

Abstract
BACKGROUND AND PURPOSE
Because the in vivo effectiveness of ligands may also be determined by the rate by which they dissociate from their target receptors, drug candidates are being increasingly screened for this kinetic property. The dissociation rate of unlabelled ligand–receptor complexes can be estimated indirectly from their ability to slow the association of subsequently added radioligand molecules.
EXPERIMENTAL APPROACH
We used the ‘two-step competition’ binding approach consisting of pre-incubating the receptor preparation with a wide range of ligand concentrations, washing off free ligand molecules, adding radioligand and monitoring its receptor binding after a fixed time. Based on the rationale that binding of both ligands is mutually exclusive and that they bind according to the law of mass action to a single class of sites, the unlabelled ligand's disociation rate can be estimated from the upward shift that the competition curve experiences after washing.
KEY RESULTS
The relevance of the ‘two-step competition’ approach was explored by computer simulations and by comparing the dissociation behaviour of unlabelled D2 dopamine and CB1 cannabinoid receptor antagonists in this and alternative approaches. Besides providing satisfactory estimations of dissociation rates, the method also detects the ability of the unlabelled ligand molecules to be released from ‘sinks’ such as the cell membrane.
CONCLUSIONS AND IMPLICATIONS
As the ‘two-step competition’ requires rapid intermediate washing steps and needs radioligand binding to be measured at only one time point, this approach is particularly suited for binding studies on intact plated cells.
Keywords: radioligand binding, dissociation, kinetics, D2 dopamine, CB1 cannabinoid, membrane partitioning, computer simulations
Introduction
Classical ‘organ bath’ experiments as well as related studies on cell lines shed light on the ability of slow dissociating antagonists to effectively prevent their receptors to be stimulated during a subsequent agonist exposure. This constitutes a major mechanism for the so-called ‘insurmountable antagonism’ (Vauquelin et al., 2002). From the clinical perspective, slow dissociation has been claimed to prolong the performance of some antagonists acting at H1 histamine, nicotinic acetylcholine, α2A adrenoceptors, 5-HT3, 5-HT7 and M3 muscarinic receptors (el Bizri and Clarke, 1994; Kukkonen et al., 1997; Anthes et al., 2002; Blower, 2003; Swinney, 2004; Smith et al., 2006). This property has been featured especially for AT1-type angiotensin II receptor antagonists (Wienen et al., 1993; Aiyar et al., 1995; De Arriba et al., 1996; Timmermans, 1999; Unger, 1999) and D2 dopamine receptor antagonists. D2 receptor blockade is of particular interest in clinical therapy as it contributes to attenuating the psychotic phases in patients suffering from schizophrenia (Seeman et al., 1975; Creese et al., 1976). In this respect, the first slow-dissociating antipsychotics were prone to produce extrapyramidal side effects due to the refractoriness of the occupied striatal D2 receptors to respond to fast fluctuations in the local dopamine concentration. (Kapur and Seeman, 2001). In line with this view, this side effect seems to be less prominent in case of the more recently developed fast-dissociating ‘atypical’ antipsychotics (Schneider et al., 2006).
In drug screening studies, ligand–receptor interactions are traditionally quantified in terms of affinity and efficacy only. Owing to the increasing awareness that the in vivo effectiveness of ligands may also be dictated by the time they reside at their receptor, there is a growing call for drug candidates to be screened for this kinetic property as well (Swinney, 2004; 2006; Copeland et al., 2006; Tummino and Copeland, 2008). To this end, the most direct procedure is to investigate the rate at which radiolabelled ligands dissociate from their receptor. While this produces the most complete and precise information about the drug's dissociation rate, it is unpractical for screening studies of drug candidates, not only because of the cost involved but also because many ligands might fail to possess an appropriate affinity and selectivity for the receptor in question. Fortunately, indirect information about the dissociation properties of unlabelled competitive ligands (further denoted as competitors) can also be obtained by methods that are based on their ability to modulate radioligand binding by co-incubation as well as by ‘two-step’ procedures involving a pre-incubation phase with the competitor only. Interpretation of the data obtained by these indirect approaches is based on the premises that the competitor and the radioligand bind to a single site at the receptor according to a bimolecular process (i.e. binding follows the law of mass action) and that they are competitive with one another (i.e. that their binding is mutually exclusive such as in the case of overlapping binding sites).
In 1984, Motulsky and Mahan described methods to obtain information about the dissociation properties of unlabelled competitive ligands based on co-incubation experiments with a well-characterized radioligand. Depending on the difference in dissociation rate of both ligands, competition binding curves may undergo a time-dependent leftward or rightward shift. In kinetic experiments, the competitor may delay the time-dependent ascent of radioligand binding or even produce a time-dependent decrease in radioligand binding after an initial ‘overshoot’. These procedures allow a highly accurate estimation of the competitor's dissociation rate (Dowling and Charlton, 2006; Heise et al., 2007) but a fairly large number of competitor concentration/incubation/time combinations need to be dealt with. As a shortcut, indications of a competitor's dissociation rate can be obtained based on the ratio of competitor IC50 values when measured at sufficiently separate times (Heise et al., 2007). Combined with a scintillation proximity assay with membrane preparations, this approach allows high-throughput semi-quantitative ranking of compounds according to their dissociation rate, at least on membrane preparations.
Alternatively, indirect information about the dissociation properties of unlabelled competitive ligands can also be obtained by two-step procedures that are based on the ability of initially added competitor to delay the association of subsequently added radioligand (or agonist molecules, in the case of initially added antagonist). As these approaches require an intermediate wash step to remove free competitor molecules from the solution, they are especially suited for intact cell binding studies, as, technically speaking, it only involves the supernatant medium to be refreshed. For such in vitro assays, the traditional procedure was until recently to pre-incubate the receptor-containing preparation with a high concentration of competitor for a sufficient amount of time to allow the occupancy of the vast majority of receptor sites (ideally, all of them should be occupied), briefly washing the preparation to remove free competitor molecules, and finally subjecting the preparation to a fixed concentration of radioligand for increasing time periods after which the amount of specific/receptor binding is measured (Hara et al., 1995; Vanderheyden et al., 2000; Packeu et al., 2008). If the above-mentioned premises are met (which is fortunately most often the case) the radioligand will be unable to bind to the receptor as long as it is occupied by the competitor and, moreover, it will be unable to affect the competitor's dissociation (as it is a first-order process). Consequently, the association of a radioligand will be delayed by the initial binding of the competitor and, as illustrated in Figure 1A and C, this delay will be more pronounced as the dissociation half-life of the competitor increases. Hence, comparing such obtained radioligand association curves with those obtained without pretreatment allows an estimation of the competitor's dissociation rate. To analyse such curves in terms of the competitor's dissociation rate, Malany et al. (2009) recently introduced a convenient non-linear regression fitting procedure based on the relevant kinetic rate equation (Equation 4). As this fitting procedure is also applicable in case of fractional receptor occupancy, it is also ideally suited for obtaining the dissociation rates of in vivo administered competitors in ex vivo radioligand binding experiments. Interestingly, the ‘delayed association’ paradigm is closely related to that of traditional ‘organ bath’ experiments. In these functional experiments, the pre-incubation with antagonist and wash steps are followed by incubating the tissue in fresh medium and, at certain time intervals, an excess of agonist is added to monitor the time-dependent recovery of the response generated by the tissue (Ojima et al., 1997; Morsing et al., 1999).
