in Bipolar Disorder. J Clin Psychiatry. 2008;69(8):1307–1318. doi: 10.4088/jcp.v69n0816. [DOI] [PubMed] [Google Scholar]
- 32.Gijsman HJ, Geddes JR, Rendell JM, Nolen WA, Goodwin GM. Antidepressants for bipolar depression: a systematic review of randomized, controlled trials. Am J Psychiatry. 2004;161(9):1537–1547. doi: 10.1176/appi.ajp.161.9.1537. [DOI] [PubMed] [Google Scholar]
- 33.Peet M. Induction of mania with selective serotonin re-uptake inhibitors and tricyclic antidepressants. Br J Psychiatry. 1994;164(4):549–550. doi: 10.1192/bjp.164.4.549. [DOI] [PubMed] [Google Scholar]
- 34.Boerlin HL, Gitlin MJ, Zoellner LA, Hammen CL. Bipolar depression and antidepressant-induced mania: a naturalistic study. J Clin Psychiatry. 1998;59(7):374–379. doi: 10.4088/jcp.v59n0706. [DOI] [PubMed] [Google Scholar]
- 35.Bunney WE, Jr, Murphy DL, Goodwin FK, Borge GF. The switch process from depression to mania: relationship to drugs which alter brain amines. Lancet. 1970;1(7655):1022–1027. doi: 10.1016/s0140-6736(70)91151-7. [DOI] [PubMed] [Google Scholar]
- 36.Wehr TA, Goodwin FK. Rapid cycling in manic-depressives induced by tricyclic antidepressants. Arch Gen Psychiatry. 1979;36(5):555–559. doi: 10.1001/archpsyc.1979.01780050065007. [DOI] [PubMed] [Google Scholar]
- 37.Bottlender R, Rudolf D, Strauss A, Moller HJ. Mood-stabilisers reduce the risk of developing antidepressant-induced maniform states in acute treatment of bipolar I depressed patients. J Affect Disord. 2001;63(1–3):79–83. doi: 10.1016/s0165-0327(00)00172-5. [DOI] [PubMed] [Google Scholar]
- 38.Lewis JL, Winokur G. The induction of mania. A natural history study with controls. Arch Gen Psychiatry. 1982;39(3):303–306. doi: 10.1001/archpsyc.1982.04290030041007. [DOI] [PubMed] [Google Scholar]
- 39.Nemeroff CB, Evans DL, Gyulai L, et al. Double-blind, placebo-controlled comparison of imipramine and paroxetine in the treatment of bipolar depression. Am J Psychiatry. 2001;158(6):906–912. doi: 10.1176/appi.ajp.158.6.906. [DOI] [PubMed] [Google Scholar]
- 40.Himmelhoch JM, Thase ME, Mallinger AG, Houck P. Tranylcypromine versus imipramine in anergic bipolar depression. Am J Psychiatry. 1991;148(7):910–916. doi: 10.1176/ajp.148.7.910. [DOI] [PubMed] [Google Scholar]
- 41.Silverstone T. Moclobemide vs. imipramine in bipolar depression: a multicentre double-blind clinical trial. Acta Psychiatr Scand. 2001;104(2):104–109. doi: 10.1034/j.1600-0447.2001.00240.x. [DOI] [PubMed] [Google Scholar]
- 42.Cohn JB, Collins G, Ashbrook E, Wernicke JF. A comparison of fluoxetine imipramine and placebo in patients with bipolar depressive disorder. Int Clin Psychopharmacol. 1989;4(4):313–322. doi: 10.1097/00004850-198910000-00006. [DOI] [PubMed] [Google Scholar]
- 43.Sachs GS, Lafer B, Stoll AL, et al. A double-blind trial of bupropion versus desipramine for bipolar depression. J Clin Psychiatry. 1994;55(9):391–393. [PubMed] [Google Scholar]
- 44.Vieta E, Colom F, Martinez-Aran A, et al. Reboxetine-induced hypomania. J Clin Psychiatry. 2001;62(8):655–656. doi: 10.4088/jcp.v62n0813c. [DOI] [PubMed] [Google Scholar]
- 45.Nolen WA, Kupka RW, Hellemann G, et al. Tranylcypromine vs. lamotrigine in the treatment of refractory bipolar depression: a failed but clinically useful study. Acta Psychiatr Scand. 2007;115(5):360–365. doi: 10.1111/j.1600-0447.2007.00993.x. [DOI] [PubMed] [Google Scholar]
- 46.Wright G, Galloway L, Kim J, Dalton M, Miller L, Stern W. Bupropion in the long-term treatment of cyclic mood disorders: mood stabilizing effects. J Clin Psychiatry. 1985;46(1):22–25. [PubMed] [Google Scholar]
- 47.Shopsin B. Bupropion's prophylactic efficacy in bipolar affective illness. J Clin Psychiatry. 1983;44(5 Pt 2):163–169. [PubMed] [Google Scholar]
