

Abstract
It is clear that the G protein-coupled receptor family play a key role in the pharmaceutical industry, with a significant proportion of approved drugs targeting this protein class. While our growing understanding of the complexity of G protein-coupled receptor pharmacology is playing a key role in the future success of these endeavours, with allosteric mechanisms now well integrated into the industrial community and G protein-independent signalling mechanisms establishing themselves as novel phenomenon to be exploited, it is still possible to underestimate the complexity of G protein signal transduction mechanisms and the impact that inappropriate study of these mechanisms can have on data interpretation. In this manuscript we review different approaches to measuring the cAMP signal transduction pathway, with particular emphasis on key parameters influencing the data quality and biological relevance.
Keywords: cAMP, signalling, G protein-coupled receptor, screen, drug discovery
Introduction
An understanding of the pathways and mechanisms of G protein-coupled receptor (GPCR) activation and their relationship to disease is key in terms of achieving success in the development of novel, efficacious drugs with limited adverse effects. Although GPCR pharmacology and disease biology are inevitably complex, the careful selection of in vitro assays throughout the screening process can considerably increase the likelihood of identifying compounds with the desired effects. Historically, small molecule modulators of GPCR function were identified through high-throughput assays that employed radiolabelled ligands, or more recently fluorescent-labelled equivalents, in membrane binding assays (Sittampalam et al., 1997; Gribbon and Sewing, 2003; Middleton and Kellam, 2005; Cordeaux et al., 2008). Ligand binding assays dominated the early stage investment in high-throughput screening (HTS) of large compound files (=1 million) as they represented a relatively cheap and rapid detection method, which contrasted the complex nature of the GPCR signal transduction pathways and the original assays to measure them. However, over the last decade, functional assays have become increasingly common in primary screening, with assays that detect changes in intracellular signalling molecules or changes in gene transcription, being cost effective, robust and eminently HTS compatible (Williams and Sewing, 2005). These technologies range from low to high information content and employ a variety of radiometric, absorbent, fluorescent and luminescent end-points (Thomsen et al., 2005; McLoughlin et al., 2007). Consequently, there appears to be an assay to study almost every GPCR target, at multiple steps of the receptor binding and activation cascade.
Classical receptor activation occurs via agonist-induced conformational changes in the receptor which, upon interaction with a heterotrimeric G protein (composed of α-, β- and γ-subunits), initiates the G protein cycle (involving GDP/GTP exchange and hydrolysis at the α-subunit; Leifert et al., 2005). There are four major types of G protein, identified by a preferential interaction with different signalling effector molecules: Gq G proteins modulate the enzyme phospholipase C β, which regulates intracellular signalling molecules such as phosphatidylinositol 4,5-bisphosphate (PIP2), inositol 1,4,5-trisphosphate (IP3) and intracellular Ca2+; the G12/13 G proteins are primarily involved in Rho-mediated responses; and the Gs and Gi/o G proteins are characterized via an increase or decrease in cAMP respectively.
The levels of intracellular cAMP are tightly regulated, with production controlled through the adenylyl cyclase family of enzymes [convert adenosine tri-phosphate (ATP) to cAMP and inorganic pyrophosphate] and degradation controlled via the cAMP phosphodiesterase (PDEs) enzymes (catalyse the hydrolysis of the 3′ ester bond of cAMP to form 5′ adenosine monophosphate (Thompson, 1991; Hanoune and Defer, 2001; Patel et al., 2001). The adenylyl cyclases are activated or inhibited via direct interaction with G protein α-subunits and, for some isoforms, with Ca2+ and calmodulin. When cAMP is produced it binds to protein kinases within the cell, initiating phosphorylation events that regulate target enzymes and transcription factors. There are a variety of cAMP PDE isoforms, which are generally activated by cAMP-dependent protein kinases, thus providing an important negative feedback system on the receptor-mediated signalling cascade and regulating the extent of changes in intracellular cAMP concentrations (Thompson, 1991).
While there are a wide variety of in vitro screening technologies that enable the measurement of changes in cAMP, to deploy them effectively in both the academic and industrial environment requires important attention to details such as kinetics and sensitivity. Therefore, in this manuscript we will review different approaches to measuring the cAMP signal transduction pathway, with particular emphasis on key parameters influencing the data and their interpretation.
3H-Adenine prelabelling approaches to monitor cyclic AMP accumulation
One of the most direct approaches to monitoring cAMP generation from ATP in response to adenylyl cyclase activation in living cells is to follow this conversion using radiolabelled precursors. In intact cells, this is best achieved by prelabelling the adenine nucleotide pool with 3H-adenine and then using sequential Dowex/Alumina column chromatography (Minneman et al., 1979) to separate 3H-cAMP from all other tritium-labelled adenine derivatives (Huang et al., 1971; Donaldson et al., 1988b; McCrea and Hill, 1996; Baker et al., 2003b). This method, although time-consuming, provides a direct read-out of 3H-cAMP accumulation. It is highly sensitive and has a large dynamic range over which cAMP responses can be measured. For example, agonist responses in excess of 100-fold over basal levels can be routinely monitored alongside partial agonist effects as low as twofold over basal (McCrea and Hill, 1996; Baker et al., 2004). This large dynamic range ensures that true maximal responses are directly measured and gives confidence in the EC50 values that are calculated from agonist concentration–response relationships obtained.
3H-adenine-labelling strategies have also proved useful in the study of the kinetics of cAMP accumulation. For example, this assay has been used to demonstrate that the synergy observed between histamine H1 and adenosine A2B receptor stimulation on cAMP accumulation is a consequence of enhanced adenylyl cyclase activity rather than decreased PDE activity in brain slices (Donaldson et al., 1988a,b;). Kinetic studies of 3H-cAMP accumulation have also provided important information on the duration of action of agonists (e.g. salmeterol on β1- and β2-adrenoceptors) at different GPCRs (McCrea and Hill, 1996) and provided information on the differing kinetics of intracellular cAMP accumulation and cAMP efflux from cells (McCrea and Hill, 1993; Baker et al., 2004). A concern that is always raised with a radioactive readout of the accumulation of any intracellular second messenger such as cAMP is that it is a measure of turnover rather than absolute levels. However in instances where not all cAMP containing compartments are responsive to the stimulus under investigation, measurement of cAMP turnover can lead to an enhanced signal-to-noise ratio. Interestingly, quantitative measurement of cAMP accumulation and cAMP efflux by liquid chromatography tandem electrospray mass spectrometry in CHO cells expressing the human β2-adrenoceptor gave similar profiles (Cordell et al., 2008) to those reported in the same cells from 3H-adenine prelabelling approaches (Baker et al., 2004).
It is also worth mentioning that the extensive efflux of cAMP from certain cell types (e.g. CHO cells; McCrea and Hill, 1993;Baker et al., 2004; Cordell et al., 2008) may compromise some of the homogenous ‘add and read’ cAMP assays (detailed below) when kinetic measurements are required and wash steps are not included in the assay. This is because extracellular cAMP appears to be largely protected from PDE activity that appears confined to the intracellular environment. The extent to which this is an issue will, however, depend on the nature of the assay concerned. For example it will not be a major problem when the cAMP sensor is confined to the intracellular environment of the cell, but will affect the interpretation of mass assays that incorporate a lysis step to enable intracellular cAMP to come in contact with an anti-cAMP antibody.