Figure 1.
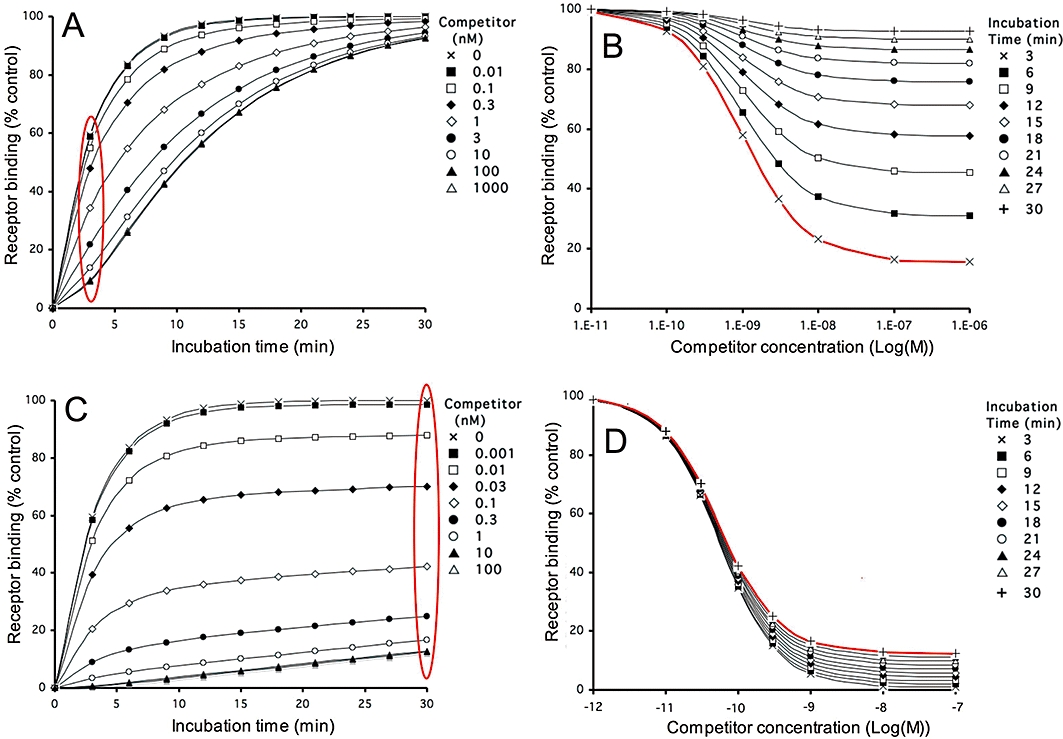
Simulations. Pre-incubation of receptors with a fast- and slow-dissociating competitor followed by a wash-step: effect on subsequent radioligand binding according to different representations. Competitor and radioligand parameters, radioligand concentration and incubation times are given in Table 1. For the simulations, receptors are pre-incubated with different concentrations of a fast- (panels A and B, dissociation t1/2= 6.9 min) and slow-dissociating (panels C and D, dissociation t1/2= 138 min) competitor till equilibrium is reached. Free competitor molecules are removed and the receptors are further incubated with a fixed concentration (2 nM) of radioligand for 3 to 30 min. Panels A and C: radioligand binding as a function of the incubation time (abscissa). Curves correspond to incubation of receptors pretreated with medium only or with the indicated concentrations of competitor. Radioligand binding is expressed in percent of control, i.e. binding after 30 min to receptors pretreated with medium only. Panels B and D: radioligand binding as a function of the competitor concentration (abscissa). Curves correspond to a transformation and subsequent normalization of the data presented in panels A and C respectively [from encircled (in red) data points to curve (in red)]. Curves correspond to incubation of competitor-pretreated receptors with radioligand for the indicated time periods. Radioligand binding is expressed in percent of control, i.e. binding after the corresponding time to receptors pretreated with medium only (i.e. [LR]c=0). Simulations are performed as described in ‘Materials and Methods’ and (Vauquelin et al., 2001; Vauquelin and Van Liefde, 2006).
The presently explored ‘two-step competition’ (TSC) binding approach is well suited for estimating the dissociation rate of unlabelled competitive ligands in in vitro studies with intact plated cells. It consists of incubating the receptor-containing preparation with a concentration range of competitor, briefly washing and finally subjecting the preparation for a single time period to a single concentration of radioligand. The dissociation rate of the competitor can then be evaluated from the upward shift of the resulting ‘competition curve’. Here again, a numerical value can be generated with the aid of a simplified version of the kinetic rate equation of Malany et al. (2009).
In this study, simulations were first performed to find out how to most conveniently estimate the competitor's dissociation rate based on the TSC approach. The relevance of this approach was then assessed by comparing the behaviour of unlabelled D2 dopamine and CB1 cannabinoid receptor antagonists with the dissociation behaviour of the radiolabelled equivalents in kinetic experiments. These examples illustrated the strengths and limitations of the TSC approach. An important characteristic of this approach is that it also allows estimation of the unlabelled ligand adsorbed by, and subsequently released from, ‘non-effector sinks’ such as the cell membrane, apart from any receptors.
Methods
Simulations
Radioligand (L) and competitor (C) receptor (R) interactions are defined to be bimolecular in nature and to be reversible:
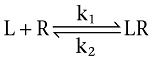 |
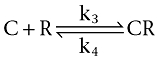 |
Computer-ssisted simulations (shown in Figures 1–3 and 7) comprise receptor pre-incubation without or with one or more concentrations of competing ligand, followed by removal of the free competing ligand molecules in solution (i.e. wash step) and subsequent incubation with one or more concentrations of radioligand for different times (t'). Integration of the differential equations (Equations 1–3) as previously described (Vauquelin et al., 2001; Vauquelin and Van Liefde, 2006) allow the receptor occupation by both ligands to be simulated as a function of t'. The differential equations yield changes in the percentage of free and occupied receptors after a very small time interval (t, typically 10 000 times less than t'). The association (k1 and k3, in M−1·min−1) and dissociation rate constants (k2 and k4, in min−1) used in the simulations (Figures 1–3 and 7) are given in Table 1.
