

Abstract
Background and purpose:
High level of plasma catecholamines is a risk factor for vascular diseases such as hypertension and atherosclerosis. Catecholamines induce hypertrophy of vascular smooth muscle through α1-adrenoceptors, which in cell culture involves the transactivation of epidermal growth factor receptor (EGFR). We hypothesized that EGFR transactivation was also involved in contractions of rat aorta mediated by α1-adrenoceptors.
Experimental approach:
Thoracic aorta was isolated from 12–14 week old male Wistar rats. In vitro aortic contractile responses to cumulative doses of phenylephrine were characterized in the absence and presence of the EGFR kinase inhibitors, AG1478 and DAPH, in intact and endothelium-denuded rings. Involvement of signal transduction pathways was investigated by using heparin and inhibitors of Src, matrix metalloproteinase (MMP), extracellular signal-regulated kinase (ERK)1/2 and phosphatidyl inositol 3-kinase (PI3K). Phosphorylation of EGFR and ERK1/2 was measured after short-term phenylephrine or EGF stimulation in aorta segments in the presence of AG1478 and the PI3K inhibitor, wortmannin.
Key results:
AG1478 and DAPH concentration dependently attenuated phenylephrine-induced contractile responses in intact or endothelium-denuded aortic rings. Inhibition of PI3K (wortmannin and LY294002) but not heparin or inhibitors of Src or MMP, prevented the effect of AG1478 on the responses to phenylephrine. Phenylephrine induced phosphorylation of EGFR, which was partially blocked by AG1478. Phenylephrine also increased phosphorylation of ERK1/2, time-dependently and was blocked by AG1478 and wortmannin.
Conclusions and implications:
Contractions of rat thoracic aorta mediated by α1-adrenoceptors involved transactivation of EGFR, mediated via a PI3K and ERK1/2 dependent pathway.
Keywords: transactivation, epidermal growth factor receptor, α1-adrenoceptor
Introduction
Prolonged elevation of plasma catecholamines contributes to arterial remodelling in several vascular diseases such as hypertension and atherosclerosis. A growing body of evidence suggests that catecholamines induce hypertrophy of the arterial wall by stimulation of α1-adrenoceptors (Chen et al., 1995; Yu et al., 1996; Xin et al., 1997; receptor nomenclature follows Alexander et al., 2009). In cultured vascular smooth muscle cells (VSMCs), transactivation of epidermal growth factor receptor (EGFR) by α1-adrenoceptors has been implicated as a major pathway involved in catecholamine induced VSMC hypertrophy. In this system, transactivation of EGFR is produced by α1-adrenoceptor evoked shedding of heparin binding EGF-like growth factor (HB-EGF) with subsequent activation of EGFR and mitogen-activated protein kinase (Zhang et al., 2004). Whereas induction of VSMC hypertrophy represents a long-term process, requiring several days of stimulation in cell culture, it is currently unknown whether the acute effects of α1-adrenoceptor stimulation on VSMC, i.e. the induction of contraction, also involve transactivation of EGFR. Should the contractile effect of catecholamines also involves EGFR transactivation, then EGFR antagonists may effectively act as anti-hypertensive agents, in addition to counteracting the hypertrophy of the arterial wall. In support of this hypothesis, chronic treatment with receptor tyrosine kinase inhibitors such as EGFR tyrosine kinase inhibitor AG1478, and EGFR antisense oligonucleotides were found to attenuate the vasoconstriction and the elevation of blood pressure in angiotensin II-induced hypertension (Kagiyama et al., 2002; Kagiyama et al., 2003).
The transactivation of EGFR by seven transmembrane (7TM) receptors other than α1-adrenoceptor has been shown previously and includes endothelin-1 type A receptor (Chansel et al., 2006), angiotensin II type 1 (AT1) (Eguchi et al., 1999), proteinase-activated receptor 1 (Kalmes et al., 2000), β2-adrenoceptors (Maudsley et al., 2000), M1 muscarinic acetylcholine (Tsai et al., 1997) and bradykinin (B2) (Zwick et al., 1997; Mukhin et al., 2003) receptors. Two major mechanisms of EGFR transactivation by 7TM receptors are put forward: (i) matrix metalloproteinase (MMP)-dependent shedding of growth factor like substances, for example HB-EGF (Prenzel et al., 1999); and (ii) the intracellular activation of Src and agonist-independent phosphorylation of EGFR (Luttrell et al., 1997). In turn, downstream of EGFR, important signal transduction routes are phosphorylation of extracellular signal-regulated kinases 1/2 (ERK1/2) and phosphatidylinositol 3-kinase (PI3K) (Schlessinger, 1993; Laffargue et al., 1999; Schlessinger, 2000).
The aim of the present study was to investigate to what extent the contractions of rat aorta VSMCs, induced by α1-adrenoceptor activation, was mediated by EGFR transactivation. To this end, we fully characterized the antagonist properties of AG1478 (a EGFR tyrosine kinase inhibitor) on phenylephrine-induced aortic contraction. As AG1478 blocked α1-adrenoceptor-mediated contractility and phenylephrine stimulation induced EGFR phosphorylation, we also aimed to dissect the signal transduction pathway(s) involved in α1-adrenoceptor dependent transactivation of EGFR.
Methods
Animals
All animal care and experimental procedures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and approved by the Committee for Animal Experiments of the University of Groningen. Experiments were performed on 67 male Wistar (12–14 weeks old, 280–320 g) rats. Animals were housed under standard conditions of temperature (21–24°C), humidity (40–60%) and 12 h light: dark cycle at the animal facilities of the University of Groningen. All animals had free access to food (standard rat chow; Hope Farms, Woerden, the Netherlands) and drinking water throughout the study.
Tissue preparation and contraction studies in rat aortic rings
Under brief anaesthesia with 2.5% isoflurane in O2, the thoracic aorta was quickly removed, cleared of fat and connective tissue and cut into segments (rings of approximately 2 mm). Rings were mounted in a 15 mL organ baths with Krebs solution (pH 7.5) containing (in mmol·L−1): NaCl (120.4), KCl (5.9), CaCl2 (2.5), MgCl2 (1.2), NaH2PO4 (1.2), glucose (11.5), NaHCO3 (25.0) which was kept at 37°C and continuously bubbled with 95% O2 and 5% CO2. Prior to isotonic measurements of vascular contractility, arteries were allowed to equilibrate for 40 min. To test for viability of smooth muscle cells, arteries were twice pre-constricted with KCl (60 mM). After washout, full characterization of the antagonist properties of AG1478 on aorta contractions mediated by phenylephrine was studied in endothelium-intact and -denuded rings. Endothelium denudation was provided by gentle removal of the intimal surface with a paper clip and confirmed by the absence of a relaxant response to acetylcholine (30 µM) following a submaximal pre-constriction with KCl (40 mM). After washout and another 30 min of stabilization, endothelium-intact and -denuded rings were incubated either with four different concentrations of AG1478 (2.5–20 µM) or dimethyl sulphoxide (DMSO, 0.5% final concentration; vehicle) for 20 min. Subsequently, aorta contractility was measured as the contractile response to cumulative concentrations of phenylephrine (1 nM–10 µM). Finally, KCl (60 mM) was added to the organ baths.