- 48.McIntyre RS, Mancini DA, McCann S, Srinivasan J, Sagman D, Kennedy SH. Topiramate versus bupropion SR when added to mood stabilizer therapy for the depressive phase of bipolar disorder: a preliminary single-blind study. Bipolar Disord. 2002;4(3):207–213. doi: 10.1034/j.1399-5618.2002.01189.x. [DOI] [PubMed] [Google Scholar]
- 49.Joffe RT, MacQueen GM, Marriott M, Robb J, Begin H, Young LT. Induction of mania and cycle acceleration in bipolar disorder: effect of different classes of antidepressant. Acta Psychiatr Scand. 2002;105(6):427–430. doi: 10.1034/j.1600-0447.2002.02360.x. [DOI] [PubMed] [Google Scholar]
- 50.Vieta E, Martinez-Aran A, Goikolea JM, et al. A randomized trial comparing paroxetine and venlafaxine in the treatment of bipolar depressed patients taking mood stabilizers. J Clin Psychiatry. 2002;63(6):508–512. doi: 10.4088/jcp.v63n0607. [DOI] [PubMed] [Google Scholar]
- 51.Amsterdam JD, Shults J. Comparison of short-term venlafaxine versus lithium monotherapy for bipolar II major depressive episode: a randomized open-label study. J Clin Psychopharmacol. 2008;28(2):171–181. doi: 10.1097/JCP.0b013e318166c4e6. [DOI] [PubMed] [Google Scholar]
- 52.Amsterdam JD, Shults J. Comparison of fluoxetine, olanzapine, and combined fluoxetine plus olanzapine initial therapy of bipolar type I and type II major depression--lack of manic induction. J Affect Disord. 2005;87(1):121–130. doi: 10.1016/j.jad.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 53.Tohen M, Vieta E, Calabrese J, et al. Efficacy of olanzapine and olanzapine-fluoxetine combination in the treatment of bipolar I depression. Arch Gen Psychiatry. 2003;60(11):1079–1088. doi: 10.1001/archpsyc.60.11.1079. [DOI] [PubMed] [Google Scholar]
- 54.Fonseca M, Soares JC, Hatch JP, Santin AP, Kapczinski F. An open trial of adjunctive escitalopram in bipolar depression. J Clin Psychiatry. 2006;67(1):81–86. doi: 10.4088/jcp.v67n0115. [DOI] [PubMed] [Google Scholar]
- 55.Schaffer A, Zuker P, Levitt A. Randomized, double-blind pilot trial comparing lamotrigine versus citalopram for the treatment of bipolar depression. J Affect Disord. 2006;96(1–2):95–99. doi: 10.1016/j.jad.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 56.Rousseva A, Henry C, van den Bulke D, et al. Antidepressant-induced mania, rapid cycling and the serotonin transporter gene polymorphism. Pharmacogenomics J. 2003;3(2):101–104. doi: 10.1038/sj.tpj.6500156. [DOI] [PubMed] [Google Scholar]
- 57.Serretti A, Artioli P, Zanardi R, et al. Genetic features of antidepressant induced mania and hypo-mania in bipolar disorder. Psychopharmacology (Berl) 2004;174(4):504–511. doi: 10.1007/s00213-004-1948-x. [DOI] [PubMed] [Google Scholar]
- 58.Hughes JH, Dunne F, Young AH. Effects of acute tryptophan depletion on mood and suicidal ideation in bipolar patients symptomatically stable on lithium. Br J Psychiatry. 2000;177:447–451. doi: 10.1192/bjp.177.5.447. [DOI] [PubMed] [Google Scholar]
- 59.Bunney WE, Jr, Goodwin FK, Murphy DL, House KM, Gordon EK. The "switch process" in manic-depressive illness. II. Relationship to catecholamines, REM sleep, and drugs. Arch Gen Psychiatry. 1972;27(3):304–309. doi: 10.1001/archpsyc.1972.01750270014002. [DOI] [PubMed] [Google Scholar]
- 60.Zis AP, Cowdry RW, Wehr TA, Muscettola G, Goodwin FK. Tricyclic-induced mania and MHPG excretion. Psychiatry Res. 1979;1(1):93–99. doi: 10.1016/0165-1781(79)90033-7. [DOI] [PubMed] [Google Scholar]
- 61.Joyce PR, Fergusson DM, Woollard G, Abbott RM, Horwood LJ, Upton J. Urinary catecholamines and plasma hormones predict mood state in rapid cycling bipolar affective disorder. J Affect Disord. 1995;33(4):233–243. doi: 10.1016/0165-0327(94)00094-p. [DOI] [PubMed] [Google Scholar]