Table 1.
Kinetic parameters of the competitors and radioligands, radioligand concentrations [L] and incubation times (t') used for the simulations
| Competitor pre-incubation | Radioligand incubation | ||||||
|---|---|---|---|---|---|---|---|
| k3 (M−1·min−1) | k4 (min−1) | Dissociation t1/2 (min) | k1 (M−1·min−1) | k2 (min−1) | [L]/KD | t' (min) | |
| Figure | |||||||
| 1A,B | 1·108 | 0.1 | 6.9 | 1·108 | 0.1 | 2 | 3–30 |
| 1C,D | 1·108 | 0.005 | 138 | 1·108 | 0.1 | 2 | 3–30 |
| 2 • | 1·108 | 0.1 | 6.9 | 1·108 | 0.1 | 2 | 30 |
| ○ | 1·108 | 0.05 | 13.8 | 1·108 | 0.1 | 2 | 30 |
| ▴ | 1·108 | 0.02 | 34.5 | 1·108 | 0.1 | 2 | 30 |
| △ | 1·108 | 0.005 | 138 | 1·108 | 0.1 | 2 | 30 |
| 3 ▪ | 1·108 | 0.1 | 6.9 | 1·108 | 0.1 | 2 | 3–30 |
| ▴ | 1·108 | 0.005 | 138 | 1·108 | 0.1 | 2 | 60–600 |
| □ | 1·108 | 0.1 | 6.9 | 2·107 | 0.02 | 1 | 3–30 |
| △ | 1·108 | 0.005 | 138 | 2·107 | 0.02 | 1 | 60–600 |
| 7A | 1·108 | 0.077 | 9.0 | 1·108 | 0.1 | 2 | 30 |
| 7B | 1·108 | 0.077 | 9.0 | 1·108 | 0.1 | 2 | 5–50 |
Radioligand pseudo first-order association rate constants (kobs, in min−1) can be calculated according to kobs= k2+ k1.[L] and KD values according to KD= k2/k1. For mono-exponential association and dissociation, the first-order rate constants (k) relate to the half-lives (t1/2) as k = 0.69/t1/2.
| (1) |
| (2) |
| (3) |
For the same experimental paradigm, the amount of radioligand–receptor complexes ([LR]) after different incubation time periods (t') can also be calculated/simulated as a function of total receptor concentration ([Rtot]) by use of the kinetic rate equation (Equation 4 below) developed by Malany et al. (2009).
| (4) |
in where:
[CR]t=0 is the amount of competitor-bound receptors at the onset on the incubation with radioligand.
[LR] values obtained by both simulation methods differ by no more than 0.2%. To avoid redundancy, only those obtained by first method are shown here.
Cell lines and culture conditions
Chinese hamster ovary cells (CHO-K1) stably transfected with the cDNA for apo-aquorin of Aquoria victoria and the GTP-binding protein Gα16 (CHO-AEQ cells) were kindly donated by Dr M. Detheux (Euroscreen s.a., Gosselies, Belgium) and further transfected with the pcDNA3.1 expression vector containing the entire coding region of the human D2 dopamine receptor (transcript variant 1 and 2; D2L and D2S) as described (Packeu et al., 2008). Transfected cells stably expressing the human D2L- and D2S-dopamine receptor (denoted as CHO-D2L and CHO-D2S cells respectively) were cultured (in 5% CO2 at 37°C) until confluence in polystyrene Costar® Corning® Cellbind® Surface 24-well plates (Elscolab, Kruibeke, Belgium) in supplemented Dulbecco's modified essential medium (DMEM), that is, with 2 mM L-glutamine, 2% of a stock solution containing 5000 I.U.·mL−1 penicillin and 5000 µg·mL−1 streptomycin (Life Technologies, Merelbeke, Belgium), and 10% fetal bovine serum (Life Technologies, Merelbeke, Belgium).
Human embryonic kidney cells stably expressing the chimeric G protein, Gqi5, together with human cannabinoid receptor 1 (denoted as HEK293-CB1r cells) (AstraZeneca, Mölndal, Sweden) were cultured in DMEM with glutamax (Invitrogen, Paisley, UK) supplemented with 10% fetal bovine serum (Hyclone, South Logan, UT, USA), 600 µg·mL geneticin and 300 µg·mL−1 hygromycin (Invitrogen, Paisley, UK) in 5% CO2 at 37°C. One day before the experiment, cells were plated (2.105 cells·well−1) in 24-well poly-D-lysine coated plates (in vitro, Stockholm, Sweden) in DMEM with glutamax + supplemented with 10% fetal bovine serum.
D2 dopamine receptor binding experiments
Prior to the experiment, cells were washed three times at room temperature with 500 µL per well of medium (HEPES/DMEM, pH 7.4 from Life Technologies, Merelbeke, Belgium). Incubations with radioligand were carried out at 37°C in a final volume of 500 µL per well in medium either alone (for [3H]-raclopride) or supplied with 0.2% (w/v) BSA (for [3H]-spiperone). Non-specific binding was determined in the presence of 1 µM spiperone (for [3H]-raclopride) or 0.3 µM (+)-butaclamol (for [3H]-spiperone).
For the TSC binding assays CHO-D2S (Figure 4A and B) and CHO-D2L cells (Figure 5) were pre-incubated for 30 min at 37°C in 500 µL medium with increasing concentrations of competitors, washed twice with an excess (1 mL) medium or not and further incubated for 30 min at 37°C with 2 nM [3H]-raclopride. To assess the inhibitory activity in the incubation medium (Figure 5), cells were pre-incubated and washed as above and further incubated for 10 min at 37°C with medium only. The supernatants were collected, added along with 2 nM [3H]-raclopride (final concentration) to naïve cells and the mixture was further incubated for 30 min at 37°C. For the delayed association assays (Figure 4C and D), CHO-D2S cells were pre-incubated with medium, 0.1 µM raclopride or 3 nM spiperone, washed twice and further incubated with 2 nM [3H]-raclopride for the indicated times. For dissociation binding assays (Figure 4E and F) CHO-D2S cells were incubated with 1 nM [3H]-spiperone or 2 nM [3H]-raclopride (30 min, 37°C), washed twice and further incubated with 2 nM raclopride for the indicated times. At the end of all experiments, wells were washed and remaining radioactivity was counted in a liquid scintillation counter as described (Packeu et al., 2008).