In additional rings, the effect of another EGFR tyrosine kinase inhibitor, 4,5-dianilinophthalimide, 5,6-bis(phenylamino)-1H-isoindole-1,3(2H)-dione (DAPH; 1–10 µM) was studied. To further dissect the signalling pathway(s) involved, the effects of heparin (an HB-EGF blocker, 10 IU·mL−1), PP1 (a Src-selective tyrosine kinase inhibitor; 10 µM), GM 6001 (a broad spectrum MMP inhibitor; 10 µM), ML5 (a broad spectrum MMP inhibitor; 10 µM), PD98059 (a specific inhibitor of ERK1/2; 10 µM), wortmannin (a PI3K inhibitor; 1 µM) and LY294002 (a specific inhibitor of PI3K; 10 µM), were also investigated alone or in combination with AG1478 (5 µM) in endothelium-intact rings. Each experimental condition was studied in duplicate rings.
Aortic segment stimulation, homogenization and immunoblotting
Endothelium-denuded segments of thoracic aorta (approximately 3 cm) isolated from 20 different rats were cut into six rings and for each experimental condition, four rings were randomly put into a tube containing 1 mL of Krebs solution, then stimulated either by EGF (1 nM) or phenylephrine (1 µM) in the absence or presence of inhibitors. After stimulation, Krebs solution was removed from the tube and rings were snap-frozen in liquid nitrogen and stored at –80°C until use. For immunoblotting, the aorta segments were homogenized in 500 µL of ice-cold homogenization buffer (1% Igepal ca-630, 1% SDS, 5 mg·mL−1 sodium deoxycholate, 1 mM sodium orthovanadate, 10 mM β-mercapto-ethanol, 40 µg·mL−1 phenylmethylsulphonyl fluoride, 100 µg·mL−1 benzamidine, 500 ng·mL−1 pepstatin A, 500 ng·mL−1 leupeptine and 500 ng·mL−1 aprotinin in PBS) with Dispomix Homogenization (L&M Biotech, Cary, NC, USA). The homogenate was clarified by centrifugation at 1000 g for 10 min at 4°C, the supernatant was collected and protein concentration was determined using Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA).
Whole aortic extracts were boiled in sample buffer for 3 min, separated by using 4–10% SDS-PAGE precise protein gel (Thermo Fisher Scientific Inc., Rockford, IL, USA) electrophoresis, and then transferred electrically onto nitrocellulose membranes. Membranes were incubated overnight with the anti-ERK1/2 (1:1000 dilution) antibody (Cell Signaling Technology, Inc., Danvers, MA, USA) or phosphospecific anti-ERK1/2 (1:1000 dilution) antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) or phosphospecific anti-EGFR (1:400 dilution) antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) at 4°C. The next day, membranes were washed with Tris-buffered saline Tween-20 (0.05 M Tris, 150 mM NaCl and 0.04% Tween-20) and re-incubated with the secondary antibodies for 2 h at room temperature. Anti-glyceraldehyde-3-phosphate dehydrogenase (1:10000 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) and anti-β-actin (1:2000 dilution; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) antibodies were used to confirm equal loading conditions. Immunoreactive bands were visualized by using SuperSignal West Pico Chemiluminescent kit (Thermo Fisher Scientific Inc., Rockford, IL, USA) and GeneGnome System (Westburg BV, Leusden, the Netherlands).
Statistical analysis
Data are expressed as mean ± SEM. SPSS 16.0 for Windows (SPSS Inc., Chicago, Illinois, USA) software was used for statistical analysis. Concentration-response curves from aortic rings were compared by anova for repeated measures followed by Bonferroni's post hoc test for multiple comparisons. Differences were considered significant at P < 0.05 (two-tailed). The rightward shifts in the phenylephrine concentration-response curves in the presence of inhibitors were evaluated by comparing pEC50 values (pEC50 = negative logarithm of the EC50) with vehicle curves by one-way anova followed by Dunnett's post hoc test. Maximal responses (Emax) induced by phenylephrine were compared with vehicle by one-way anova followed by Dunnett's post hoc test. Schild analysis was used to investigate the EGFR antagonism and calculated by plotting the log (dose ratio-1) against the log of the molar concentration of AG1478 or DAPH for individual rats (GraphPad Software, CA, USA). Schild slopes were tested statistically for a significant difference from unity by using Z-score (GraphPad Prism Software, CA, USA). Semi-quantitative comparison of phosphorylated epidermal growth factor receptor (pEGFR), and phosphorylated extracellular signal-regulated kinases 1/2 (pERK1/2) bands was carried out with the Mann–Whitney U-test.
Materials
All compounds for Krebs solution and all other molecules were purchased from Sigma-Aldrich (Sigma-Aldrich Chemie BV, Zwijndrecht, the Netherlands) except PP1 (BIOMOL International, PA, USA), and GM 6001 (Calbiochem, Darmstadt, Germany). ML5 was kindly provided by Prof Dr Rainer Bischoff (Department of Analytical Biochemistry, Centre of Pharmacy, University of Groningen, Groningen, the Netherlands).
Results
Antagonism of phenylephrine-induced contraction by AG1478 and DAPH
To characterize the involvement of EGFR in α1-adrenoceptor mediated contraction, full concentration-response curves of phenylephrine were obtained in the presence of different concentrations of AG1478 (2.5–20 µM). In the presence of AG1478, the concentration-response curve to phenylephrine shifted to the right in a concentration-dependent manner both in endothelium-intact and -denuded rings (Figure 1A,C, Table 1). In addition, the two highest concentrations of AG1478 (10 and 20 µM) caused a decrease of the Emax induced by phenylephrine (Table 1, P < 0.05). Schild analysis was used to investigate the antagonism of AG1478 and similar plots were obtained from endothelium-intact and -denuded rings (Figure 1B,D). Clearly, slopes of Schild plots differed significantly from unity, precluding simple competitive antagonism on α1-adrenoceptors as the mode of action of AG1478. These findings demonstrated that the antagonist action of AG1478 on phenylephrine-evoked contractions was endothelium-independent.
Figure 1.