- 62.Paul MI, Cramer H, Bunney WE., J Urinary adenosine 3',5'-monophosphate in the switch process from depression to mania. Science. 1971;171(968):300–303. doi: 10.1126/science.171.3968.300. [DOI] [PubMed] [Google Scholar]
- 63.Juckel G, Hegerl U, Mavrogiorgou P, et al. Clinical and biological findings in a case with 48-hour bipolar ultrarapid cycling before and during valproate treatment. J Clin Psychiatry. 2000;61(8):585–593. doi: 10.4088/jcp.v61n0808. [DOI] [PubMed] [Google Scholar]
- 64.Post RM, Stoddard FJ, Gillin JC, et al. Alterations in motor activity, sleep, and biochemistry in a cycling manic-depressive patient. Arch Gen Psychiatry. 1977;34(4):470–477. doi: 10.1001/archpsyc.1977.01770160104009. [DOI] [PubMed] [Google Scholar]
- 65.Jones FD, Maas JW, Dekirmenjian H, Fawcett JA. Urinary catecholamine metabolites during behavioral changes in a patient with manic-depressive cycles. Science. 1973;179(70):300–302. doi: 10.1126/science.179.4070.300. [DOI] [PubMed] [Google Scholar]
- 66.Bunney WE, Jr, Goodwin FK, Murphy DL. The "switch process" in manic-depressive illness. 3. Theoretical implications. Arch Gen Psychiatry. 1972;27(3):312–317. doi: 10.1001/archpsyc.1972.01750270022003. [DOI] [PubMed] [Google Scholar]
- 67.Serretti A, Lilli R, Smeraldi E. Pharmacogenetics in affective disorders. Eur J Pharmacol. 2002;438(3):117–128. doi: 10.1016/s0014-2999(02)01309-2. [DOI] [PubMed] [Google Scholar]
- 68.Pryor JC, Sulser F. Evolution of monoamine hypotheses of depression. In: Horton RW, Katona C, editors. Biological Aspects of Affective Disorders. London: Academic Press; 1991. pp. 77–94. [Google Scholar]
- 69.de Montigny CCY, Blier P. Modification of serotonergic neuron properties by long-term treatment with serotonin reuptake blockers. J Clin Psychiatry. 1990;51 Suppl B:4–8. [PubMed] [Google Scholar]
- 70.Butkerait P, Wang HY, Friedman E. Increases in guanine nucleotide binding to striatal G proteins is associated with dopamine receptor supersensitivity. J Pharmacol Exp Ther. 1994;271(1):422–428. [PubMed] [Google Scholar]
- 71.Zarate CA, Jr, Singh J, Manji HK. Cellular plasticity cascades: targets for the development of novel therapeutics for bipolar disorder. Biol Psychiatry. 2006;59(11):1006–1020. doi: 10.1016/j.biopsych.2005.10.021. [DOI] [PubMed] [Google Scholar]
- 72.Du J, Creson TK, Wu LJ, et al. The role of hippocampal GluR1 and GluR2 receptors in manic-like behavior. J Neurosci. 2008;28(1):68–79. doi: 10.1523/JNEUROSCI.3080-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shaltiel G, Maeng S, Malkesman O, et al. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol Psychiatry. 2008;13(9):858–872. doi: 10.1038/mp.2008.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ketter TA, Manji HK, Post RM. Potential mechanisms of action of lamotrigine in the treatment of bipolar disorders. J Clin Psychopharmacol. 2003;23(5):484–495. doi: 10.1097/01.jcp.0000088915.02635.e8. [DOI] [PubMed] [Google Scholar]
- 75.Calabrese JR, Rapport DJ, Kimmel SE, Shelton MD. Controlled trials in bipolar I depression: focus on switch rates and efficacy. Eur Neuropsychopharmacol. 1999;9 Suppl 4:S109–S112. doi: 10.1016/s0924-977x(99)00023-1. [DOI] [PubMed] [Google Scholar]
- 76.Zarate CA, Jr, Quiroz JA, Singh JB, et al. An open-label trial of the glutamate-modulating agent riluzole in combination with lithium for the treatment of bipolar depression. Biol Psychiatry. 2005;57(4):430–432. doi: 10.1016/j.biopsych.2004.11.023. [DOI] [PubMed] [Google Scholar]
- 77.Teng CT, Demetrio FN. Memantine may acutely improve cognition and have a mood stabilizing effect in treatment-resistant bipolar disorder. Rev Bras Psiquiatr. 2006;28(3):252–254. doi: 10.1590/s1516-44462006000300020. [DOI] [PubMed] [Google Scholar]
- 78.Krystal JH, Perry EB, Jr, Gueorguieva R, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry. 2005;62(9):985–994. doi: 10.1001/archpsyc.62.9.985. [DOI] [PubMed] [Google Scholar]