Mass-assay competition assays for cyclic AMP detection
There are a plethora of technologies, ranging from radiometric to enzymatic, that take advantage of a simple approach to measuring cAMP accumulation via competition between cellular cAMP and labelled cAMP moieties for binding to an anti-cAMP antibody (reviewed extensively within Williams, 2004). Radiometric cAMP accumulation assays, such as elisas and scintillation proximity assays (SPA, GE healthcare, Chalfont St Giles, Bucks, UK & Flashplates, Perkin Elmer, Waltham, MA, USA), are readily available and are employed by many academic laboratories. The latter are homogeneous formats, which enable the direct detection of [125I]-labelled cAMP once it is in close proximity to a solid scintillant surface (Kariv et al., 1999). These have been successfully employed in HTS and offer a distinct advantage over more traditional elisa methods in terms of convenience (stimulation and detection can be carried out in the same well), time and reproducibility. However, the majority of pharmaceutical company laboratories prefer to employ fluorescent or luminescent assays due to safety, cost and throughput considerations. One of the first fluorescent technologies to emerge in this area was the fluorescence polarization (FP) technology, in which the emission from ‘free’ labelled cAMP, when excited with polarized light, is depolarized due to the rotation of the molecule in the time between excitation and emission. FP cAMP accumulation assays are very simple, do not require bespoke equipment and can be employed with whole cells (where the cytoplasmic cAMP content is exposed by cell lysis), or cell membranes. However, these assays are often susceptible to compound interference, particularly with the commonly employed green dyes (e.g. fluorescein). The use of more red-shifted dyes can minimize this, as demonstrated by Banks et al. who highlighted that sensitivity to the highly coloured tartrazine molecule was reduced using Bodipy-TMR (Banks et al., 2000). Furthermore, while use of this red-shifted assay was clearly enabling for the melanocortin receptors studied, the low quantum yields obtained with further red-shifted dyes can make these assays technically more challenging. In addition to these compound artefacts, FP assays are relatively insensitive to low levels of cAMP, with detection limits in the range of 50–100 fmol (or worse). To overcome these issues a number of fluorescent and luminescent technologies emerged, including chemiluminescent proximity assays (ALPHAScreen, Perkin Elmer), enzyme complementation assays (e.g. Hit Hunter, DiscoveRx), electro-chemiluminescence detection (Meso Scale Discovery) and time resolved fluorescence resonance energy transfer (FRET) (HTRF®, Cis Bio & LANCE®, DELFIA®, Perkin Elmer). These technologies vary in terms of their costs, reader compatibility and impact of compound interference (Williams, 2004; McLoughlin et al., 2007).
The ratiometric read-out obtained with FRET technologies enables correction for well to well variation, can minimize short-lived background fluorescence and can limit quenching and autofluorescence effects, providing the interference is observed at both fluorescence wavelengths (Gabriel et al., 2003). Furthermore, all of these FRET-based techniques demonstrate a high cAMP sensitivity, particularly for the DELFIA method that is reported to achieve levels as low as 0.1 fmol (although this technology does require wash steps, which is a significant disadvantage in the HTS arena). While the broad linear range and high signal-to-background of these FRET-based techniques is also shared with ALPHAScreen (Golla and Seethala, 2002; Gabriel et al., 2003), the key disadvantages of the latter are the impact of light and temperature fluctuations on data quality, robustness and ease of use in the industrial setting (Golla and Seethala, 2002; Williams, 2004). Furthermore, all of these technologies share the disadvantage that increases in cAMP produce a decrease in signal, making them liable to false positives.
This is a factor that differentiates the enzyme complementation technology, in which an increase in cAMP leads to a positive signal. However, it is important to emphasize that this enzyme complementation approach like the other techniques described above is based on competition between the endogenous cAMP and a labelled cAMP probe. While there is a greater temptation to use the positive readout from the enzyme complementation as a direct indicator of cAMP levels, unless the data are transformed through a standard curve, the outcomes in terms of EC50 and apparent efficacy can potentially be very misleading, a factor true of all of these technologies (Figure 1).
Figure 1.
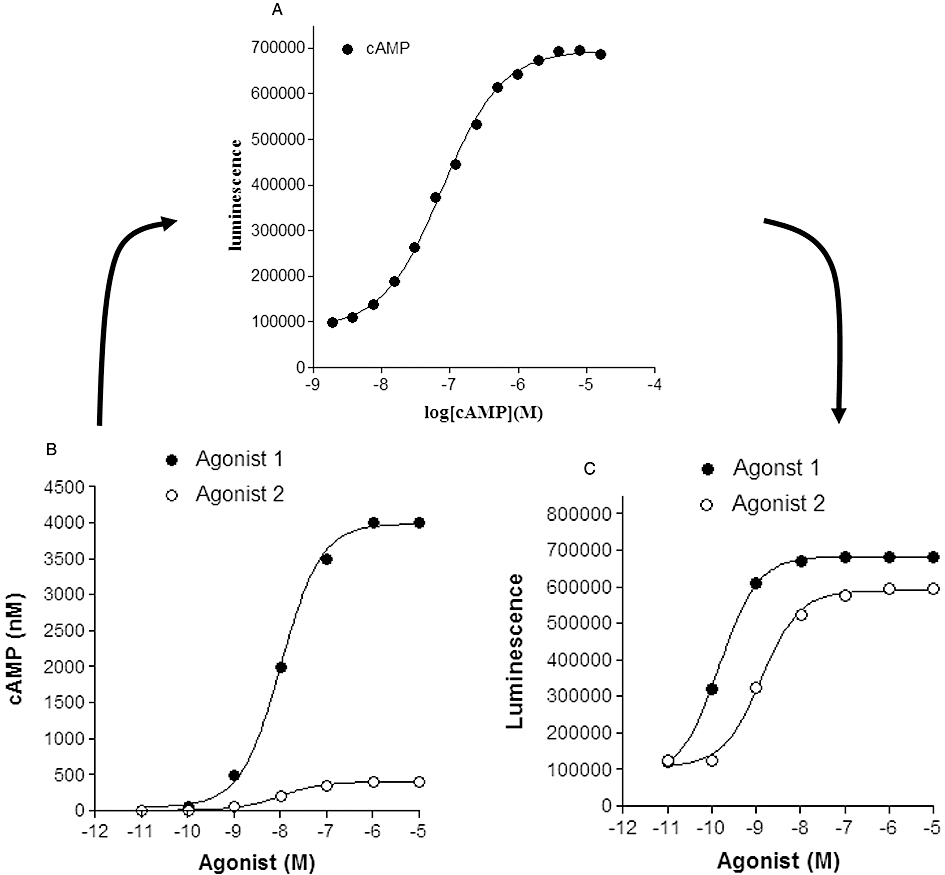
Influence of cAMP standard curves on the relationship between agonist concentration and measured response (cAMP or luminescence activity). (A) A cAMP standard curve determined for a competitive immunoassay-based enzyme complementation assay for cAMP (HitHunter, DiscoveRx). In this assay, cAMP competes for antibody binding against a cAMP analogue that is conjugated to a small peptide fragment of β-galactosidase. In the absence of free cAMP, the majority of this galactosidase conjugate of cAMP is captured by the antibody and is therefore unavailable for complementation to a larger fragment of β-galactosidase resulting in a low signal output. In the presence of free cAMP, antibody sites are occupied, leaving the cAMP conjugate free to complement with the large fragment of β-galactosidase, forming an active β-galactosidase enzyme that leads to substrate hydrolysis and the production of a chemiluminescent signal. Data points (Dr J.G. Baker, unpubl. data) are the mean of 6 replicates at each concentration of cAMP. (B) Simulated concentration–response curves for two agonists with a log EC50 of −8.0 and maximal responses that differ by one order of magnitude (i.e. generating cAMP levels in each well of 400 and 4000 nM). (C) Resultant concentration–response curves generated from the concentration–response curves in (B) when converted to luminescence activity via interpolation from the cAMP standard curve in (A). Log EC50 values obtained in (C) were −8.9 and −9.8 for agonists 2 and 1 respectively.