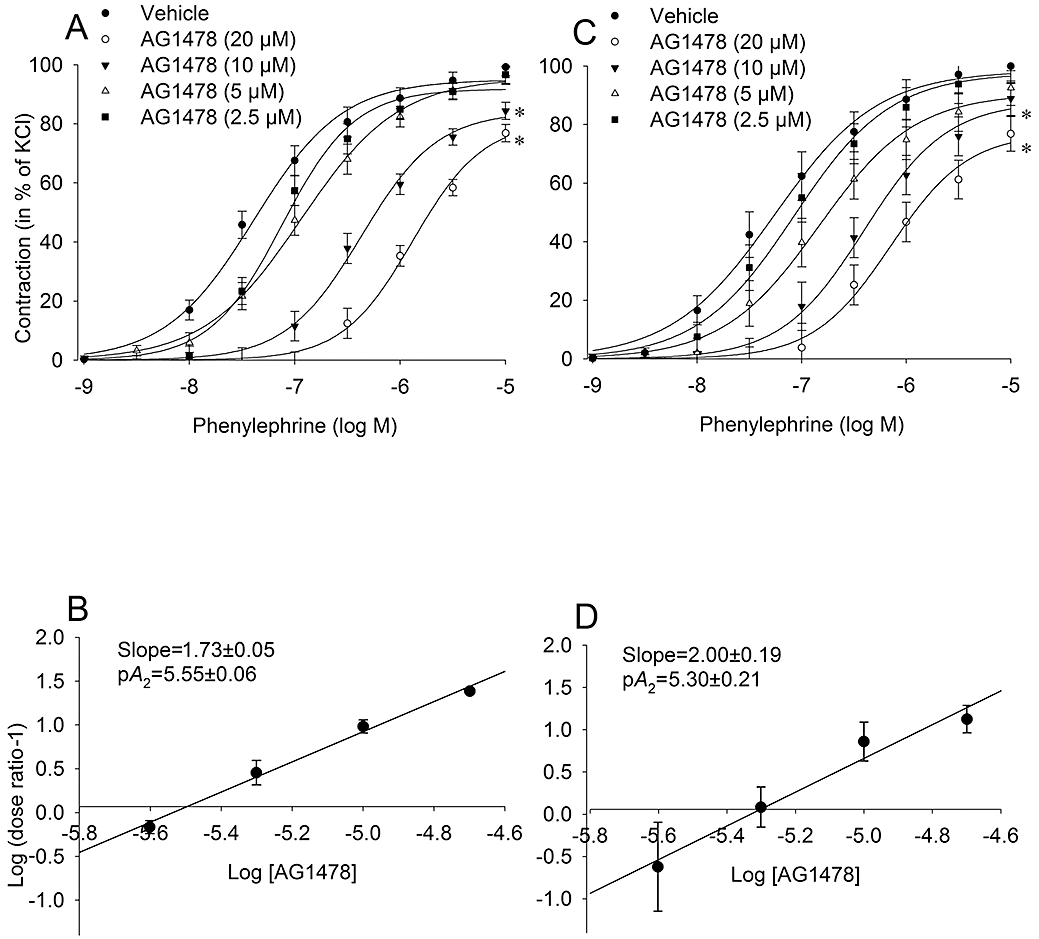
Characterization of the inhibitory effect of the EGFR tyrosine kinase inhibitor, AG1478, on α1-adrenoceptor mediated contraction in rat thoracic aortic rings with intact (A, n = 6) and denuded endothelium (C, n = 6). Rings were pre-incubated with indicated concentrations of AG1478 (20 min) or vehicle (DMSO, 0.5% final concentration), prior to construction of cumulative concentration-response curves. Absolute tension values (in mN) after the highest dose of phenylephrine (10 µM) in the rings with intact endothelium were as follows; vehicle: 333 ± 38, AG1478 (20 µM): 165 ± 26, AG1478 (10 µM): 258 ± 38, AG1478 (5 µM): 363 ± 62, and AG1478 (2.5 µM): 386 ± 50. In the endothelium-denuded rings absolute tension values (in mN) were as follows; vehicle: 408 ± 52, AG1478 (20 µM): 177 ± 36, AG1478 (10 µM): 254 ± 25, AG1478 (5 µM): 398 ± 57, and AG1478 (2.5 µM): 475 ± 58. Schild analysis was used to investigate the EGFR antagonism and calculated by plotting the log (dose ratio-1) against the log of the molar concentration of AG1478 (B, D). The slope was calculated both in endothelium-intact and -denuded rings and found to be significantly larger than unity. Data are expressed as mean ± SEM *P < 0.05 versus vehicle curves (repeated measures anova). EGFR, epidermal growth factor receptor.
Table 1.
The effect of AG1478 and DAPH on phenylephrine concentration-response curves
| Vehicle | AG1478 (2.5 µM) | AG1478 (5 µM) | AG1478 (10 µM) | AG1478 (20 µM) | |
|---|---|---|---|---|---|
| Endothelium-intact rings | |||||
| Emax (%) | 99.2 ± 3.1 | 96.6 ± 0.4 | 96.3 ± 2.3 | 84.4 ± 3.7a | 76.8 ± 3.6a |
| pEC50 | 7.39 ± 0.05 | 7.16 ± 0.06 | 6.79 ± 0.12a | 6.36 ± 0.08a | 5.99 ± 0.04a |
| Endothelium-denuded rings | |||||
| Emax (%) | 99.1 ± 5.1 | 99.8 ± 2.6 | 92.5 ± 5.1 | 89.0 ± 5.1a | 76.9 ± 9.5a |
| pEC50 | 7.43 ± 0.16 | 7.15 ± 0.13 | 7.01 ± 0.15 | 6.48 ± 0.15a | 6.26 ± 0.06a |
| Vehicle | DAPH (1 µM) | DAPH (5 µM) | DAPH (10 µM) | ||
| Endothelium-denuded rings | |||||
| Emax (%) | 93.1 ± 2.3 | 77.8 ± 13.9 | 70.3 ± 12.5 | 46.8 ± 11.2a | |
| pEC50 | 7.01 ± 0.25 | 6.86 ± 0.25 | 6.60 ± 0.20 | 6.20 ± 0.16a | |
Data are given as mean ± SEM.
P < 0.05 versus vehicle.
DAPH, 4,5-dianilinophthalimide, 5,6-bis(phenylamino)-1H-isoindole-1,3(2H)-dione; Emax, maximal contraction response in % of KCl; pEC50, negative logarithm of the EC50.
To substantiate that the antagonistic effect of AG1478 was dependent on its blockade of EGFR, the experiment was repeated using a structurally unrelated inhibitor of EGFR phosphorylation, DAPH. Pretreatment of endothelium-denuded rings with the highest concentration of DAPH (10 µM) also shifted the concentration-response curve to phenylephrine to the right (Figure 2A, Table 1), as did AG1478. Moreover, the highest concentration of DAPH (10 µM) also caused a significant attenuation of Emax (Table 1, P < 0.05). Albeit in small number of animals, the slope of the Schild plot also differed significantly from unity (Figure 2B, P < 0.05). Thus, these data collectively showed that blockade of EGFR attenuated α1-adrenoceptor mediated contraction.
Figure 2.