- 79.Murphy DL, Brodie HK, Goodwin FK, Bunney WE., Jr Regular induction of hypomania by L-dopa in "bipolar" manic-depressive patients. Nature. 1971;229(5280):135–136. doi: 10.1038/229135a0. [DOI] [PubMed] [Google Scholar]
- 80.Asghar SJ, Tanay VA, Baker GB, Greenshaw A, Silverstone PH. Relationship of plasma amphetamine levels to physiological, subjective, cognitive and biochemical measures in healthy volunteers. Hum Psychopharmacol. 2003;18(4):291–299. doi: 10.1002/hup.480. [DOI] [PubMed] [Google Scholar]
- 81.Jacobs D, Silverstone T. Dextroamphetamine-induced arousal in human subjects as a model for mania. Psychol Med. 1986;16(2):323–329. doi: 10.1017/s0033291700009132. [DOI] [PubMed] [Google Scholar]
- 82.Masand PS, Pickett P, Murray GB. Hypomania precipitated by psychostimulant use in depressed medically ill patients. Psychosomatics. 1995;36(2):145–147. doi: 10.1016/S0033-3182(95)71684-X. [DOI] [PubMed] [Google Scholar]
- 83.Drevets WC, Gautier C, Price JC, et al. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry. 2001;49(2):81–96. doi: 10.1016/s0006-3223(00)01038-6. [DOI] [PubMed] [Google Scholar]
- 84.Dlugos A, Freitag C, Hohoff C, et al. Norepinephrine transporter gene variation modulates acute response to D-amphetamine. Biol Psychiatry. 2007;61(11):1296–1305. doi: 10.1016/j.biopsych.2006.09.031. [DOI] [PubMed] [Google Scholar]
- 85.Lott DC, Kim SJ, Cook EH, Jr, de Wit H. Dopamine transporter gene associated with diminished subjective response to amphetamine. Neuropsychopharmacology. 2005;30(3):602–609. doi: 10.1038/sj.npp.1300637. [DOI] [PubMed] [Google Scholar]
- 86.Anand A, Darnell A, Miller HL, et al. Effect of catecholamine depletion on lithium-induced long-term remission of bipolar disorder. Biol Psychiatry. 1999;45(8):972–978. doi: 10.1016/s0006-3223(98)00293-5. [DOI] [PubMed] [Google Scholar]
- 87.McTavish SF, McPherson MH, Harmer CJ, et al. Antidopaminergic effects of dietary tyrosine depletion in healthy subjects and patients with manic illness. Br J Psychiatry. 2001;179:356–360. doi: 10.1192/bjp.179.4.356. [DOI] [PubMed] [Google Scholar]
- 88.D'Aquila PS, Peana AT, Panin F, Grixoni C, Cossu M, Serra G. Reversal of antidepressant-induced dopaminergic behavioural supersensitivity after long-term chronic imipramine withdrawal. Eur J Pharmacol. 2003;458(1–2):129–134. doi: 10.1016/s0014-2999(02)02731-0. [DOI] [PubMed] [Google Scholar]
- 89.Ichikawa J, Meltzer HY. Effect of antidepressants on striatal and accumbens extracellular dopamine levels. Eur J Pharmacol. 1995;281(3):255–261. doi: 10.1016/0014-2999(95)00264-l. [DOI] [PubMed] [Google Scholar]
- 90.Papp M, Klimek V, Willner P. Parallel changes in dopamine D2 receptor binding in limbic forebrain associated with chronic mild stress-induced anhedonia and its reversal by imipramine. Psychopharmacology (Berl) 1994;115(4):441–446. doi: 10.1007/BF02245566. [DOI] [PubMed] [Google Scholar]
- 91.Anisman H, Irwin J, Sklar LS. Deficits of escape performance following catecholamine depletion: implications for behavioral deficits induced by uncontrollable stress. Psychopharmacology (Berl) 1979;64(2):163–170. doi: 10.1007/BF00496057. [DOI] [PubMed] [Google Scholar]
- 92.Machado-Vieira R, Kapczinski F, Soares JC. Perspectives for the development of animal models of bipolar disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2004;28(2):209–224. doi: 10.1016/j.pnpbp.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 93.Shaldubina A, Einat H, Szechtman H, Shimon H, Belmaker RH. Preliminary evaluation of oral anticonvulsant treatment in the quinpirole model of bipolar disorder. J Neural Transm. 2002;109(3):433–440. doi: 10.1007/s007020200035. [DOI] [PubMed] [Google Scholar]