Figure 1 shows the impact of the dynamic range of a cAMP standard curve (Figure 1A; for a competitive immunoassay-based cAMP assay) on the relationship between agonist concentration and response when directly read as a positive signal from the cAMP assay (in this case as measured luminescence activity; Figure 1C) rather than converting to absolute cAMP levels via the standard curve. In the particular example shown in Figure 1, the partial agonist nature of agonist 2 is lost in Figure 1C and the EC50 of agonist 1 is wrongly reported. These artefacts are caused because the true maximum response to agonist 1 measured in the well (seen in Figure 1B) exceeds the maximal range of the cAMP standard curve. To achieve a true reading, the cAMP content of the test well needs to be diluted and re-probed in order to ensure that the cAMP levels are within the dynamic range of the assay. This is a particular problem with homogenous ‘add-and-read’ assays where the total cAMP in the well is normally exposed to the cAMP antibody.
The sensitivity of enzyme complementation to coloured artefacts can be highly dependent on the use of adherent versus suspended cells, as noted by Golla and Seethala when they investigated these two different approaches in the study of the GLP-1 receptor (Golla and Seethala, 2002). Each of these next generation mass-assay competition methods are more sensitive than radiometric and FP assays, with levels of ≤10 fmol cAMP per well being quoted (Williams, 2004). However, it is important to note that the sensitivity of the assay within the cell-free environment, used to generate the standard curve, is not necessarily the same as that for the cell-based assay environment, required to generate the response of interest (Gabriel et al., 2003).
One critical factor to consider when adopting any of these mass-assay competition technologies is the fact that there is a non-linear relationship between receptor activation and signal detection. Therefore relatively large changes in absolute cAMP concentration can equate to small changes in read-out. This in turn can impact upon agonist potency, the differentiation between full and partial agonism and assay robustness. This was clearly highlighted by Allen et al. who investigated the influence of the non-linear relationship between the signal detected and cAMP concentration on the stimulation of two Gs-coupled systems, the human vasopressin V2 receptor and β2-adrenoceptor, using a membrane-based FP assay (Allen et al., 2002). At high agonist concentrations, the relative change in the degree of polarization was low, as would be expected being at the bottom of the measured dose–response curve. However, these relatively small changes in polarization equated to large differences in cAMP (as measured from the standard curve). This led to an increase in the potency of vasopressin when measured directly from the polarization signal (the signal being compressed compared with the absolute levels of cAMP) and a higher level of variation within the cAMP estimation resulting in a lower assay robustness. Another factor to consider in these assays is the impact of temperature, particularly for assays utilizing the shorter incubation times (e.g. signals can be detected with as little as 15 min incubation with some of these techniques), where differences in temperature can make a significant difference to the dynamics of the reaction. Furthermore, all of these assays detect the levels of cAMP accumulated across a population of cells during the time course of the assay, which means that the kinetics and temporal nature of the cAMP changes are lost.
cAMP response element-based reporter genes
The downstream effects of cAMP can also be utilized to monitor changes in the intracellular levels of this cyclic nucleotide. One of the easiest readouts of this downstream signalling is provided by reporter genes that contain a cAMP response element (CRE) that regulates the transcription of an enzyme or a fluorescent/bioluminescent protein (Hill et al., 2001). cAMP activates this process by stimulating protein kinase A (PKA) activity. The catalytic unit of PKA then translocates to the nucleus where it phosphorylates a CRE-binding protein (CREB) allowing it to bind to CRE sequences in target genes. The CRE in the promoters of the genes for somatostatin and corticotrophin-releasing hormone contain the sequence TGACGTCA, but other variations do occur. For example, in the c-fos promoter the sequence is TGAACGTTT (Montminy et al., 1990; Guardiola-Diaz et al., 1994; Hill et al., 2001). Synthetic promoters made up of multiple copies of these sequences are routinely employed in reporter genes and have been extensively used to study GPCRs (George et al., 1997; McDonnell et al., 1998; Baker et al., 2003a,b;).
The reporter protein needs to be easily distinguishable from other cell products [e.g. a heat-stable secreted placental alkaline phosphatase, firefly luciferase or green fluorescent protein (GFP)] but also to have a short half-life to minimize basal accumulation of reporter proteins that can restrict the sensitivity of the final readout. In the case of GFP, this can only be achieved by creating a destabilized version of GFP by fusing the PEST degradation domain from mouse ornithine decarboxylase to the C-terminal of GFP (Li et al., 1998). β-Lactamase (penicillin amido-β-lactamhydrolase) has also been used increasingly as a reporter gene because of the synthesis of highly fluorescent substrates in membrane-permeant ester forms that can readily be taken up into living cells (Zlokarnik et al., 1998). This can potentially be used to monitor gene expression in single living cells. The substrate consists of two fluorophores that, when attached to the β-lactam ring of cephalosporin, are held sufficiently close together for FRET to occur. Generation of the β-lactamase reporter enzyme in cells in response to an elevation of intracellular cAMP breaks open the β-lactam ring leading to a decreased FRET signal.
Reporter assays have been used extensively to monitor the pharmacological characteristics of Gs- and Gi-coupled GPCRs but they do involve substantial signal amplification between ligand binding and the final measured response (Hill et al., 2001; Baker and Hill, 2007b). This is most marked when evaluating ligands with partial agonist activity. In most reporter gene systems these molecules are likely to manifest themselves as full agonists because of the increased signal amplification (Baker et al., 2003b). The level of signal amplification achievable within reporter assays is probably best illustrated by the observation that many β-blockers in common clinical practice produce substantial agonist effects at β1- and β2-adrenoceptors when measured at the level of gene expression (Baker et al., 2003a,b; Baker and Hill, 2007b). However, given the fact that many antagonists are prescribed to patients in order to achieve prolonged receptor antagonism over months and years, this is a property of these ligands that should be investigated further.