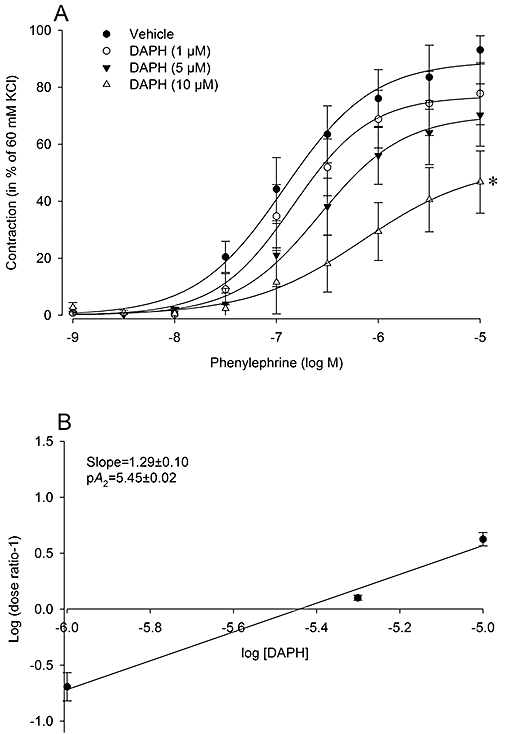
Characterization of the inhibitory effect of the EGFR tyrosine kinase inhibitor, DAPH, on α1-adrenoceptor mediated contraction in endothelium-denuded rat thoracic aortic rings (A). Rings were pre-incubated with indicated concentrations of DAPH (20 min) or vehicle (DMSO), prior to construction of cumulative concentration-response curves. Absolute tension values (in mN) after the highest dose of phenylephrine (10 µM) were as follows; vehicle: 349 ± 29, DAPH (10 µM): 161 ± 54, DAPH (5 µM): 201 ± 51, DAPH (1 µM): 229 ± 50. Schild analysis was used to investigate the EGFR antagonism and calculated by plotting the log (dose ratio-1) against the log of the molar concentration of DAPH (B). The slope value was calculated and found to be significantly larger than unity. Data are expressed as mean ± SEM n = 3 rats. *P < 0.05 versus vehicle curve (repeated measures anova). DAPH, 4,5-dianilinophthalimide, 5,6-bis(phenylamino)-1H-isoindole-1,3(2H)-dione; EGFR, epidermal growth factor receptor.
Dissection of the pathways involved in antagonistic effect of AG1478
In order to dissect the involvement of different signalling pathways in the antagonistic effect of AG1478 on α1-adrenoceptor mediated aorta contraction, we studied whether the action of AG1478 was influenced by heparin (to block HB-EGF) and inhibitors of MMP, Src, ERK1/2 and PI3K.
The characteristics of the inhibitors on phenylephrine concentration-response curves are presented in Figure 3 and Table 2. Heparin pretreatment (10 IU·mL−1) did not alter concentration-response curve to phenylephrine, whereas its combination with AG1478 (5 µM) caused a further attenuation of Emax induced by phenylephrine (Figure 3A). Broad spectrum MMP inhibitors GM 6001 and ML5 (10 µM; Figure 3B,C) did not have any effect on phenylephrine induced contractions either in the absence or presence of AG1478. PD98059 (10 µM; Figure 3D), an inhibitor of ERK1/2, slightly (non-significantly) right-shifted the concentration-response curve to phenylephrine in vehicle incubated rings, whereas it had no additional effect on AG1478 incubated rings. Similar to GM6001 and ML5, the Src inhibitor PP1 (10 µM; Figure 3E) did not have any effect on phenylephrine induced contractions either in absence or presence of AG1478. However, the PI3K inhibitors, wortmannin (1 µM; Figure 3F) and LY294002 (10 µM; Figure 3G) suppressed its Emax (P < 0.05). In contrast to vehicle incubated rings, AG1478 did not affect the phenylephrine curve in rings pretreated either with wortmannin or LY294002.
Figure 3.
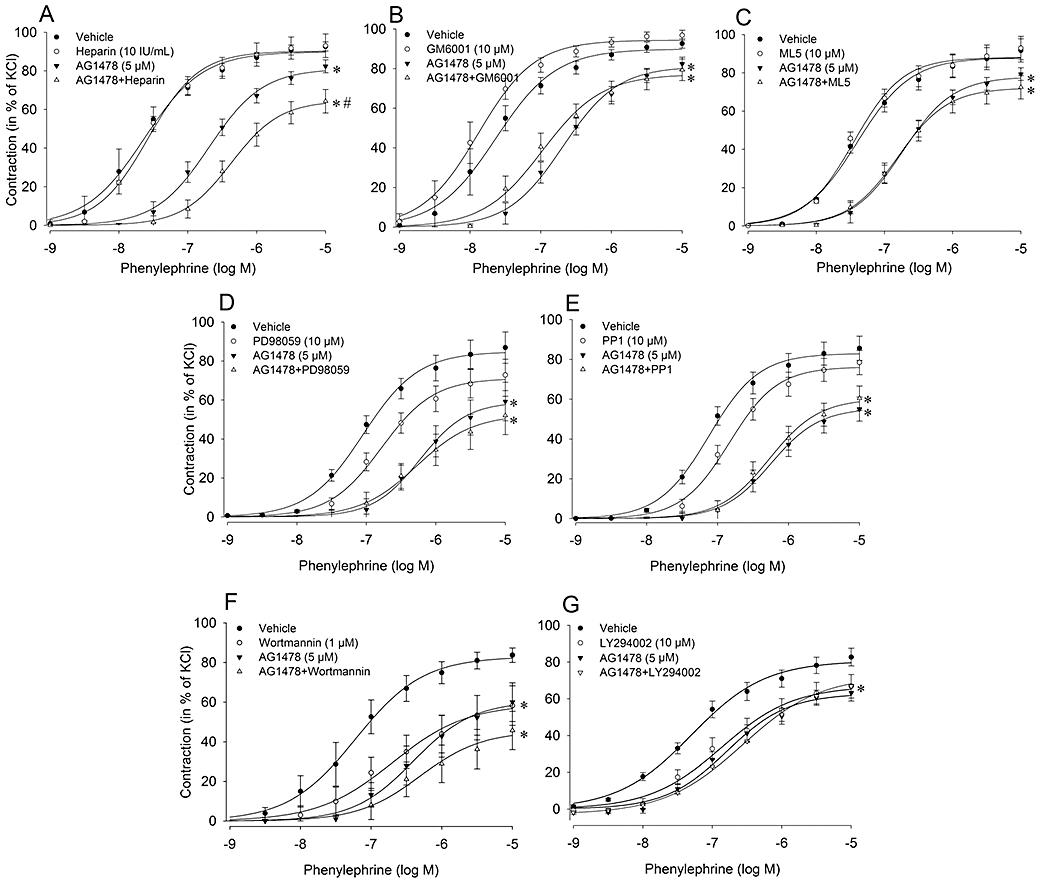
Characterization of signalling pathways involved in α1-adrenoceptor mediated transactivation of EGFR in rat thoracic aortic rings with intact endothelium. Concentration-response curves to phenylephrine were constructed in the absence and the presence of heparin (A, n = 6), broad spectrum MMP inhibitors; GM 6001 (B, n = 6) and ML5 (C, n = 4), ERK inhibitor; PD98059 (D, n = 6), Src inhibitor; PP1 (E, n = 5), and PI3K inhibitors; wortmannin (F, n = 6) and LY294002 (G, n = 3). Data are expressed as mean ± SEM *P < 0.05 versus DMSO (Vehicle control), #P < 0.05 versus AG1478 (repeated measures anova). EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinases; MMP, matrix metalloproteinase.
Table 2.