- 94.Eilam D, Szechtman H. Biphasic effect of D-2 agonist quinpirole on locomotion and movements. Eur J Pharmacol. 1989;161(2–3):151–157. doi: 10.1016/0014-2999(89)90837-6. [DOI] [PubMed] [Google Scholar]
- 95.Beaulieu JM, Sotnikova TD, Yao WD, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci U S A. 2004;101(14):5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Einat H, Manji HK. Cellular plasticity cascades: genes-to-behavior pathways in animal models of bipolar disorder. Biol Psychiatry. 2006;59(12):1160–1171. doi: 10.1016/j.biopsych.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 97.Chen R, Furman CA, Zhang M, et al. Protein kinase C{beta} is a critical regulator of dopamine transporter trafficking and regulates the behavioral response to amphetamine in mice. J Pharmacol Exp Ther. 2008 doi: 10.1124/jpet.108.147959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yildiz A, Guleryuz S, Ankerst DP, Ongur D, Renshaw PF. Protein kinase C inhibition in the treatment of mania: a double-blind, placebo-controlled trial of tamoxifen. Arch Gen Psychiatry. 2008;65(3):255–263. doi: 10.1001/archgenpsychiatry.2007.43. [DOI] [PubMed] [Google Scholar]
- 99.Zarate CA, Jr, Singh JB, Carlson PJ, et al. Efficacy of a protein kinase C inhibitor (tamoxifen) in the treatment of acute mania: a pilot study. Bipolar Disord. 2007;9(6):561–570. doi: 10.1111/j.1399-5618.2007.00530.x. [DOI] [PubMed] [Google Scholar]
- 100.Ritchie EA. Toxic psychosis under cortisone and corticotrophin. J Ment Sci. 1956;102(429):830–837. doi: 10.1192/bjp.102.429.830. [DOI] [PubMed] [Google Scholar]
- 101.Clark LD, Quarton GC, Cobb S, Bauer W. Further observations on mental disturbances associated with cortisone and ACTH therapy. N Engl J Med. 1953;249(5):178–183. doi: 10.1056/NEJM195307302490502. [DOI] [PubMed] [Google Scholar]
- 102.Lewis DA, Smith RE. Steroid-induced psychiatric syndromes. A report of 14 cases and a review of the literature. J Affect Disord. 1983;5(4):319–332. doi: 10.1016/0165-0327(83)90022-8. [DOI] [PubMed] [Google Scholar]
- 103.Lewis LD, Cochrane GM. Psychosis in a child inhaling budesonide. Lancet. 1983;2(8350):634. doi: 10.1016/s0140-6736(83)90725-0. [DOI] [PubMed] [Google Scholar]
- 104.Boston Collaborative Drug Surveillance Program. Acute adverse reactions to prednisone in relation to dosage. Clin Pharmacol Ther. 1972;13:694–698. doi: 10.1002/cpt1972135part1694. [DOI] [PubMed] [Google Scholar]
- 105.Minden SL, Orav J, Schildkraut JJ. Hypomanic reactions to ACTH and prednisone treatment for multiple sclerosis. Neurology. 1988;38(10):1631–1634. doi: 10.1212/wnl.38.10.1631. [DOI] [PubMed] [Google Scholar]
- 106.Naber D, Sand P, Heigl B. Psychopathological and neuropsychological effects of 8-days' corticosteroid treatment. A prospective study. Psychoneuroendocrinology. 1996;21(1):25–31. doi: 10.1016/0306-4530(95)00031-3. [DOI] [PubMed] [Google Scholar]
- 107.Brown ES, Suppes T, Khan DA, Carmody TJ., 3rd Mood changes during prednisone bursts in outpatients with asthma. J Clin Psychopharmacol. 2002;22(1):55–61. doi: 10.1097/00004714-200202000-00009. [DOI] [PubMed] [Google Scholar]
- 108.Turktas L, Gucuyener K, Ozden A. Medication-induced psychotic reaction. J Am Acad Child Adolesc Psychiatry. 1997;36(8):1017–1018. doi: 10.1097/00004583-199708000-00006. [DOI] [PubMed] [Google Scholar]
- 109.Wolkowitz OM, Rubinow D, Doran AR, et al. Prednisone effects on neurochemistry and behavior. Preliminary findings. Arch Gen Psychiatry. 1990;47(10):963–968. doi: 10.1001/archpsyc.1990.01810220079010. [DOI] [PubMed] [Google Scholar]
- 110.Wada K, Yamada N, Suzuki H, Lee Y, Kuroda S. Recurrent cases of corticosteroid-induced mood disorder: clinical characteristics and treatment. J Clin Psychiatry. 2000;61(4):261–267. doi: 10.4088/jcp.v61n0404. [DOI] [PubMed] [Google Scholar]