Kinetic studies of the time course of agonist-stimulated gene expression have been undertaken using reporter genes (Baker et al., 2004). These studies have shown that a minimum of 30 min of agonist exposure is required to detect a measureable change in reporter gene activity and that it is the duration of cAMP elevation rather than the total quantity of cAMP produced that is the major determinant of the final response (Baker et al., 2004). This is likely due to the rate-limiting nature of the nuclear entry of the PKA-catalytic unit (Hagiwara et al., 1993; Mayr and Montminy, 2001) and the saturation of this process at relatively low subunit concentrations. The requirement for sustained stimulation needs to be taken into account when designing an assay and the potential for receptor desensitization with highly efficacious agonists during the time course of the assay means that lower efficacy agonists should be employed whenever possible. Interestingly, agonist efficacy and the state of receptor phosphorylation have been implicated in the observed difference in antagonist affinity obtained from measurements of cAMP accumulation and reporter gene assays (Baker et al., 2003c).
A final issue to bear in mind in the interpretation of reporter gene responses is the potential for a response that is considerably downstream of the initial ligand receptor interaction to be generated by more than one signalling pathway. For example, the generally accepted route between cAMP and CRE-mediated reporter genes via PKA can be by-passed by a signalling pathway that involves another cAMP responsive protein, exchange protein directly activated by cAMP (EPAC) and the intermediacy of B-Raf, p42/44 ERK and Rsk-2 leading to CREB phosphorylation (Baker et al., 2003b). As a consequence, reporter gene assays designed to pick up Gs- and cAMP-mediated responses can also reveal responses mediated via the p42/44 ERK pathway, potentially through G protein-independent mechanisms. This latter property has been used to provide evidence for agonist-directed signalling by the human β2-adrenoceptor (Baker et al., 2003b).
Firefly luciferase-based biosensors
To date, the majority of population-based cAMP readouts require cell lysis prior to cAMP detection. As such, the advantage of the live cell firefly luciferase-based cAMP biosensor, which can continually track the generation of cAMP-mediated luminescence over time, is clear when investigating the kinetics of cAMP accumulation. This intact cell multi-well microplate assay is based on the oxidation of firefly luciferin by firefly luciferase, a by-product of which is the emission of light at 550–570 nM, according to the following reaction:
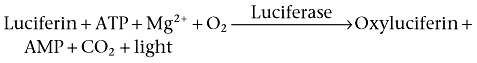 |
Genetic manipulation of firefly luciferase into a reversible biosensor of cAMP generation involved the insertion of the cAMP binding domain B from the regulatory subunit type IIβ into a circularly permuted form of firefly luciferase (Fan et al., 2008; Binkowski et al., 2009). In vitro, the firefly luciferase-based cAMP biosensor has a pEC50 for cAMP of 6.3 and a large signal-to-noise window of approximately 70-fold (Fan et al., 2008). When expressed in HEK293 cells, the addition of 10 µM forskolin, a direct activator of adenylyl cyclase, can mediate a 25-fold increase in the luminescent signal within 3.5 min (Fan et al., 2008). As such, this sensor represents a powerful method to detect the kinetics of cAMP generation and degradation/removal in vivo. However when interpreting kinetics of the luminescent signal, consideration must be given towards the potential for a delay between the real-time cAMP dynamics and the generation of the active form of firefly luciferase.
Single cell biosensors
Currently, most studies investigating the spatial and temporal characteristics of cAMP signalling at a single cell level rely on FRET-based biosensors. FRET refers to the transfer of energy from a donor fluorophore to an acceptor fluorophore that has a lower, but overlapping excitation spectrum. The efficiency of the energy transfer is highly dependent on a number of factors including the spectral characteristics of the FRET pair, the distance between the donor and acceptor fluorophores and the relative orientation of the fluorescent proteins. The following equation describes FRET efficiency (E) in terms of the distance (r) between the fluorescent proteins and the Förster radius (R0):
The Förster radius represents the distance between the two fluorophores at which half maximal energy transfer occurs. This factor is specific for each FRET pair and takes into account the relative orientation of each fluorophore and the spectral characteristics of the FRET pair. Additional factors that need to be taken into account when developing FRET biosensors include the folding efficiency of the fluorophores at 37°C, their sensitivity to changes in the cellular microenvironment such as pH and ionic strength, their fluorescent intensity and the resistance of the FRET pair to bleaching. The FRET sensors should have minimal impact on intracellular signalling; however, by their very nature, the expression of the biosensor will cause a degree of buffering of the pathway of interest. Additional factors that could lead to an alteration in intracellular signalling include the size of the FRET pair, the propensity of the fluorescent proteins to dimerize or aggregate and the level of expression (van der Krogt et al., 2008).
Fluorescence imaging of cAMP dynamics was first reported using FlCRhR, a FRET sensor consisting of fluorescein-tagged PKA-catalytic subunits and rhodamine-tagged regulatory subunits, that could be microinjected into living cells (Adams et al., 1991). In the absence of cAMP, the PKA-regulatory and catalytic subunits form a holoenzyme complex, consisting of two PKA-regulatory and two PKA-catalytic subunits. Prior to dissociation of the complex, four cAMP molecules are required to bind. The potency of cAMP at FlCRhR is approximately 90 nM with slight positive cooperatively and a dynamic range of approximately 0.01 to 1 µM (Adams et al., 1991). In the presence of low concentrations of cAMP, the formation of FlCRhR into the holoenzyme complex allows transfer of energy from the fluorescein donor to the rhodamine acceptor upon excitation of fluorescein at 480–495 nm. Under these conditions, emission at 500–570 nm and 570–620 nM that are characteristic of fluorescein and rhodamine, respectively, can be detected. Upon binding cAMP, the PKA holoenzyme complex dissociates causing an increase in the distance between the two fluorophores and a subsequent decrease in the energy transfer.
FlCRhR has been used successfully to study cAMP compartmentalization within neurons (Bacskai et al., 1993; Hempel et al., 1996). Bacskai et al. demonstrated that exposure of Aplysia sensory neurons to serotonin stimulated a greater level of cAMP accumulation within the neuronal processes as compared with the cell body. The kinetics of cAMP accumulation increased with the concentration of neuromodulator (Bacskai et al., 1993). In addition, Gorbunova and Spitzer used FlCRhR to investigate the dynamic interplay between calcium oscillations and transient increases in the intracellular cAMP concentration in embryonic spinal neurons (Gorbunova and Spitzer, 2002). These studies demonstrate some of the advantages of using fluorescent sensors to image cAMP fluctuations in live cells. In contrast to population assays, FlCRhR can provide information on the kinetics of cAMP accumulation within the various subcellular domains. However, the requirement to microinject FlCRhR into cells is technically demanding and has limited utility within a range of cells (Zaccolo et al., 2000).