The effect of inhibitors used on phenylephrine concentration-response curves in endothelium intact rings
| Vehicle | AG1478 | Inhibitor | AG1478+ inhibitor | |
|---|---|---|---|---|
| Heparin | ||||
| Emax (%) | 91.2 ± 2.8 | 76.8 ± 5.2a | 93.0 ± 2.6 | 64.2 ± 5.7a |
| pEC50 | 7.61 ± 0.14 | 6.64 ± 0.09a | 7.62 ± 0.12 | 6.40 ± 0.03a |
| GM6001 | ||||
| Emax (%) | 94.1 ± 1.7 | 85.1 ± 1.7a | 96.8 ± 1.8 | 79.3 ± 3.8a |
| pEC50 | 7.79 ± 0.15 | 6.92 ± 0.06a | 7.93 ± 0.12 | 7.02 ± 0.10a |
| ML5 | ||||
| Emax (%) | 91.7 ± 6.1 | 79.3 ± 3.8a | 92.9 ± 1.7 | 72.5 ± 1.6a |
| pEC50 | 7.41 ± 0.06 | 6.64 ± 0.09a | 7.48 ± 0.06 | 6.80 ± 0.12a |
| PD98059 | ||||
| Emax (%) | 88.4 ± 1.2 | 54.7 ± 10.6a | 69.8 ± 7.8 | 52.0 ± 9.2a |
| pEC50 | 7.06 ± 0.09 | 6.28 ± 0.06a | 6.80 ± 0.06 | 6.28 ± 0.10a |
| PP1 | ||||
| Emax (%) | 85.6 ± 2.9 | 55.0 ± 6.8a | 78.4 ± 5.3 | 60.5 ± 8.1a |
| pEC50 | 7.17 ± 0.10 | 6.36 ± 0.14a | 6.85 ± 0.11 | 6.30 ± 0.10a |
| Wortmannin | ||||
| Emax (%) | 85.6 ± 3.1 | 59.6 ± 8.3a | 58.3 ± 7.0a | 42.7 ± 6.6a |
| pEC50 | 7.26 ± 0.20 | 6.38 ± 0.15a | 6.83 ± 0.13 | 6.41 ± 0.17a |
| LY294002 | ||||
| Emax (%) | 82.7 ± 2.5 | 63.2 ± 7.3a | 66.8 ± 0.9a | 66.9 ± 3.1a |
| pEC50 | 7.25 ± 0.10 | 6.85 ± 0.07a | 7.00 ± 0.10 | 6.63 ± 0.08a |
Data are given as mean ± SEM.
P < 0.01 versus vehicle.
Emax, maximal contraction response in % of KCl; pEC50, negative logarithm of the EC50.
Phosphorylation of EGFR by phenylephrine
To further substantiate EGFR transactivation by α1-adrenoceptor agonists, we measured phosphorylation of EGFR after stimulation with phenylephrine (1 µM; 5 and 10 min) or EGF (1 nM; 10 min) in the absence and in the presence of AG1478 (10 µM) in endothelium-denuded aorta segments (Figure 4). Phenylephrine and EGF stimulation evoked an increase in the pEGFR signal, which was partially blocked by AG1478, confirming α1-adrenoceptor agonist-mediated EGFR transactivation in the rat thoracic aorta.
Figure 4.
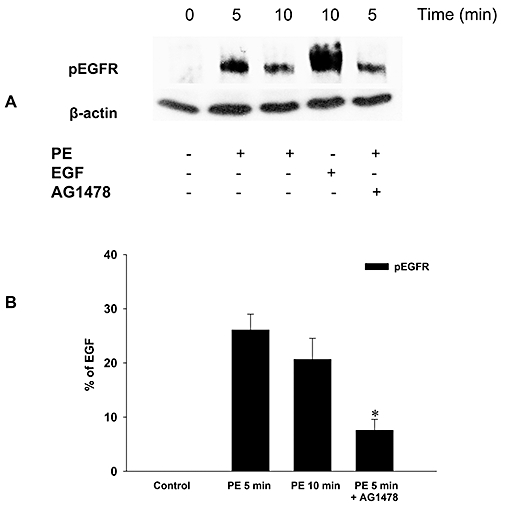
Transactivation of EGFR after stimulation of α1-adrenoceptor in endothelium-denuded rat thoracic aorta segments. Phosphorylation of EGFR (pEGFR) after phenylephrine (PE; 5 and 10 min) or EGF (10 min) stimulation in the absence and in the presence of EGFR tyrosine kinase inhibitor AG1478 (20 min pre-incubation) was determined by immunoblotting (A). The results from all of the experiments shown are representative of four independent experiments in isolated endothelium-denuded thoracic aorta segments from different rats. pEGFR was normalized to β-actin, quantified as the percentage of EGF (10 min)-induced pEGFR and expressed as mean ± SEM (B). *P < 0.05 versus phenylephrine alone at 5 min (Mann–Whitney U-test). pEGFR, phosphorylated epidermal growth factor receptor.
Effects of AG1478 on α1-adrenoceptor mediated phosphorylation of ERK1/2
Because inhibition of ERK1/2 activation was reported to attenuate contraction evoked by phenylephrine (Dessy et al., 1998) and in our study ERK1/2 inhibition slightly shifted the concentration-response curve to phenylephrine, pERK1/2 activity after EGF (1 nM; positive control) and phenylephrine (1 µM) stimulation was also measured in endothelium-denuded aorta segments. The EGF induced increase in pERK1/2 signal was blocked by AG1478 (10 µM; Figure 5A,B) and to a lesser extent by wortmannin (1 µM; Figure 5C). Phenylephrine also increased pERK1/2, which was time dependent with a maximum at 10 min and blocked by AG1478 (Figure 5A,B) and wortmannin (Figure 5C) in a similar manner. Collectively, these data support our findings in organ bath experiments that the transactivation of EGFR in rat aorta, mediated by α1-adrenoceptor agonists, involves mainly activation of PI3K. In addition, ERK1/2 activation seems to be partially involved, as indicated by the inhibitory effect of AG1478 and wortmannin on the phenylephrine-evoked increase in pERK1/2.
Figure 5.
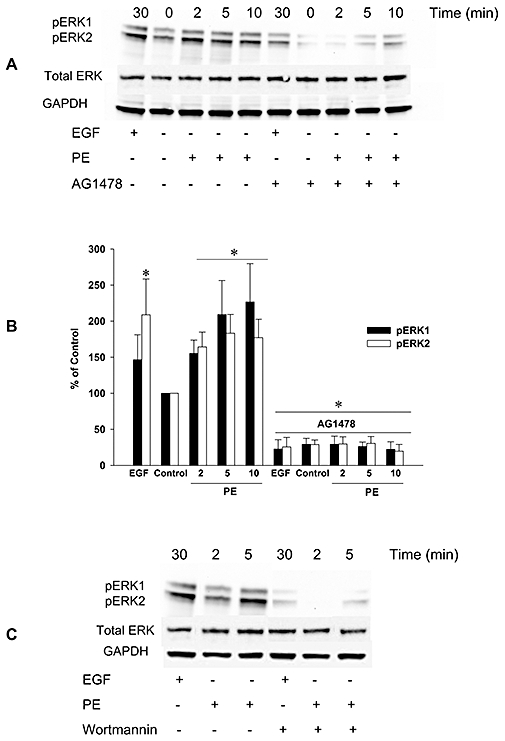
Involvement of EGFR in α1-adrenoceptor mediated stimulation of phosphorylation of ERK1/2 in endothelium-denuded rat thoracic aorta segments. Phosphorylation of ERK1/2 (pERK1, pERK2) after stimulation with epidermal growth factor (EGF; 30 min) or phenylephrine (PE; 2, 5, and 10 min) was determined by immunoblotting (A). The results from all of the experiments shown are representative of four independent experiments in isolated endothelium-denuded thoracic aorta segments from different rats. Phosphorylation of ERK1/2 was normalized to GAPDH, quantified as the percentage of control (PE, 0 min) and expressed as mean ± SEM (B). *P < 0.05 versus Control (Mann–Whitney U-test). EGFR tyrosine kinase inhibitor, AG1478 (A) and PI3K inhibitor, wortmannin (C) blocked the phosphorylation of ERK1/2 after stimulation with EGF or phenylephrine. EGFR, epidermal growth factor receptor; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; pERK, phosphorylated extracellular signal-regulated kinases.