- 111.Phelan MC. Beclomethasone mania. Br J Psychiatry. 1989;155:871–872. doi: 10.1192/bjp.155.6.871. [DOI] [PubMed] [Google Scholar]
- 112.Goldstein ET, Preskorn SH. Mania triggered by a steroid nasal spray in a patient with stable bipolar disorder. Am J Psychiatry. 1989;146(8):1076–1077. doi: 10.1176/ajp.146.8.1076. [DOI] [PubMed] [Google Scholar]
- 113.Vacheron-Trystram MN, Cheref S, Gauillard J, Plas J. [A case report of mania precipitated by use of DHEA] Encephale. 2002;28(6 Pt 1):563–566. [PubMed] [Google Scholar]
- 114.Fishman SM, Catarau EM, Sachs G, Stojanovic M, Borsook D. Corticosteroid-induced mania after single regional application at the celiac plexus. Anesthesiology. 1996;85(5):1194–1196. doi: 10.1097/00000542-199611000-00030. [DOI] [PubMed] [Google Scholar]
- 115.Sharfstein SS, Sack DS, Fauci AS. Relationship between alternate-day corticosteroid therapy and behavioral abnormalities. JAMA. 1982;248(22):2987–2989. [PubMed] [Google Scholar]
- 116.Daban C, Vieta E, Mackin P, Young AH. Hypothalamic-pituitary-adrenal axis and bipolar disorder. Psychiatr Clin North Am. 2005;28(2):469–480. doi: 10.1016/j.psc.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 117.Krishnan RR, Maltbie AA, Davidson JR. Abnormal cortisol suppression in bipolar patients with simultaneous manic and depressive symptoms. Am J Psychiatry. 1983;140(2):203–205. doi: 10.1176/ajp.140.2.203. [DOI] [PubMed] [Google Scholar]
- 118.Swann AC, Stokes PE, Casper R, et al. Hypothalamic-pituitary-adrenocortical function in mixed and pure mania. Acta Psychiatr Scand. 1992;85(4):270–274. doi: 10.1111/j.1600-0447.1992.tb01468.x. [DOI] [PubMed] [Google Scholar]
- 119.Gann H, Riemann D, Hohagen F, et al. 48-hour rapid cycling: results of psychopathometric, polysomnographic, PET imaging and neuro-endocrine longitudinal investigations in a single case. J Affect Disord. 1993;28(2):133–140. doi: 10.1016/0165-0327(93)90042-i. [DOI] [PubMed] [Google Scholar]
- 120.Doerr P, von Zerssen D, Fischler M, Schulz H. Relationship between mood changes and adrenal cortical activity in a patient with 48-hour unipolar-depressive cycles. J Affect Disord. 1979;1(2):93–104. doi: 10.1016/0165-0327(79)90028-4. [DOI] [PubMed] [Google Scholar]
- 121.Bunney WE, Jr, Hartmann EL, Mason JW. Study of a Patient with 48-Hour Manic-Depressive Cycles. Ii. Strong Positive Correlation between Endocrine Factors and Manic Defense Patterns. Arch Gen Psychiatry. 1965;12:619–625. doi: 10.1001/archpsyc.1965.01720360091015. [DOI] [PubMed] [Google Scholar]
- 122.Wei Q, Lu XY, Liu L, et al. Glucocorticoid receptor overexpression in forebrain: a mouse model of increased emotional lability. Proc Natl Acad Sci U S A. 2004;101(32):11851–11856. doi: 10.1073/pnas.0402208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wehr TA, Sack DA, Rosenthal NE. Sleep reduction as a final common pathway in the genesis of mania. Am J Psychiatry. 1987;144(2):201–204. doi: 10.1176/ajp.144.2.201. [DOI] [PubMed] [Google Scholar]
- 124.Barbini B, Colombo C, Benedetti F, Campori E, Bellodi L, Smeraldi E. The unipolar-bipolar dichotomy and the response to sleep deprivation. Psychiatry Res. 1998;79(1):43–50. doi: 10.1016/s0165-1781(98)00020-1. [DOI] [PubMed] [Google Scholar]
- 125.Colombo C, Benedetti F, Barbini B, Campori E, Smeraldi E. Rate of switch from depression into mania after therapeutic sleep deprivation in bipolar depression. Psychiatry Res. 1999;86:267–270. doi: 10.1016/s0165-1781(99)00036-0. [DOI] [PubMed] [Google Scholar]
- 126.Leibenluft E, Albert PS, Rosenthal NE, Wehr TA. Relationship between sleep and mood in patients with rapid-cycling bipolar disorder. Psychiatry Res. 1996;63(2–3):161–168. doi: 10.1016/0165-1781(96)02854-5. [DOI] [PubMed] [Google Scholar]