Genetically encoded FRET sensors have increased versatility as cAMP probes. Currently, most cAMP FRET sensors use cyan fluorescent protein (CFP) as the donor fluorophore and yellow fluorescent protein (YFP) as the acceptor fluorophore. The emission spectrum of CFP is relatively wide and has considerable overlap with the excitation spectrum of YFP. The initial genetically encoded CFP/YFP-based cAMP FRET sensor consisted of CFP-tagged regulatory RII PKA subunits and YFP-tagged catalytic subunits (Zaccolo and Pozzan, 2002). Similar to FlCRhR, the FRET generated by this sensor is inversely proportional to the cAMP concentration. When expressed in rat neonatal cardiac myocytes, the PKA-based cAMP sensor was shown to be restricted to sarcomeric Z lines as a result of anchoring by A-kinase anchoring proteins. Upon direct stimulation of adenylyl cyclase activity or slowing of cAMP metabolism, the CFP-regulatory subunit remained localized to the sarcomeric Z lines; whereas the distribution of the YFP-catalytic subunit was more diffuse, reflecting subunit dissociation. Stimulation of β-adrenoceptors with a relatively high concentration of agonist typically caused a transient decrease in FRET that was localized to discrete microdomains along the striations, had a t1/2 of approximately 11 s, was maximal at approximately 45 s and reversed within 100–300 s (Zaccolo and Pozzan, 2002).
Exchange protein directly activated by cAMP is another protein that has been employed to generate a CFP/YFP-based cAMP FRET sensor (Figure 2; DiPilato et al., 2004; Nikolaev et al., 2004; Ponsioen et al., 2004). However in contrast to PKA, the EPAC-based sensor is a unimolecular probe and as such has the advantage of expressing the CFP and YFP proteins at equivalent levels. The EPAC-based sensors consist of either the complete EPAC protein or a region of the protein containing the cAMP binding domain, with a fluorophore attached to each end (DiPilato et al., 2004; Nikolaev et al., 2004; Ponsioen et al., 2004). A direct comparison of PKA- and EPAC-based FRET sensors by Nikolaev et al. found that the PKA probe had slower kinetics than that of the EPAC sensor. This has been suggested to be due to the requirement of the PKA-based sensor to bind four cAMP molecules prior to dissociation as opposed to the EPAC-based sensor that undergoes a conformational change upon the binding of one molecule of cAMP (Nikolaev et al., 2004). The PKA- and EPAC-based FRET sensors have a similar affinity for cAMP of approximately 0.3–3 µM and a dynamic range of approximately 0.1–10 µM (Mongillo et al., 2004; Nikolaev et al., 2004). As a consequence of their relatively high sensitivity, these biosensors are likely to be quickly saturated in cell types, such as adult cardiomyocytes, that have particularly high concentrations of cAMP (Nikolaev et al., 2006). In these cases it may be more appropriate to use a probe with lower sensitivity such as the FRET sensor based on the cAMP binding domain of the hyperpolarization-activated cyclic nucleotide-gated channel 2 (HCN2) that has an affinity of approximately 6 µM and a dynamic range of approximately 1–100 µM (Nikolaev et al., 2006). When expressed in HEK cells, the EPAC-based cAMP probe had a uniform, cytosolic distribution (Figure 2B). However, to further define the compartmentalization of cAMP responses, FRET sensors have been targeted to discrete microcellular domains. For example, EPAC and PKA-based cAMP sensors have been targeted to the plasma membrane through polybasic sequences and/or a farnesylation, myristoylation or palmitoylation motif. Generally, the activation kinetics of plasma membrane targeted cAMP sensors are more rapid and of greater amplitude than those of their cytosolically distributed equivalents (DiPilato et al., 2004; Dyachok et al., 2006; Terrin et al., 2006). This may reflect the fact that the plasma membrane is the site of cAMP production and/or the restriction of cAMP diffusion within microdomains located near the plasma membrane. Mitochondria and nuclear targeted cAMP FRET sensors have also been developed (DiPilato et al., 2004). These sensors can provide a wealth of information with regards to subcellular cAMP signalling; however, any alteration in the properties of the sensor due to the addition of a targeting motif should be taken into consideration.
Figure 2.
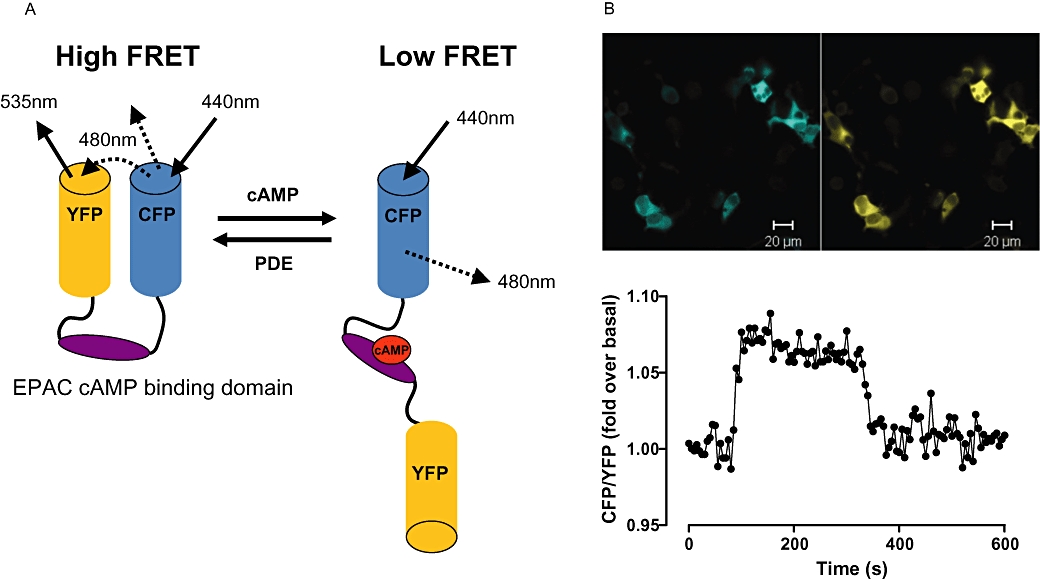
EPAC-based cAMP FRET sensor. (A) A schematic showing the basic design of an EPAC-based cAMP FRET sensor. In the absence of cAMP the conformation of the sensor is such that the fluorophores are in close proximity, generating FRET. Upon cAMP binding, conformational changes result in a decrease in FRET. (B) Top panel: Fluorescent images of HEK293T cells transiently transfected with a CFP/YFP FRET sensor containing the EPAC2 cAMP binding domain. CFP and YFP fluorescence are shown in blue and yellow respectively. Lower panel: A single cell trace plotted as a ratio of CFP: YFP intensity from HEK293T cells transiently tranfected with the EPAC2-based cAMP sensor. Exposure of cells to 5′-N-ethyl carboxamide adenosine at 30 s stimulates cAMP accumulation through an endogenously expressed adenosine A2B receptor. Inhibition of cAMP accumulation results from the addition of the adenosine receptor antagonist xanthine amine congener at 300 s. CFP, cyan fluorescent protein; EPAC, exchange protein directly activated by cAMP; FRET, fluorescence resonance energy transfer; PDE, phosphodiesterase; YFP, yellow fluorescent protein.
As such, cAMP FRET sensors can provide the temporal and spatial resolution required to study cAMP accumulation at the single cell level. These sensors are well suited for live cell imaging as they are readily reversible and generally, variations in fluorescent intensity due to the probe concentration and/or the laser power is largely corrected for upon transformation of the results into a FRET ratio. However, many of the cAMP FRET sensors have a limited dynamic range that may cause low levels of cAMP to go undetected and saturation of the probe at high cAMP concentrations. In addition, cAMP FRET sensors have a more restricted signal-to-noise ratio when compared with many of the alternative approaches for measuring cAMP accumulation.