Discussion and conclusions
This study shows that transactivation of the EGFR mediated, in part, the contraction of isolated rat thoracic aorta rings induced by α1-adrenoceptor stimulation, as it was blocked by different EGFR tyrosine kinase inhibitors. Stimulation of α1-adrenoceptors also induced an acute, time-dependent increase in the phosphorylation of EGFR and ERK1/2, both of which were blocked by AG1478 and wortmannin. As a range of blockers of pathways implicated in EGFR transactivation by 7TM receptors did not affect α1-adrenoceptor mediated contraction, our data provide strong evidence that α1-adrenoceptor mediated transactivation of EGFR is mainly dependent on PI3K activation. Our study extends evidence of EGFR transactivation governing the response to long-term α1-adrenoceptor stimulation in cultured cells (Chen et al., 1995; Yu et al., 1996; Bleeke et al., 2004) by showing that transactivation of EGFR is also involved in mediating the acute effects of α1-adrenoceptor stimulation on VSMC. To our knowledge this is the first study reporting on the role of EGFR transactivation in α1-adrenoceptor mediated aorta contractility.
The EGFR tyrosine kinase inhibitors, AG1478 and DAPH, inhibited Emax of the α1-adrenoceptor mediated contractions at the higher concentrations. Moreover, AG1478 also blocked α1-adrenoceptor induced phosphorylation of EGFR. Collectively, these data exclude the simple explanation of the antagonist action of AG1478 and DAPH to be a straightforward competitive antagonism at the α1-adrenoceptor. Interactions between both receptor types involved may offer other possible explanations, such as allosteric antagonism by the non-phosphorylated EGFR, or positive cooperativity of the activated EGFR or components of its downstream signalling route on the α1-adrenoceptors. However, testing these hypotheses will require various forms of tagged receptors and an adequate model system expressing the receptor types. Thus, blockade of the transactivation of EGFR is the most obvious explanation for the antagonist action of AG1478 and DAPH on α1-adrenoceptor mediated contraction. To the best of our knowledge this is the first report on Schild analysis of the inhibitory action of AG1478 or DAPH on 7TM receptor mediated responses.
In recent years, a link between vascular pathologies such as hypertension-associated oxidative stress, cardiovascular hypertrophy, vascular remodelling and the actions of elevated levels of agonists for various vasoconstrictor 7TM receptors has been shown (Asakura et al., 2002; Fortuno et al., 2005; Fernandez-Patron, 2007). The most striking of this link is the involvement of transactivation of the EGFR by many different agonists of 7TM receptors (Fernandez-Patron, 2007). In this way, in the absence of direct growth factor signalling, the EGFR can still be activated in a wide array of biological signalling processes, which is generally known as ‘transactivation’ (Schafer et al., 2004; Ohtsu et al., 2006). Vascular pathology is typically brought about by multiple 7TM receptor agonists. Therefore, instead of targeting several 7TM receptors with combination treatment, it is conceivable that blockade of EGFR transactivation may have a significant potential in vascular disorders, particularly in hypertension.
Whereas EGFR transactivation is known to have a very rapid kinetics (Daub et al., 1997), its mechanism has not completely been explored yet. Both ligand-independent and nonclassical ligand-dependent pathways have been put forward to explain the transactivation of the EGFR (Zwick et al., 1999). The ligand-independent mechanism (also called the intracellular mechanism) hypothesizes that EGFR transactivation is exclusively mediated through intracellular signals. These signals may be cytoplasmic kinases and second messengers such as Src, Pyk2, protein kinase C, PI3K, reactive oxygen species (ROS) or Ca2+ (Dikic et al., 1996; Luttrell et al., 1997; Tsai et al., 1997; Zwick et al., 1997; Zwick et al., 1999; Zhang et al., 2004). Unfortunately, so far none of the ligand-independent pathways could fully account for transactivation of EGFR. As a result, the non-classical extracellular ligand-dependent pathway was put forward to explain EGFR transactivation. Prenzel et al. (1999) reported that 7TM receptors activate the MMP-mediated, extracellular shedding of HB-EGF, which binds to the ectodomain of the EGFR and activates its intracellular mitogenic signals. Their findings were further corroborated by studies in which MMP mediated shedding of HB-EGF was linked to the generation of NADPH oxidase–dependent ROS (Bleeke et al., 2004) and EGFR-dependent ERK1/2 activation (Zhang et al., 2004). Because of these multiple mechanisms involved, 7TM receptor mediated EGFR transactivation is most likely to vary with the cell and receptor types involved.
Our data on vascular contractility obtained by using compounds that block the implicated pathways of α1-adrenoceptor mediated EGFR transactivation suggest that transactivation of EGFR in rat aorta is mainly dependent on activation of PI3K and partially dependent on ERK1/2 activation. In addition, α1-adrenoceptor induced phosphorylation of ERK1/2 was significantly decreased by AG1478 and wortmannin. Finally, this is the first study in which increased phosphorylation of EGFR is demonstrated in α1-adrenoceptor stimulated intact vascular tissue. In accord with our findings it was shown in a previous study that α1-adrenoceptor stimulation induces a transient ERK1/2 activation, which was attenuated by the ERK1/2 inhibitor U0126 in rat tail artery (Tsai and Jiang, 2005). Moreover, α1-adrenoceptor induced constriction of isolated rat mesenteric artery was reported to be inhibited by EGFR inhibitors AG1478, and PD153035 (Hao et al., 2004), while it was counteracted by wortmannin infusion, also suggesting a PI3K-dependent mechanism. These findings have been further supported by a very recent study in which it was shown that the maintenance of adrenergic vascular tone by the MMP-EGFR pathway requires PI3K activation (Nagareddy et al., 2009). The main difference between our findings and the latter study is that Nagareddy et al. reported a MMP-dependent transactivation of the EGFR, whereas in our study we demonstrated the lack of involvement of MMPs in α1-adrenoceptor-mediated EGFR transactivation in rat thoracic aorta. However, in contrast to our findings in rat thoracic aorta which suggest an intracellular EGFR transactivation pathway, the pan-MMP inhibitor GM6001 has been shown to block EGFR transactivation (Hao et al., 2004), which points to an extracellular EGFR transactivation mechanism in the mesenteric vascular bed. Therefore, these findings collectively suggest that the mechanism of EGFR transactivation by α1-adrenoceptors also depends on the vascular bed and possibly on α1-adrenoceptor subtypes involved.