- 127.Albert I, Cicala GA, Siegel J. The behavioral effects of REM sleep deprivation in rats. Psychophysiology. 1970;6:550–560. doi: 10.1111/j.1469-8986.1970.tb02244.x. [DOI] [PubMed] [Google Scholar]
- 128.Gessa GL, Pani L, Fadda P, Fratta W. Sleep deprivation in the rat: an animal model of mania. Eur Neuropsychopharmacol. 1995;(5 Suppl):89–93. doi: 10.1016/0924-977x(95)00023-i. [DOI] [PubMed] [Google Scholar]
- 129.Hicks RA, Moore JD, Hayes C, Phillips N, Hawkins J. REM sleep deprivation increases aggressiveness in male rats. Physiol Behav. 1979;22(6):1097–1100. doi: 10.1016/0031-9384(79)90263-4. [DOI] [PubMed] [Google Scholar]
- 130.Moore JD, Hayes C, Hicks RA. REM sleep deprivation increases preference for novelty in rats. Physiol Behav. 1979;23(5):975–976. doi: 10.1016/0031-9384(79)90211-7. [DOI] [PubMed] [Google Scholar]
- 131.Ferraz MR, Ferraz MM, Santos R. How REM sleep deprivation and amantadine affects male rat sexual behavior. Pharmacol Biochem Behav. 2001;69(3–4):325–332. doi: 10.1016/s0091-3057(01)00508-1. [DOI] [PubMed] [Google Scholar]
- 132.Benedetti F, Fresi F, Maccioni P, Smeraldi E. Behavioural sensitization to repeated sleep deprivation in a mice model of mania. Behav Brain Res. 2008;187(2):221–227. doi: 10.1016/j.bbr.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 133.Siegel JM, Rogawski MA. A function for REM sleep: regulation of noradrenergic receptor sensitivity. Brain Res. 1988;472(3):213–233. doi: 10.1016/0165-0173(88)90007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Demontis MG, Fadda P, Devoto P, Martellotta MC, Fratta W. Sleep deprivation increases dopamine D1 receptor antagonist [3H]SCH 23390 binding and dopamine-stimulated adenylate cyclase in the rat limbic system. Neurosci Lett. 1990;117(1–2):224–227. doi: 10.1016/0304-3940(90)90148-3. [DOI] [PubMed] [Google Scholar]
- 135.Schreiber W, Opper C, Dickhaus B, Heiser P, Wesemann W, Krieg JC. Alterations of blood platelet MAO-B activity and LSD-binding in humans after sleep deprivation and recovery sleep. J Psychiatr Res. 1997;31(3):323–331. doi: 10.1016/s0022-3956(96)00062-3. [DOI] [PubMed] [Google Scholar]
- 136.Ebert D, Ebmeier KP. The role of the cingulate gyrus in depression: from functional anatomy to neurochemistry. Biol Psychiatry. 1996;39(12):1044–1050. doi: 10.1016/0006-3223(95)00320-7. [DOI] [PubMed] [Google Scholar]
- 137.Gerner RH, Post RM, Gillin JC, Bunney WE., Jr Biological and behavioral effects of one night's sleep deprivation in depressed patients and normals. J Psychiatr Res. 1979;15(1):21–40. doi: 10.1016/0022-3956(79)90004-9. [DOI] [PubMed] [Google Scholar]
- 138.Post RM, Kotin J, Goodwin FK. Effects of sleep deprivation on mood and central amine metabolism in depressed patients. Arch Gen Psychiatry. 1976;33(5):627–632. doi: 10.1001/archpsyc.1976.01770050077012. [DOI] [PubMed] [Google Scholar]
- 139.Cirelli C, Tononi G. Differential expression of plasticity-related genes in waking and sleep and their regulation by the noradrenergic system. J Neurosci. 2000;20(24):9187–9194. doi: 10.1523/JNEUROSCI.20-24-09187.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tononi G, Cirelli C. Modulation of brain gene expression during sleep and wakefulness: a review of recent findings. Neuropsychopharmacology. 2001;25(5 Suppl):S28–S35. doi: 10.1016/S0893-133X(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 141.Payne JL, Quiroz JA, Zarate CA, Jr, Manji HK. Timing is everything: does the robust upregulation of noradrenergically regulated plasticity genes underlie the rapid antidepressant effects of sleep deprivation? Biol Psychiatry. 2002;52(10):921–926. doi: 10.1016/s0006-3223(02)01676-1. [DOI] [PubMed] [Google Scholar]
- 142.Gillin JCMC, Post RM, Jimerson D, Bunney WE., Jr An EEG sleep study of a bipolar (manic-depressive) patient with a noctural switch. Biol Psychiatry. 1977;12(6):711–718. [PubMed] [Google Scholar]