Figure 4.
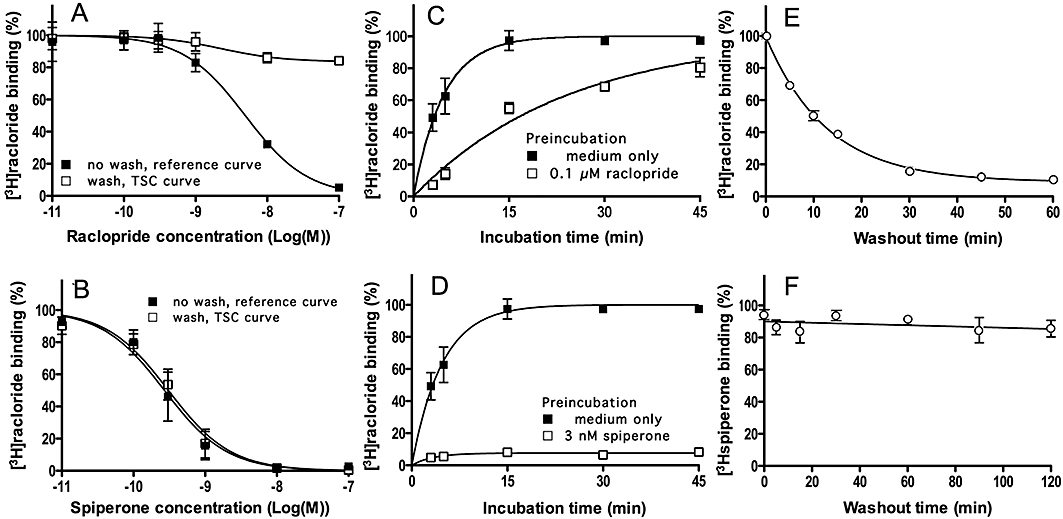
Experimens. Raclopride and spiperone dissociation from D2S dopamine receptors: different approaches. (A, B): TSC experiments. CHO-D2S cells were pre-incubated for 30 min at 37°C with increasing concentrations of raclopride (A) or spiperone (B) followed by a brief wash or not. [3H]-Raclopride (2 nM, final concentration) was then added and the incubation was continued for 30 min. Data refer to specific binding expressed as percentage of control binding (i.e. specific binding to cells pre-incubated with medium only) and are presented as means ± SEM of three individual experiments with three determinations each. (C, D): Delayed association experiments. Cells were pre-incubated for 30 min at 37°C with medium only or with 0.1 µM raclopride (C) or 3 nM spiperone (D), washed and further incubated for the indicated time periods at 37°C with 2 nM [3H]-raclopride. Data refer to specific binding expressed as percentage of control binding (i.e. specific binding after 30 min to cells pre-incubated with medium only) and presented as above. (E, F): Radioligand dissociation experiments. Cells were pre-incubated for 30 min at 37°C with 2 nM [3H]-raclopride (C) or 1 nM [3H]-spiperone (D), washed and further incubated for the indicated time periods at 37°C with 2 nM raclopride. Data refer to specific binding expressed as percentage of control binding (i.e. specific binding at the very onset of the washout) and are presented as above. The curve in (E) is monoexponential without constraint to zero at long times. TSC, two-step competition.
Figure 5.
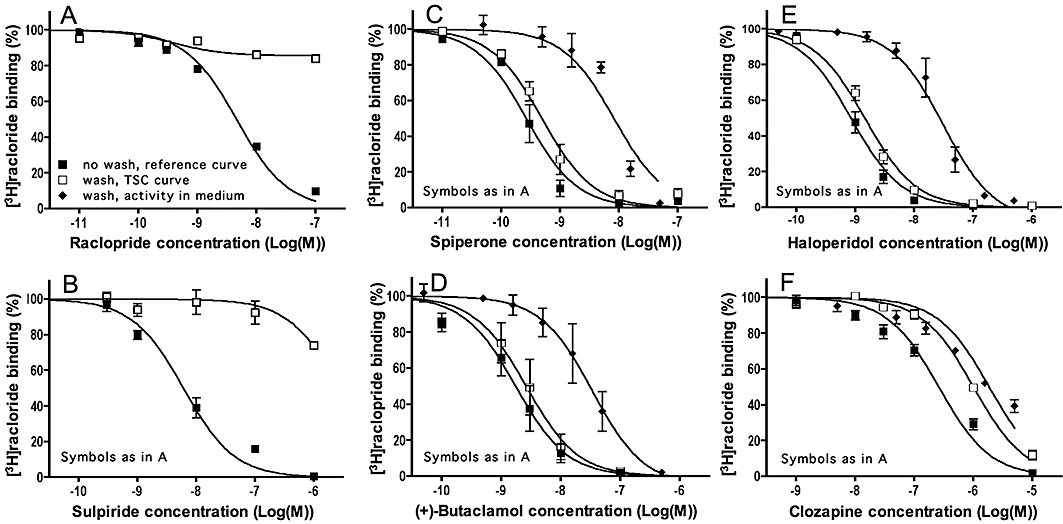
Experimens. Antagonist dissociation from D2L dopamine receptors: TSC approach. CHO-D2L cells were pre-incubated for 30 min at 37°C with increasing concentrations of raclopride (A) sulpiride (B), spiperone (C) (+)-butaclamol (D), haloperidol (E) or clozapine (F) followed by a brief wash or not. For the reference and TSC curves, 2 nM [3H]-Raclopride (final concentration) was then added and the incubation was continued for 30 min. Alternatively, to check for cellular uptake and subsequent release of unlabelled antagonist molecules in the medium, washed cells were incubated with fresh medium for 10 min, the supernatants were collected and added along with 2 nM [3H]-raclopride (final concentration) to naïve cells. The mixture was further incubated for 30 min at 37°C. For comparison with the TSC curves, the inhibitory activity in these supernatants is expressed as a function of the initial antagonist concentration in the pre-incubation step. Data refer to specific binding expressed as percentage of control binding [i.e. specific binding to naïve cells (when testing activity in medium) or cells pre-incubated with medium only (for reference and TSC curves)] and are presented as means ± SEM of three individual experiments with three determinations each. TSC, two-step competition.