Effects of including forskolin
In order to detect negative regulation of adenylyl cyclase by Gi-coupled GPCRs, the system must first be active. To achieve this, investigators have normally employed the diterpine forskolin as a direct activator of adenylyl cyclase. The EC50 value for forskolin can vary dependent upon the cell type employed and also upon the type of assay format used (e.g. accumulation vs. reporter gene). It is therefore important to determine the potency of forskolin for the system of choice. For example, the study of adenosine A1 receptor activation on Gi-mediated inhibition of adenylyl cyclase activity utilized 10 µM forskolin for 3H-cyclic AMP accumulation but 3 µM forskolin to demonstrate the same effect in reporter gene assays (Baker and Hill, 2007a). It is important to note, however, that direct activation of adenylyl cyclase is not the only consequence of forskolin administration and there are well documented reports of its effect to enhance the activation of adenylyl cyclase via Gs-coupled receptors (Jasper et al., 1990; Insel and Ostrom, 2003). Thus, forskolin can markedly potentiate the agonist actions of β-blockers such as celiprolol and pindolol at the β2-adrenoceptor on cAMP generation in S49 lymphoma cells (Jasper et al., 1990). Furthermore, the presence of forskolin can also reveal the Gs-stimulatory effects of adenosine A1 receptor stimulation in both cAMP and reporter gene assays (Baker and Hill, 2007a).
Influence of PDE inhibitors
Breakdown of cAMP via PDE enzymes can also impact upon the data generated in assays monitoring the adenylyl cyclase pathway. For example, PDE inhibitors could appear as false positives in a screen for agonists of a Gs-coupled GPCR. To control for such effects, the non-selective PDE inhibitor isobutyl methyl-xanthine (IBMX) can be included in the buffer provided that the receptor under study is not an adenosine receptor. Dependent upon the system this may significantly increase basal levels of cAMP, but may also improve the observed potency of forskolin therefore the impact of its inclusion on the assay sensitivity should be fully evaluated.
A common misconception in studies involving PDE inhibitors in cAMP assays, however, is that cAMP breakdown is no longer occurring. This is clearly not correct, particularly for the majority of PDE inhibitors where their mode of action is by competition with cAMP for the active site of the target PDE isoform (e.g. rolipram; Xu et al., 2004). In this case, the effect of the competitive PDE inhibitor will be to decrease cAMP metabolism and allow the steady-state levels of cAMP (achieved at equilibrium) to rise until the concentration of cAMP is sufficient to compete with the PDE inhibitor for the active site of the enzyme. Under these conditions the rate of cAMP synthesis will once again match its removal by PDE activity (Donaldson et al., 1988a). The increase in steady-state levels of cAMP can be expressed by the following equation:
Where [A]S and [A]'S are the steady-state levels of cAMP in the presence and absence of PDE inhibitor, [I] is the intracellular concentration of PDE inhibitor, and Ki is the Michaelis constant for the inhibitor (Donaldson et al., 1988a). Figure 3 illustrates how cAMP metabolism can be demonstrated to be rapid and extensive when the agonist stimulus is removed in the continued presence of a PDE inhibitor, even when the increases in steady-state cAMP levels are considerable. The impact of the PDE inhibitor, however, can be seen in the slower return to basal levels of cAMP.
Figure 3.
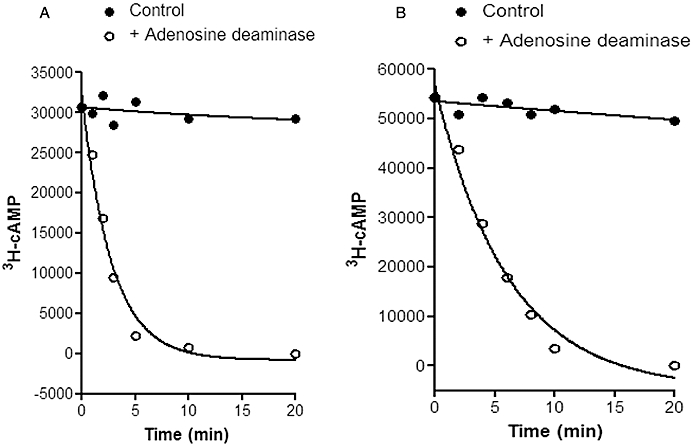
Effect of the phosphodiesterase (PDE) inhibitor rolipram on 3H-cAMP breakdown and steady-state levels of cAMP in 3H-adenine-labelled brain slices from guinea-pig cerebral cortical slices. Data are taken from Donaldson et al. (1988a). In (A) brain slices were pre-incubated with 0.1 mM adenosine for 10 min to achieve a steady-state level. At time zero adenosine deaminase was added (1.2 U·mL−1) to rapidly remove adenosine and cAMP levels then rapidly fell under the influence of endogenous PDEs. In (B) the same experiment was undertaken in the presence of the PDE inhibitor rolipram (0.1 mM). In this case a new and much higher steady-state level of cAMP was achieved. This is because the reduced PDE activity caused by rolipram allows the cAMP levels to rise until they achieve a new equilibrium at which cAMP synthesis is matched again by cAMP metabolism. The continued ability to metabolize cAMP under these conditions is clearly evident when adenosine deaminase is once again applied (B). The influence of the competitive PDE inhibitor rolipram is evident, however, in the reduced rate at which cAMP levels fall in (B). In both (A) and (B) the solid symbols show that the steady-state level of cAMP is well maintained in the absence of adenosine deaminase.
Conclusion
The choice of a method to follow the influence of GPCRs on cAMP accumulation in intact cells needs to be undertaken with care and be tailored to the particular scientific question being addressed. A wide range of techniques are available, but in many cases they are complementary and address different aspects of the Gs-adenylyl cyclase-cAMP pathway. Collectively, they can provide novel insights into GPCR pharmacology and the new opportunities to follow the kinetics of cAMP changes in microdomains of single living cells will almost certainly take our knowledge of this ‘basic’ functional response to new levels.
Acknowledgments
We thank the Medical Research Council for financial support (G0800006). LTM is a NHMRC Postdoctoral Fellow.