The specificity of the inhibitory effects of AG1478 and DAPH on EGFR kinase may be questioned. To date, AG1478 is known to be a very potent and selective inhibitor of EGFR kinase with IC50 of 3 nM (Levitzki and Gazit, 1995). Similar to AG1478, DAPH is also known to be a potent and selective inhibitor of EGFR kinase with IC50 of 1 µM (Buchdunger et al., 1994). Although AG1478 and DAPH are widely accepted as specific EGFR kinase inhibitors, the specificity of wortmannin at the concentration used in this study may also be questioned, as wortmannin was originally reported to be an inhibitor of smooth muscle myosin light chain kinase (Nakanishi et al., 1992). Therefore, in addition to wortmannin we also obtained phenylephrine dose-response curves in endothelium-denuded aortic rings by using another specific PI3K inhibitor, LY294002. Our data employing LY294002 were identical to those obtained with wortmannin. Furthermore, phenylephrine-induced phosphorylation of ERK1/2 was inhibited by wortmannin, which offered additional support to the involvement of the PI3K dependent pathway in EGFR transactivation following α1-adrenoceptor stimulation.
By acknowledging the importance of EGFR transactivation in α1-adrenoceptor-induced vascular contractility, a connection between hypertension and EGFR signalling may be postulated. In fact, a number of studies provide strong clues to this possibility, as both an enhanced expression level of EGFR and a significant correlation between maximal aortic EGF binding capacity and blood pressure was shown in experimental (Northcott et al., 2001) and genetic (Swaminathan et al., 1996) models of hypertension. Subsequent reports in another model of spontaneous hypertension (SHR rat) substantiated these findings by demonstrating EGF to act as a potent vasoconstrictor of arteries (Florian and Watts, 1999) and by increased EGFR expression levels in the hypertrophied left ventricle (Fujino et al., 1998). Furthermore, chronic treatment with AG1478, and EGFR antisense oligonucleotides successfully prevented angiotensin II-induced hypertension and attenuated the associated vasoconstriction (Kagiyama et al., 2002; Kagiyama et al., 2003). Nevertheless, the mechanism of the development and progression of hypertension and EGFR transactivation remains unclear. However, because MMP inhibitors successfully prevented cardiac remodelling induced by chronic infusion of angiotensin II and phenylephrine, 7TM receptor-induced HB-EGF shedding and subsequent EGFR transactivation seems likely to play an important role in hypertension (Asakura et al., 2002).
In conclusion, EGFR transactivation significantly contributes to α1-adrenoceptor mediated contractile responses in rat aorta, most likely via activation of the PI3K and ERK1/2 pathways, and independent from the MMP-HB-EGF pathway. Interruption of EGFR transactivation by targeting dissected key components may provide a novel approach for the development of new classes of drugs in the treatment of vascular pathologies, particularly of hypertension.
Acknowledgments
This study was supported by a research grant from The Scientific and Technological Research Council of Turkey (SBAG-109S125).
Glossary
Abbreviations:
- AG1478
N-(3-Chlorophenyl)-6,7-dimethoxy-4-quinazolinamine
- DAPH
4,5-dianilinophthalimide, 5,6-bis(phenylamino)-1H-isoindole-1,3(2H)-dione
- EGFR
epidermal growth factor receptor
- ERK1/2
extracellular signal-regulated kinases 1/2
- HB-EGF
heparin binding EGF-like growth factor
- MMP
matrix metalloproteinase
- pEGFR
phosphorylated epidermal growth factor receptor
- pERK1/2
phosphorylated extracellular signal-regulated kinases 1/2
- PI3K
phosphatidylinositol 3-kinase
- ROS
reactive oxygen species
- VSMC
vascular smooth muscle cell
Conflicts of interest
None.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, et al. Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat Med. 2002;8:35–40. doi: 10.1038/nm0102-35. [DOI] [PubMed] [Google Scholar]
- Bleeke T, Zhang H, Madamanchi N, Patterson C, Faber JE. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ Res. 2004;94:37–45. doi: 10.1161/01.RES.0000109412.80157.7D. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, Trinks U, Mett H, Regenass U, Muller M, Meyer T, et al. 4,5-Dianilinophthalimide: a protein-tyrosine kinase inhibitor with selectivity for the epidermal growth factor receptor signal transduction pathway and potent in vivo antitumor activity. Proc Natl Acad Sci U S A. 1994;91:2334–2338. doi: 10.1073/pnas.91.6.2334. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chansel D, Ciroldi M, Vandermeersch S, Jackson LF, Gomez AM, Henrion D, et al. Heparin binding EGF is necessary for vasospastic response to endothelin. FASEB J. 2006;20:1936–1938. doi: 10.1096/fj.05-5328fje. [DOI] [PubMed] [Google Scholar]
- Chen L, Xin X, Eckhart AD, Yang N, Faber JE. Regulation of vascular smooth muscle growth by alpha 1-adrenoreceptor subtypes in vitro and in situ. J Biol Chem. 1995;270:30980–30988. doi: 10.1074/jbc.270.52.30980. [DOI] [PubMed] [Google Scholar]
- Daub H, Wallasch C, Lankenau A, Herrlich A, Ullrich A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997;16:7032–7044. doi: 10.1093/emboj/16.23.7032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessy C, Kim I, Sougnez CL, Laporte R, Morgan KG. A role for MAP kinase in differentiated smooth muscle contraction evoked by alpha-adrenoceptor stimulation. Am J Physiol. 1998;275:C1081–C1086. doi: 10.1152/ajpcell.1998.275.4.C1081. [DOI] [PubMed] [Google Scholar]
- Dikic I, Tokiwa G, Lev S, Courtneidge SA, Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- Eguchi S, Iwasaki H, Ueno H, Frank GD, Motley ED, Eguchi K, et al. Intracellular signaling of angiotensin II-induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal-regulated kinase, and Akt. J Biol Chem. 1999;274:36843–36851. doi: 10.1074/jbc.274.52.36843. [DOI] [PubMed] [Google Scholar]
- Fernandez-Patron C. Therapeutic potential of the epidermal growth factor receptor transactivation in hypertension: a convergent signaling pathway of vascular tone, oxidative stress, and hypertrophic growth downstream of vasoactive G-protein-coupled receptors? Can J Physiol Pharmacol. 2007;85:97–104. doi: 10.1139/y06-097. [DOI] [PubMed] [Google Scholar]
- Florian JA, Watts SW. Epidermal growth factor: a potent vasoconstrictor in experimental hypertension. Am J Physiol. 1999;276:H976–H983. doi: 10.1152/ajpheart.1999.276.3.H976. [DOI] [PubMed] [Google Scholar]