CB1 cannabinoid receptor binding experiments
Prior to the experiment, cells were washed three times at room temperature with 500 µL per well of medium (Leibowitz with glutamax, pH 7.4 from Invitrogen, Paisley, UK). Incubations with radioligand were carried out at 37°C in a final volume of 500 µL per well in medium supplied with 1% (w/v) fatty acid-free BSA. Non-specific binding was determined in the presence of 0.1 µM AM 251.
For the TSC binding assays (Figure 6A and B) HEK293-CB1r cells were pre-incubated for 30 min at 37°C in 500 µl medium with increasing concentrations of competitors, rapidly washed three times with 750 µL per well Dulbecco's phosphate buffered saline (Invitrogen, Paisley, UK) with 1% fatty acid-free BSA (Sigma-Aldrich, St. Louis, MO, USA) or not and further incubated for 60 min at 37°C with 5 nM [3H]-rimonabant. To assess the inhibitory activity in the incubation medium (Figure 6), cells were pre-incubated and washed as above and further incubated for 60 min at 37°C with medium only. The supernatants were collected, added along with 5 nM [3H]-rimonabant (final concentration) to naïve cells and the mixture was further incubated for 60 min at 37°C. For the radioligand dissociation experiments (Figure 6C and D) cells were pre-incubated for 60 min 2 nM [3H]-taranabant or 5 nM [3H]-rimonabant, washed three times and further incubated with medium containing 5 nM rimonabant for the indicated time periods. At the end of all experiments, cells were scraped off the wells in 250 µL PBS (Invitrogen, Paisley, UK) and transferred to scintilation vials. The radioactivity was counted for 3 min in presence of 3 mL scintillation liquid (Optiphase Hisafe from PerkinElmer; Boston, MA, USA) in a liquid scintillation counter.
Figure 6.
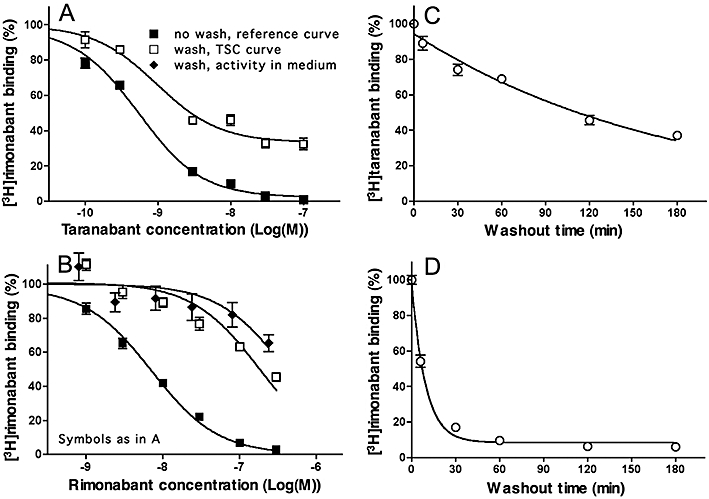
Experiments. Taranabant and rimonabant dissociation from CB1 cannabinoid receptors: different approaches. (A, B): TSC approach. HEK293-CB1r cells were pre-incubated for 30 min at 37°C with increasing concentrations of taranabant (A) or rimonabant (B) followed by a brief wash or not. For the reference and TSC curves, [3H]-Rimonabant (5 nM, final concentration) was then added and the incubation was continued for 60 min. Alternatively, to check for cellular uptake and subsequent release of rimonabant in the medium, washed cells were incubated with fresh medium for 30 min, the supernatants were collected and added along with 5 nM [3H]-rimonabant (final concentration) to naïve cells. The mixture was further incubated for 60 min at 37°C. For comparison with the TSC curves, the inhibitory activity in these supernatants is expressed as a function of the initial antagonist concentration in the pre-incubation step. Data refer to specific binding expressed as percentage of control binding (i.e. specific binding to cells pre-incubated with medium only) and are presented as means ± SEM of at least three individual experiments with three determinations each. (C, D): Radioligand dissociation experiments. Cells were pre-incubated for 60 min at 37°C with 2 nM [3H]-taranabant (C) or 5 nM [3H]-rimonabant (D), washed and further incubated for the indicated time periods at 37°C with 5 nM unlabelled rimonabant (washout phase). Data refer to specific binding expressed as percentage of control binding (i.e. specific binding at the very onset of the washout) and are presented as means ± SD of two individual experiments with three determinations each. Curves are monoexponential and constrained to zero at long times (C) or not (D). TSC, two-step competition.
Data analysis
The half maximal inhibitory concentration values (IC50) from competition binding experiments and kinetic constants from time curves were calculated by non-linear regression analysis by GraphPad Prism™ (San Diego, CA, USA) based on a one-site bimolecular reaction obeying the Law of Mass Action. For curve fitting of the competition binding experiments, a one-site model with free-floating parameters was used for the experimental data and a two-site model for the simulated TSC binding data in Figure 7A.
Figure 7.
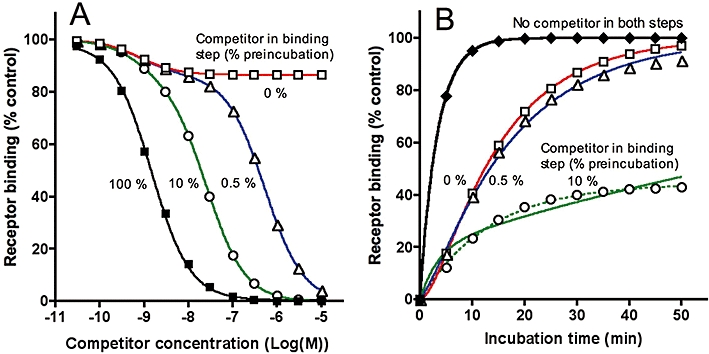
Simulations. Pre-incubation of receptors with a fast-dissociating competitor followed by a wash-step: effect of the release of adsorbed competitor in solution on subsequent radioligand binding. Competitor and radioligand parameters, radioligand concentration and incubation times are given in Table 1. Calculations of k4 (given below as the corresponding t1/2) were based on Equation 5 (A) and 4 (B). (A) For simulating the TSC data, receptors are pre-incubated for 30 min with different concentrations of competitor, washed and further incubated for 30 min with a fixed concentration (2 nM) of radioligand either in the absence (0%) or presence of still remaining competitor at the indicated fractions of its initial concentration. Radioligand binding is expressed in percent of control, i.e. binding to receptors pretreated with medium only (i.e. [LR]c=0). [LR]cmax/[LR]c=0 ratios obtained by non-linear regression analysis varied little: that is, 0.866 (red curve, for 1-site competition), 0.867 (blue curve, for 2-site competition) and 0.863 (green curve, for 2-site competition). The calculated competitor dissociation t1/2 values (8.9–9.1 min) according to Equation 5 were close to the input value (9.0 min). The lower potency component of the green and blue TSC curves were shifted 17- and 360-fold to the right of the reference (black) curve. (B) For simulating the ‘delayed association’ data, receptors are pre-incubated for 30 min in medium only or with a fixed concentration (3.10−8 M) of competitor (open symbols) and further incubated for increasing time periods with a fixed concentration (2 nM) of radioligand either in the absence or presence of competitor released at 0.5 or 10% of its initial concentration. Radioligand binding is expressed in percent of control, i.e. binding after 30 min to receptors pretreated with medium only. The calculated competitor dissociation t1/2 and initial receptor occupancy ([CR]t=0) values according to Equation 4 were identical to the input values (9.0 min and 97.5% of [Rtot]) in case of no competitor release (red curve) but diverged more and more with increasing competitor release (blue curve for 0.5% release: 11.3 min and 90.3%; green solid curve for 10% release: 82 min and 78.2%). In case of 10% competitor release, the data fitted better with a mono-exponential association paradigm (dotted green curve) in where only 44.5% of the control radioligand binding was recoverable. TSC, two-step competition.