Glossary
Abbreviations:
- ATP
adenosine tri-phosphate
- CFP
cyan fluorescent protein
- CRE
cAMP response element
- CREB
CRE-binding protein
- EPAC
exchange protein directly activated by cAMP
- FP
fluorescence polarization
- FRET
fluorescence resonance energy transfer
- GFP
green fluorescent protein
- GPCR
G protein-coupled receptor
- HCN2
hyperpolarization-activated cyclic nucleotide-gated channel 2
- HTS
high-throughput screening
- IBMX
isobutyl methyl-xanthine
- IP3
inositol 1,4,5-trisphosphate
- PDE
phosphodiesterase
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PKA
protein kinase A
- YFP
yellow fluorescent protein
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- Adams SR, Harootunian AT, Buechler YJ, Taylor SS, Tsien RY. Fluorescence ratio imaging of cyclic AMP in single cells. Nature. 1991;349:694–697. doi: 10.1038/349694a0. [DOI] [PubMed] [Google Scholar]
- Allen M, Hall D, Collins B, Moore K. A homogeneous high throughput nonradioactive method for measurement of functional activity of Gs-coupled receptors in membranes. J Biomol Screen. 2002;7:35–44. doi: 10.1177/108705710200700106. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Hochner B, Mahaut-Smith M, Adams SR, Kaang BK, Kandel ER, et al. Spatially resolved dynamics of cAMP and protein kinase A subunits in Aplysia sensory neurons. Science. 1993;260:222–226. doi: 10.1126/science.7682336. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. A comparison of the antagonist affinities for the Gi- and Gs-coupled states of the human adenosine A1-receptor. J Pharmacol Exp Ther. 2007a;320:218–228. doi: 10.1124/jpet.106.113589. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hill SJ. Multiple GPCR conformations and signalling pathways: implications for antagonist affinity estimates. Trends Pharmacol Sci. 2007b;28:374–381. doi: 10.1016/j.tips.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist actions of ‘beta-blockers’ provide evidence for two agonist activation sites or conformations of the human beta1-adrenoceptor. Mol Pharmacol. 2003a;63:1312–1321. doi: 10.1124/mol.63.6.1312. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Agonist and inverse agonist actions of beta-blockers at the human beta 2-adrenoceptor provide evidence for agonist-directed signaling. Mol Pharmacol. 2003b;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Influence of agonist efficacy and receptor phosphorylation on antagonist affinity measurements: differences between second messenger and reporter gene responses. Mol Pharmacol. 2003c;64:679–688. doi: 10.1124/mol.64.3.679. [DOI] [PubMed] [Google Scholar]
- Baker JG, Hall IP, Hill SJ. Temporal characteristics of cAMP response element-mediated gene transcription: requirement for sustained cAMP production. Mol Pharmacol. 2004;65:986–998. doi: 10.1124/mol.65.4.986. [DOI] [PubMed] [Google Scholar]
- Banks P, Gosselin M, Prystay L. Impact of a red-shifted dye label for high throughput fluorescence polarization assays of G protein-coupled receptors. J Biomol Screen. 2000;5:329–334. doi: 10.1177/108705710000500504. [DOI] [PubMed] [Google Scholar]
- Binkowski B, Fan F, Wood K. Engineered luciferases for molecular sensing in living cells. Curr Opin Biotechnol. 2009;20:14–18. doi: 10.1016/j.copbio.2009.02.013. [DOI] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Alexander SP, Kellam B, Hill SJ. Agonist-occupied A3 adenosine receptors exist within heterogeneous complexes in membrane microdomains of individual living cells. FASEB J. 2008;22:850–860. doi: 10.1096/fj.07-8180com. [DOI] [PubMed] [Google Scholar]
- Cordell RL, Hill SJ, Ortori CA, Barrett DA. Quantitative profiling of nucleotides and related phosphate-containing metabolites in cultured mammalian cells by liquid chromatography tandem electrospray mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;871:115–124. doi: 10.1016/j.jchromb.2008.07.005. [DOI] [PubMed] [Google Scholar]
- DiPilato LM, Cheng X, Zhang J. Fluorescent indicators of cAMP and Epac activation reveal differential dynamics of cAMP signaling within discrete subcellular compartments. Proc Natl Acad Sci USA. 2004;101:16513–16518. doi: 10.1073/pnas.0405973101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaldson J, Brown AM, Hill SJ. Influence of rolipram on the cyclic 3′,5′-adenosine monophosphate response to histamine and adenosine in slices of guinea-pig cerebral cortex. Biochem Pharmacol. 1988a;37:715–723. doi: 10.1016/0006-2952(88)90146-3. [DOI] [PubMed] [Google Scholar]
- Donaldson J, Hill SJ, Brown AM. Kinetic studies on the mechanism by which histamine H1 receptors potentiate cyclic AMP accumulation in guinea pig cerebral cortical slices. Mol Pharmacol. 1988b;33:626–633. [PubMed] [Google Scholar]
- Dyachok O, Isakov Y, Sagetorp J, Tengholm A. Oscillations of cyclic AMP in hormone-stimulated insulin-secreting beta-cells. Nature. 2006;439:349–352. doi: 10.1038/nature04410. [DOI] [PubMed] [Google Scholar]
- Fan F, Binkowski BF, Butler BL, Stecha PF, Lewis MK, Wood KV. Novel genetically encoded biosensors using firefly luciferase. ACS Chem Biol. 2008;3:346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- Gabriel D, Vernier M, Pfeifer MJ, Dasen B, Tenaillon L, Bouhelal R. High throughput screening technologies for direct cyclic AMP measurement. Assay Drug Dev Technol. 2003;1:291–303. doi: 10.1089/15406580360545107. [DOI] [PubMed] [Google Scholar]
- George SE, Bungay PJ, Naylor LH. Functional coupling of endogenous serotonin (5-HT1B) and calcitonin (C1a) receptors in CHO cells to a cyclic AMP-responsive luciferase reporter gene. J Neurochem. 1997;69:1278–1285. doi: 10.1046/j.1471-4159.1997.69031278.x. [DOI] [PubMed] [Google Scholar]
- Golla R, Seethala R. A homogeneous enzyme fragment complementation cyclic AMP screen for GPCR agonists. J Biomol Screen. 2002;7:515–525. doi: 10.1177/1087057102238625. [DOI] [PubMed] [Google Scholar]
- Gorbunova YV, Spitzer NC. Dynamic interactions of cyclic AMP transients and spontaneous Ca(2+) spikes. Nature. 2002;418:93–96. doi: 10.1038/nature00835. [DOI] [PubMed] [Google Scholar]
- Gribbon P, Sewing A. Fluorescence readouts in HTS: no gain without pain? Drug Discov Today. 2003;8:1035–1043. doi: 10.1016/s1359-6446(03)02895-2. [DOI] [PubMed] [Google Scholar]
- Guardiola-Diaz HM, Boswell C, Seasholtz AF. The cAMP-responsive element in the corticotropin-releasing hormone gene mediates transcriptional regulation by depolarization. J Biol Chem. 1994;269:14784–14791. [PubMed] [Google Scholar]
- Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, et al. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanoune J, Defer N. Regulation and role of adenylyl cyclase isoforms. Annu Rev Pharmacol Toxicol. 2001;41:145–174. doi: 10.1146/annurev.pharmtox.41.1.145. [DOI] [PubMed] [Google Scholar]