- 143.Einat H, Manji HK, Gould TD, Du J, Chen G. Possible involvement of the ERK signaling cascade in bipolar disorder: behavioral leads from the study of mutant mice. Drug News Perspect. 2003;16(7):453–463. doi: 10.1358/dnp.2003.16.7.829357. [DOI] [PubMed] [Google Scholar]
- 144.Sitaram N, Gillin JC, Bunney WE., Jr The switch process in manic-depressive illness. Circadian variation in time of switch and sleep and manic ratings before and after switch. Acta Psychiatr Scand. 1978;58(3):267–278. doi: 10.1111/j.1600-0447.1978.tb06938.x. [DOI] [PubMed] [Google Scholar]
- 145.Wirz-Justice A. Biological rhythm disturbances in mood disorders. Int Clin Psychopharmacol. 2006;21 Suppl 1:S11–S15. doi: 10.1097/01.yic.0000195660.37267.cf. [DOI] [PubMed] [Google Scholar]
- 146.Gould TD, Zarate CA, Manji HK. Glycogen synthase kinase-3: a target for novel bipolar disorder treatments. J Clin Psychiatry. 2004;65(1):10–21. [PubMed] [Google Scholar]
- 147.Lee KY, Ahn YM, Joo EJ, et al. No association of two common SNPs at position −1727 A/T, −50 C/T of GSK-3 beta polymorphisms with schizophrenia and bipolar disorder of Korean population. Neurosci Lett. 2006;395(2):175–178. doi: 10.1016/j.neulet.2005.10.059. [DOI] [PubMed] [Google Scholar]
- 148.Michelon L, Meira-Lima I, Cordeiro Q, et al. Association study of the INPP1, 5HTT, BDNF, AP-2beta and GSK-3beta GENE variants and restrospectively scored response to lithium prophylaxis in bipolar disorder. Neurosci Lett. 2006;403(3):288–293. doi: 10.1016/j.neulet.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 149.Benedetti F, Bernasconi A, Lorenzi C, et al. A single nucleotide polymorphism in glycogen synthase kinase 3-beta promoter gene influences onset of illness in patients affected by bipolar disorder. Neurosci Lett. 2004;355(1–2):37–40. doi: 10.1016/j.neulet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 150.Roybal K, Theobold D, Graham A, et al. Mania-like behavior induced by disruption of CLOCK. Proc Natl Acad Sci U S A. 2007;104(15):6406–6411. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu L, Foroud T, Xuei X, et al. Evidence of association between brain-derived neurotrophic factor gene and bipolar disorder. Psychiatr Genet. 2008;18(6):267–274. doi: 10.1097/YPG.0b013e3283060f59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. doi: 10.1016/s0092-8674(03)00035-7. [DOI] [PubMed] [Google Scholar]
- 153.Sklar P, Gabriel SB, McInnis MG, et al. Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry. 2002;7(6):579–593. doi: 10.1038/sj.mp.4001058. [DOI] [PubMed] [Google Scholar]
- 154.Rybakowski JK, Borkowska A, Czerski PM, Skibinska M, Hauser J. Polymorphism of the brain-derived neurotrophic factor gene and performance on a cognitive prefrontal test in bipolar patients. Bipolar Disord. 2003;5(6):468–472. doi: 10.1046/j.1399-5618.2003.00071.x. [DOI] [PubMed] [Google Scholar]
- 155.Muller DJ, de Luca V, Sicard T, King N, Strauss J, Kennedy JL. Brain-derived neurotrophic factor (BDNF) gene and rapid-cycling bipolar disorder: family-based association study. Br J Psychiatry. 2006;189:317–323. doi: 10.1192/bjp.bp.105.010587. [DOI] [PubMed] [Google Scholar]
- 156.Dmitrzak-Weglarz M, Rybakowski JK, Suwalska A, et al. Association studies of the BDNF and the NTRK2 gene polymorphisms with prophylactic lithium response in bipolar patients. Pharmacogenomics. 2008;9(11):1595–1603. doi: 10.2217/14622416.9.11.1595. [DOI] [PubMed] [Google Scholar]
- 157.Sarchiapone M, Carli V, Roy A, et al. Association of polymorphism (Val66Met) of brain-derived neurotrophic factor with suicide attempts in depressed patients. Neuropsychobiology. 2008;57(3):139–145. doi: 10.1159/000142361. [DOI] [PubMed] [Google Scholar]