Equation 4 is very useful for calculating competitor dissociation rates (k4) with TSC experiments as concurrent control experiments (either co-incubation experiments or TSC experiments without intermediate wash step) allow competitor concentrations to be chosen at which near-maximal saturaturation of the receptor takes place. The corresponding ‘recovered’ amount of radioligand binding ([LR]cmax) is then equal to (a + b).[Rtot]. Note that radioligand binding after pretreatment of medium only (i.e. [LR]c=0, the control binding habitually used in competition binding experiments) equals a.[Rtot]. Hence:
| (5) |
With earlier knowledge of k1 and k2 (obtained from radioligand association and dissociation binding experiments), [L], t' and the experimentally obtained values of [LR]c=0 and [LR]cmax, k4 is easily calculated by subjecting Equation 5 to, for example, the Microsoft® Excel Solver function.
Materials
The tritium labelled D2 dopamine receptor antagonists [3H]-raclopride (60–63 Ci·mmol−1) and [3H]-spiperone (79–113 Ci·mmol−1) and the CB1 cannabinoid receptor antagonist [3H]-rimonabant (44 Ci·mmol−1) were purchased from PerkinElmer (Boston, MA, USA). The unlabelled D2 receptor antagonists sulpiride, clozapine, haloperidol, raclopride and spiperone were purchased from Tocris (Avonmouth, UK) and (+)-butaclamol was from Sigma-Aldrich (St. Louis, MO, USA). The unlabelled CB1 receptor antagonists rimonabant and taranabant were from AstraZeneca (Mölndal, Sweden) and AM 251 was from Tocris (Avonmouth, UK). Drug/molecular target nomenclature follows Alexander et al. (2009).
Results
Simulations
The dissociation rate of unlabelled ligand/competitor–receptor complexes can be estimated indirectly by measuring their ability to slow the association of a subsequently added radiologand molecule in a sequential competitor pre-incubation–wash–radioligand incubation paradigm (Hara et al., 1995; Vanderheyden et al., 2000; Packeu et al., 2008; Malany et al., 2009). Panels A and C in Figure 1 simulate typical outcomes of such experiments for a fast (k4= 0.1 min−1, t1/2= 6.9 min) and slow (k4= 0.005 min−1, t1/2= 138 min) dissociating competitors respectively. The curves refer to radioligand binding for increasing times to receptor preparations that have been pretreated with medium only (top curve) or with different concentrations of competitor and then briefly washed to remove free competitor molecules. The wash is assumed to remove all free competitor molecules without affecting its receptor occupancy. Gradually increasing the competitor concentration will produce corresponding delays in radioligand association till a limit is attained; that is, when the competitor occupies all the receptor sites at the onset of the incubation with radioligand. Comparing these latter curves in Figure 1A and C clearly reveals the capability of the slow dissociating competitor to delay the radioligand association more severely than the fast-dissociating one (in all cases radioligand binding will reach the control value corresponding to the maximal binding to medium-pretreated preparations, provided that the incubation lasts long enough). Radioligand association curves may acquire complex shapes when the receptors were pretreated with competitor (including a delayed onset in binding such as in panel A and biphasic curves such as in panel C). Despite their complexity, such binding data can now adequately be analysed by the fitting procedure developed by Malany et al. (2009) to yield the competitor's dissociation rate (k4) and the amount of competitor-bound receptors at the onset of the radioligand incubation step, provided that the radioligand's k1 and k2 values are known.
An interesting situation arises when the same binding data are plotted as a function of the initial free competitor concentration instead of the incubation time (Figure 1B and D). In conventional competition binding experiments, involving the concomitant presence of free radioligand and competitor molecules, the latter will produce a concentration-dependent decline in radioligand binding till it is completely abolished. Here, however, the competitor will continue to produce a concentration-dependent decline in radioligand binding even when free competitor molecules have been washed away, but with this peculiarity that the maximal decline gradually fades as the incubation time increases. This fading process is much swifter for the fast dissociating competitor (Figure 1B) than for the slow dissociating one (Figure 1D). Note that, while the incubation time has a profound impact on the difference in the maximal effect of both types of competitors, it does not substantially affect their IC50 value.
Information about the competitor's dissociation rate can thus be acquired by pre-incubating the receptor-containing preparation with increasing concentrations of competitor, washing out free competitor molecules and further incubating the preparation with a constant concentration of radioligand for a constant time period. This is clearly illustrated in Figure 2, which compares the so-obtained ‘two-step’ competition (TSC) curves for competitors with dissociation rates (k4) ranging between 0.005 and 0.1 min−1 (t1/2 between 138 and 6.9 min). The ‘recovered’ radioligand binding after near-saturation of the receptors by the competitors during the pre-incubation step (i.e. [LR]cmax) constitutes the most contrasting parameter for ordering them according to their dissociation rate (Figure 2, insert). [LR]cmax is also important for the calculation of k4. To find out the relevant (i.e. receptor-saturating) competitor concentrations, a concurrent, control competition binding experiment (either a co-incubation experiment or a TSC experiment without intermediate wash step) is recommended. Alternatively, [LR]cmax can also be obtained by non-regression analysis of the TSC curve according to a one-site (or even a two-site, see Figure 7) competition model without constraint of the lower limit. Based on the obtained values of [LR]cmax and [LR]c=0 (i.e. radioligand binding after pretreatment of the receptors with medium only) and knowledge of the radioligand's association and dissociation rates (k1 and k2 respectively), k4 is easily calculated by using Equation 5 (see Methods). Indeed, this equation constitutes a special case of the kinetic rate equation (Equation 4) wherein k4 is the only unknown parameter. Accordingly, additional simulations also showed that the [LR]cmax values were independent of the competitor's input k3 values (data not shown). Also, for the simulated data presented in Figure 2, the calculated k4 values based on [LR]cmax after pre-incubation with a receptor-saturating concentration of each competitor are only marginally different from the input values (see figure legend).