- Hempel CM, Vincent P, Adams SR, Tsien RY, Selverston AI. Spatio-temporal dynamics of cyclic AMP signals in an intact neural circuitm. Nature. 1996;384:166–169. doi: 10.1038/384166a0. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Baker JG, Rees S. Reporter-gene systems for the study of G-protein-coupled receptors. Curr Opin Pharmacol. 2001;1:526–532. doi: 10.1016/s1471-4892(01)00091-1. [DOI] [PubMed] [Google Scholar]
- Huang M, Shimizu H, Daly J. Regulation of adenosine cyclic 3′,5′-phosphate formation in cerebral cortical slices. Interaction among norepinephrine, histamine, serotonin. Mol Pharmacol. 1971;7:155–162. [PubMed] [Google Scholar]
- Insel PA, Ostrom RS. Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol. 2003;23:305–314. doi: 10.1023/a:1023684503883. [DOI] [PubMed] [Google Scholar]
- Jasper JR, Michel MC, Insel PA. Amplification of cyclic AMP generation reveals agonistic effects of certain beta-adrenergic antagonists. Mol Pharmacol. 1990;37:44–49. [PubMed] [Google Scholar]
- Kariv II, Stevens ME, Behrens DL, Oldenburg KR. High throughput quantitation of cAMP production mediated by activation of seven transmembrane domain receptors. J Biomol Screen. 1999;4:27–32. doi: 10.1177/108705719900400105. [DOI] [PubMed] [Google Scholar]
- van der Krogt GN, Ogink J, Ponsioen B, Jalink K. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. Plos ONE. 2008;3:e1916. doi: 10.1371/journal.pone.0001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifert WR, Aloia AL, Bucco O, McMurchie EJ. GPCR-induced dissociation of G-protein subunits in early stage signal transduction. Mol Membr Biol. 2005;22:507–517. doi: 10.1080/09687860500370604. [DOI] [PubMed] [Google Scholar]
- Li X, Zhao X, Fang Y, Jiang X, Duong T, Fan C, et al. Generation of destabilized green fluorescent protein as a transcription reporter. J Biol Chem. 1998;273:34970–34975. doi: 10.1074/jbc.273.52.34970. [DOI] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ. Salmeterol, a long-acting beta 2-adrenoceptor agonist mediating cyclic AMP accumulation in a neuronal cell line. Br J Pharmacol. 1993;110:619–626. doi: 10.1111/j.1476-5381.1993.tb13856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCrea KE, Hill SJ. Comparison of duration of agonist action at beta 1- and beta 2- adrenoceptors in C6 glioma cells: evidence that the long duration of action of salmeterol is specific to the beta 2-adrenoceptor. Mol Pharmacol. 1996;49:927–937. [PubMed] [Google Scholar]
- McDonnell J, Latif ML, Rees ES, Bevan NJ, Hill SJ. Influence of receptor number on the stimulation by salmeterol of gene transcription in CHO-K1 cells transfected with the human beta2-adrenoceptor. Br J Pharmacol. 1998;125:717–726. doi: 10.1038/sj.bjp.0702139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLoughlin DJ, Bertelli F, Williams C. The A, B, C's of G protein coupled receptor pharmacology in assay development for HTS. Expert Opin Drug Discov. 2007;2:1–17. doi: 10.1517/17460441.2.5.603. [DOI] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Middleton RJ, Kellam B. Fluorophore-tagged GPCR ligands. Curr Opin Chem Biol. 2005;9:517–525. doi: 10.1016/j.cbpa.2005.08.016. [DOI] [PubMed] [Google Scholar]
- Minneman KP, Hegstrand LR, Molinoff PB. The pharmacological specificity of beta-1 and beta-2 adrenergic receptors in rat heart and lung in vitro. Mol Pharmacol. 1979;16:21–33. [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95:67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- Montminy MR, Gonzalez GA, Yamamoto KK. Regulation of cAMP-inducible genes by CREB. Trends Neurosci. 1990;13:184–188. doi: 10.1016/0166-2236(90)90045-c. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Hein L, Hannawacker A, Lohse MJ. Novel single chain cAMP sensors for receptor-induced signal propagation. J Biol Chem. 2004;279:37215–37218. doi: 10.1074/jbc.C400302200. [DOI] [PubMed] [Google Scholar]
- Nikolaev VO, Bunemann M, Schmitteckert E, Lohse MJ, Engelhardt S. Cyclic AMP imaging in adult cardiac myocytes reveals far-reaching beta1-adrenergic but locally confined beta2-adrenergic receptor-mediated signaling. Circ Res. 2006;99:1084–1091. doi: 10.1161/01.RES.0000250046.69918.d5. [DOI] [PubMed] [Google Scholar]
- Patel TB, Du Z, Pierre S, Cartin L, Scholich K. Molecular biological approaches to unravel adenylyl cyclase signaling and function. Gene. 2001;269:13–25. doi: 10.1016/s0378-1119(01)00448-6. [DOI] [PubMed] [Google Scholar]
- Ponsioen B, Zhao J, Riedl J, Zwartkruis F, van der Krogt G, Zaccolo M, et al. Detecting cAMP-induced Epac activation by fluorescence resonance energy transfer: Epac as a novel cAMP indicator. EMBO Rep. 2004;5:1176–1180. doi: 10.1038/sj.embor.7400290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittampalam GS, Kahl SD, Janzen WP. High-throughput screening: advances in assay technologies. Curr Opin Chem Biol. 1997;1:384–391. doi: 10.1016/s1367-5931(97)80078-6. [DOI] [PubMed] [Google Scholar]
- Terrin A, Di Benedetto G, Pertegato V, Cheung YF, Baillie G, Lynch MJ, et al. PGE(1) stimulation of HEK293 cells generates multiple contiguous domains with different [cAMP]: role of compartmentalized phosphodiesterases. J Cell Biol. 2006;175:441–451. doi: 10.1083/jcb.200605050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WJ. Cyclic nucleotide phosphodiesterases: pharmacology, biochemistry and function. Pharmacol Ther. 1991;51:13–33. doi: 10.1016/0163-7258(91)90039-o. [DOI] [PubMed] [Google Scholar]
- Thomsen W, Frazer J, Unett D. Functional assays for screening GPCR targets. Curr Opin Biotechnol. 2005;16:655–665. doi: 10.1016/j.copbio.2005.10.008. [DOI] [PubMed] [Google Scholar]
- Williams C. cAMP detection methods in HTS: selecting the best from the rest. Nat Rev Drug Discov. 2004;3:125–135. doi: 10.1038/nrd1306. [DOI] [PubMed] [Google Scholar]
- Williams C, Sewing A. G-protein coupled receptor assays: to measure affinity or efficacy that is the question. Comb Chem High Throughput Screen. 2005;8:285–292. doi: 10.2174/1386207054020778. [DOI] [PubMed] [Google Scholar]
- Xu RX, Rocque WJ, Lambert MH, Vanderwall DE, Luther MA, Nolte RT. Crystal structures of the catalytic domain of phosphodiesterase 4B complexed with AMP, 8-Br-AMP, and rolipram. J Mol Biol. 2004;337:355–365. doi: 10.1016/j.jmb.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Pozzan T. Discrete microdomains with high concentration of cAMP in stimulated rat neonatal cardiac myocytes. Science. 2002;295:1711–1715. doi: 10.1126/science.1069982. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, De Giorgi F, Cho CY, Feng L, Knapp T, Negulescu PA, et al. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat Cell Biol. 2000;2:25–29. doi: 10.1038/71345. [DOI] [PubMed] [Google Scholar]
- Zlokarnik G, Negulescu PA, Knapp TE, Mere L, Burres N, Feng L, et al. Quantitation of transcription and clonal selection of single living cells with beta-lactamase as reporter. Science. 1998;279:84–88. doi: 10.1126/science.279.5347.84. [DOI] [PubMed] [Google Scholar]