- Fortuno A, San Jose G, Moreno MU, Diez J, Zalba G. Oxidative stress and vascular remodelling. Exp Physiol. 2005;90:457–462. doi: 10.1113/expphysiol.2005.030098. [DOI] [PubMed] [Google Scholar]
- Fujino T, Hasebe N, Fujita M, Takeuchi K, Kawabe J, Tobise K, et al. Enhanced expression of heparin-binding EGF-like growth factor and its receptor in hypertrophied left ventricle of spontaneously hypertensive rats. Cardiovasc Res. 1998;38:365–374. doi: 10.1016/s0008-6363(98)00010-8. [DOI] [PubMed] [Google Scholar]
- Hao L, Du M, Lopez-Campistrous A, Fernandez-Patron C. Agonist-induced activation of matrix metalloproteinase-7 promotes vasoconstriction through the epidermal growth factor-receptor pathway. Circ Res. 2004;94:68–76. doi: 10.1161/01.RES.0000109413.57726.91. [DOI] [PubMed] [Google Scholar]
- Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips MI. Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation. 2002;106:909–912. doi: 10.1161/01.cir.0000030181.63741.56. [DOI] [PubMed] [Google Scholar]
- Kagiyama S, Qian K, Kagiyama T, Phillips MI. Antisense to epidermal growth factor receptor prevents the development of left ventricular hypertrophy. Hypertension. 2003;41:824–829. doi: 10.1161/01.HYP.0000047104.42047.9B. [DOI] [PubMed] [Google Scholar]
- Kalmes A, Vesti BR, Daum G, Abraham JA, Clowes AW. Heparin blockade of thrombin-induced smooth muscle cell migration involves inhibition of epidermal growth factor (EGF) receptor transactivation by heparin-binding EGF-like growth factor. Circ Res. 2000;87:92–98. doi: 10.1161/01.res.87.2.92. [DOI] [PubMed] [Google Scholar]
- Laffargue M, Raynal P, Yart A, Peres C, Wetzker R, Roche S, et al. An epidermal growth factor receptor/Gab1 signaling pathway is required for activation of phosphoinositide 3-kinase by lysophosphatidic acid. J Biol Chem. 1999;274:32835–32841. doi: 10.1074/jbc.274.46.32835. [DOI] [PubMed] [Google Scholar]
- Levitzki A, Gazit A. Tyrosine kinase inhibition: an approach to drug development. Science. 1995;267:1782–1788. doi: 10.1126/science.7892601. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Della Rocca GJ, van Biesen T, Luttrell DK, Lefkowitz RJ. Gbetagamma subunits mediate Src-dependent phosphorylation of the epidermal growth factor receptor. A scaffold for G protein-coupled receptor-mediated Ras activation. J Biol Chem. 1997;272:4637–4644. doi: 10.1074/jbc.272.7.4637. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, et al. The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem. 2000;275:9572–9580. doi: 10.1074/jbc.275.13.9572. [DOI] [PubMed] [Google Scholar]
- Mukhin YV, Garnovsky EA, Ullian ME, Garnovskaya MN. Bradykinin B2 receptor activates extracellular signal-regulated protein kinase in mIMCD-3 cells via epidermal growth factor receptor transactivation. J Pharmacol Exp Ther. 2003;304:968–977. doi: 10.1124/jpet.102.043943. [DOI] [PubMed] [Google Scholar]
- Nagareddy PR, Chow FL, Hao L, Wang X, Nishimura T, Macleod KM, et al. Maintenance of adrenergic vascular tone by MMP transactivation of the EGFR requires PI3K and mitochondrial ATP synthesis. Cardiovasc Res. 2009;84:368–377. doi: 10.1093/cvr/cvp230. [DOI] [PubMed] [Google Scholar]
- Nakanishi S, Kakita S, Takahashi I, Kawahara K, Tsukuda E, Sano T, et al. Wortmannin, a microbial product inhibitor of myosin light chain kinase. J Biol Chem. 1992;267:2157–2163. [PubMed] [Google Scholar]
- Northcott C, Florian JA, Dorrance A, Watts SW. Arterial epidermal growth factor receptor expression in deoxycorticosterone acetate-salt hypertension. Hypertension. 2001;38:1337–1341. doi: 10.1161/hy1201.096815. [DOI] [PubMed] [Google Scholar]
- Ohtsu H, Dempsey PJ, Eguchi S. ADAMs as mediators of EGF receptor transactivation by G protein-coupled receptors. Am J Physiol Cell Physiol. 2006;291:C1–C10. doi: 10.1152/ajpcell.00620.2005. [DOI] [PubMed] [Google Scholar]
- Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, et al. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999;402:884–888. doi: 10.1038/47260. [DOI] [PubMed] [Google Scholar]
- Schafer B, Gschwind A, Ullrich A. Multiple G-protein-coupled receptor signals converge on the epidermal growth factor receptor to promote migration and invasion. Oncogene. 2004;23:991–999. doi: 10.1038/sj.onc.1207278. [DOI] [PubMed] [Google Scholar]
- Schlessinger J. Cellular signaling by receptor tyrosine kinases. Harvey Lect. 1993;89:105–123. [PubMed] [Google Scholar]
- Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- Swaminathan N, Vincent M, Sassard J, Sambhi MP. Elevated epidermal growth factor receptor levels in hypertensive Lyon rat kidney and aorta. Clin Exp Pharmacol Physiol. 1996;23:793–796. doi: 10.1111/j.1440-1681.1996.tb01181.x. [DOI] [PubMed] [Google Scholar]
- Tsai MH, Jiang MJ. Extracellular signal-regulated kinase1/2 in contraction of vascular smooth muscle. Life Sci. 2005;76:877–888. doi: 10.1016/j.lfs.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Tsai W, Morielli AD, Peralta EG. The m1 muscarinic acetylcholine receptor transactivates the EGF receptor to modulate ion channel activity. EMBO J. 1997;16:4597–4605. doi: 10.1093/emboj/16.15.4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin X, Yang N, Eckhart AD, Faber JE. Alpha1D-adrenergic receptors and mitogen-activated protein kinase mediate increased protein synthesis by arterial smooth muscle. Mol Pharmacol. 1997;51:764–775. doi: 10.1124/mol.51.5.764. [DOI] [PubMed] [Google Scholar]
- Yu SM, Tsai SY, Guh JH, Ko FN, Teng CM, Ou JT. Mechanism of catecholamine-induced proliferation of vascular smooth muscle cells. Circulation. 1996;94:547–554. doi: 10.1161/01.cir.94.3.547. [DOI] [PubMed] [Google Scholar]
- Zhang H, Chalothorn D, Jackson LF, Lee DC, Faber JE. Transactivation of epidermal growth factor receptor mediates catecholamine-induced growth of vascular smooth muscle. Circ Res. 2004;95:989–997. doi: 10.1161/01.RES.0000147962.01036.bb. [DOI] [PubMed] [Google Scholar]
- Zwick E, Daub H, Aoki N, Yamaguchi-Aoki Y, Tinhofer I, Maly K, et al. Critical role of calcium-dependent epidermal growth factor receptor transactivation in PC12 cell membrane depolarization and bradykinin signaling. J Biol Chem. 1997;272:24767–24770. doi: 10.1074/jbc.272.40.24767. [DOI] [PubMed] [Google Scholar]
- Zwick E, Hackel PO, Prenzel N, Ullrich A. The EGF receptor as central transducer of heterologous signalling systems. Trends Pharmacol Sci. 1999;20:408–412. doi: 10.1016/s0165-6147(99)01373-5. [DOI] [PubMed] [Google Scholar]