Figure 2.
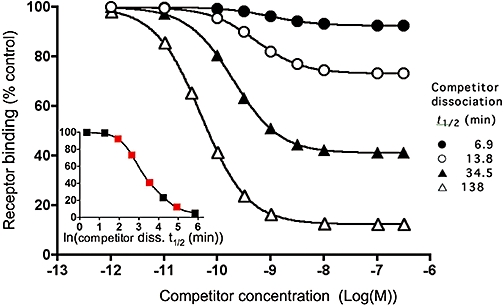
Simulations. Effect of the competitor dissociation rate on the recovery of radioligand binding after a fixed-time incubation. Competitor and radioligand parameters, radioligand concentration and incubation time are given in Table 1. For the simulations, the receptor-containing preparation is pre-incubated with different concentrations (abscissa) of competitors with the indicated dissociation t1/2 till equilibrium is reached. Free competitor molecules are removed and the preparation is further incubated with a fixed concentration (2 nM = 2.KD) of radioligand for 30 min. Radioligand binding is expressed in percent of control, i.e. binding to receptors pretreated with medium only (i.e. [LR]c=0). Competitor dissociation t1/2 values (=0.69/k4), calculated according to Equation 5 with [LR]cmax (radioligand binding to receptors pretreated with the highest concentration of each competitor) and the radioligand k1 and k2 values (Table 1) are close to the input values: i.e. 6.9, 13.7, 34.4 and 137.6 min respectively. Insert: [LR]cmax as a function of the dissociation t1/2 (abscissa, natural logarithmic scale) of distinct competitors (symbols are in red for the competitors displayed in the main panel).
Simulations were also performed to evaluate the possibility of acquiring the competitor's k4 values based on the incubation time (t') and the upward shift of the competition curve (i.e. [LR]cmax/[LR]c=0 ratio) only. To this end, simulated ratios were plotted as a function of t', expressed in units of dissociation t1/2 of each competitor to facilitate their comparison. Figure 3A compares the situation of the fastest- and slowest-dissociating competitors from Figure 2. Although both curves do not fully coincide when binding is performed with the fast-associating radioligand (kobs= 0.3 M−1·min−1, t1/2= 2.3 min), the difference remains certainly acceptable for the semi-quantitative approximation of a wide range of competitor k4 values based on their [LR]cmax/[LR]c=0 ratio (the competitor's dissociation t1/2 values are obtained by dividing t' by the extrapolated X values). However, the curves diverge more significantly when binding is performed with a slow-associating radioligand (kobs= 0.04 M−1·min−1, t1/2= 17 min). Additional simulations (data not shown) confirmed that any move to increase the radioligand kobs (such as by increasing its concentration) yields a better convergence of the curves and, hence, a better approximation of the competitor k4.
Figure 3.
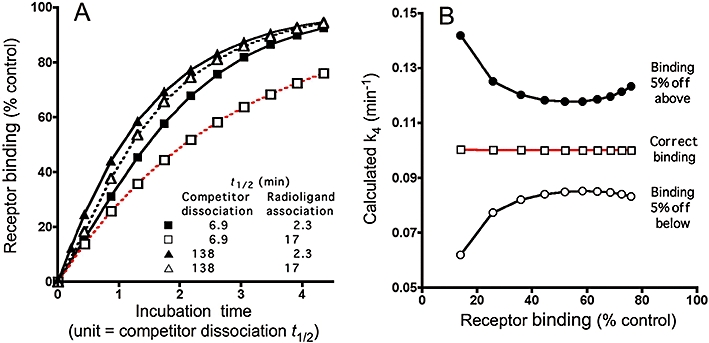
Simulations. Effect of the competitor dissociation rate on the recovery of radioligand binding and the calculated competitor k4 values, after increasing incubation times. (A) For the simulations, the receptor-containing preparation is pre-incubated with a receptor-saturating concentration (0.33 µM) of competitors with the indicated dissociation t1/2 till equilibrium is reached. Free competitor molecules are removed and the preparation is further incubated with a fast- (closed symbols and solid line, kobs= 0.3 M−1·min−1, t1/2= 2.3 min) or slow-associating radioligand (open symbols and dotted line, kobs= 0.04 M−1·min−1, t1/2= 17 min) for increasing time periods (abscissa, expressed in units of dissociation half-life of each individual competitor). Radioligand binding is expressed in percent of control, i.e. binding after the same time periods to receptors pretreated with medium only (i.e. [LR]c=0). (B) k4 values calculated according to Equation 5 for the fast dissociating competitor/slow associating radioligand combination (red curve in panel A) based on the simulated receptor binding values and for situations in where receptor binding should be off by 5% either below or above. Competitor and radioligand parameters, radioligand concentrations and incubation times are given in Table 1.
Nevertheless, calculations by using Equation 5 provide much more accurate estimates of k4 (i.e. varying less than 0.5% of the input values for all the conditions shown in Figure 3A). Figure 3B illustrates one example (i.e. the least accurate situation in Figure 3A) of the lack of variance of the calculated k4 values with the incubation time along with the potential outcomes if the [LR]cmax/[LR]c=0 ratios diverge from the correct value by 5%. In this respect, while experimental binding values in the steep part of the competition curves show the greatest variability (due to the important contribution of the IC50), SEMs of plateau binding values rarely exceed 5%. As clearly illustrated in Figure 3B, this error margin brings about the highest variations in estimates of k4 at the extreme (high as well as low) [LR]cmax/[LR]c=0 ratios. Based on this potential error margin, we will also provide a range of k4 (and t1/2) values when analysing the experimental TSC curves presented below.
Raclopride and spiperone dissociation from D2S receptors
The dissociation rate of the antagonists raclopride and spiperone from the D2S dopamine receptors was indirectly estimated by the TSC approach described above, the ‘delayed radioligand association’ approach as well as by direct dissociation experiments using the radiolabelled antagonists.
For the TSC approach, plated CHO-D2S cells were pre-incubated for 30 min at 37°C with increasing concentrations of raclopride or spiperone, rapidly washed or not (as reference) and further incubated with 2 nM [3H]-raclopride for 30 min after which the amount of D2S receptor-associated (i.e. specifically bound) radioligand was determined. Figure 4A and B compares the binding under both conditions as a function of the initial competitor concentration. Compared with the reference curve, the TSC curve of raclopride underwent a marked upward shift when cells were washed before the radioligand binding step (Figure 4A). For cells that were pre-incubated with a receptor-saturating concentration of raclopride (0.1 µM, obtained from the reference curve) and washed, [3H]-raclopride binding amounted 79% [74–84% with 5% error margin] of control binding. The corresponding k4 value for raclopride was 0.067 [0.057–0.079] min−1 (t1/2= 10.3 [8.7–12.1] min) when calculated according to Equation 5 (and based on the kinetic parameters of [3H]-raclopride obtained by the association and dissociation experiments, i.e. k1= 4.9 107 M−1·min−1 and k2= 0.078 min−1). In contrast to raclopride, the TSC curve of spiperone did not undergo a perceptible shift in washed cells (Figure 4B). This should point at extremely slow spiperone dissociation with a t1/2 well above the incubation time of 30 min. As summarized in Table 2, these results fit well with those obtained by the two alternative approaches described below.
Table 2.
Antagonist dissociation half-lives calculated by different approaches
| Dissociation t1/2 (in min) | |||
|---|---|---|---|
| Antagonist | ‘Two-step competition’ | Delayed association | Radioligand dissociation |
| D2S receptors (Figure 4) | |||
| Raclopride | 10.3 (8.7–12.1) | 13.5 | 8.9 |
| Spiperone | ≫30 | ≫45 | ≫120 |
| D2L receptors (Figure 5) | |||
| Raclopride | 10.2 (8.4–12) | 9.2–9.5* | 5.8* |
| Spiperone | ≫30 | ≫45* | ≫120* |
| CB1 receptors (Figure 6) | |||
| Taranabant | 90 (75–109) | 122 | |
| Rimonabant | <<60 | 6.4 | |
Experiments are shown in Figures 4 and 5 or published
(Packeu et al., 2008) for the D2 receptor antagonists raclopride and spiperone and in Figure 6 for the CB1 receptor antagonists taranabant and rimonabant. Data from the TSC approach were calculated according to Equation 5 based on the obtained values of [LR]cmax and [LR]c=0 and knowledge of [L], t', k1 and k2 (see Methods). Values in brackets account for a 5% error margin of the [LR]cmax/[LR]c=0 ratio. Data from the delayed radioligand association approach were calculated according to Equation 4 by the non-linear regression fitting procedure developed by Malany et al. (2009). Data from radioligand dissociation curves were calculated by non-regression analysis of the curve based on a mono-exponential decline.
TSC, two-step competition.
The dissociation rate of unlabelled raclopride and spiperone were also estimated indirectly by the delayed radioligand association approach. In these experiments, cells were pre-incubated with medium only (for control curves) or a high, receptor-saturating concentration of competitor, washed and finally incubated with 2 nM [3H]-raclopride for increasing time periods. Pre-incubation with 0.1 µM raclopride did not affect the maximal extent of the subsequent [3H]-raclopride binding but it effectively delayed its association (Figure 4C). Making use of the non-linear regression fitting procedure (Malany et al., 2009), the k4 value for raclopride was 0.051 min−1 (t1/2= 13.5 min). In contrast, following pre-incubation with a receptor near-saturating concentration of spiperone (3 nM) and washing, almost no recovery of [3H]-raclopride binding was found to take place (Figure 4D). This points again at very slow spiperone dissociation.
For direct [3H]-raclopride dissociation experiments, cells were pre-incubated for 30 min with 2 nM [3H]-raclopride, rapidly washed and further incubated for different periods of time with 2 nM unlabelled raclopride (for conditions to match those of the two previous approaches). As depicted in Figure 4E, [3H]-raclopride binding decreased exponentially and the vast majority of specific binding sites were freed at the end of the experiment, i.e. after 60 min. The corresponding dissociation t1/2 was 8.9 min. In equivalent experiments, in where cells were pre-incubated for 30 min with 1 nM [3H]-spiperone, almost no disociation of this specifically bound radioligand could be perceived (Figure 4F).
Antagonist dissociation from D2L receptors
Identical TSC binding experiments with raclopride, spiperone and the additional antagonists (+)-butaclamol, haloperidol, clozapine and sulpiride were also performed with CHO-D2L cells to yield their k4 values for the long D2 dopamine receptor isoform (Figure 5). When appropriate k4 values were calculated according to Equation 5 based on the kinetic parameters of [3H]-raclopride for this receptor isoform (i.e. k1= 1.25 108 M−1·min−1 and k2= 0.12 min−1; calculated from Figures 4 and 5 in Packeu et al., 2008). Compared with the reference curve for experiments without wash-step, the raclopride and sulpiride TSC curves were markedly shifted upward (Figure 5A and B), indicating that both antagonists dissociate rapidly from the D2L receptor. At a saturating concentration (0.1 µM) of raclopride, radioligand binding amounted 84% of control binding. The corresponding k4 value for raclopride [0.068 (0.058–0.082) min−1, t1/2= 10.2 (8.4–12.0) min] is in good agreement with data previously obtained by the direct [3H]-raclopride dissociation and indirect delayed [3H]-raclopride association approaches (Table 2) (Packeu et al., 2008). It is more difficult to attribute a [LR]cmax/[LR]c=0 ratio to sulpiride as the binding of [3H]-raclopride already drops when this antagonist attains a receptor-saturation concentration (1 µM). Yet, based on the 92% radioligand binding at 0.1 µM sulpiride, its k4 value can be approximated to range between 0.075 and 0.13 min−1 (t1/2= 5.3–9.1 min). In contrast, the TSC curve curve of spiperone (+)-butaclamol and haloperidol did not undergo a perceptible upward shift in washed cells (Figure 5C–E). This could point to extremely slow dissociation of these antagonists from the D2L receptor with a t1/2 well above the incubation time of 30 min.
Interestingly, the TSC curve of clozapine experienced a fourfold rightward shift when compared with the reference curve (Figure 5F). As further outlined in the Discussion section, this could point to the release of (initially adsorbed) clozapine molecules from the cells during the radioligand binding step. To check for this possibility, cells were pretreated with the same concentrations of clozapine as in the TSC curve experiment, washed and finally incubated for 10 min with fresh medium. The presence of released clozapine in the resulting washout medium was then evaluated by monitoring its ability to decrease [3H]-raclopride binding to fresh CHO-D2L cells in a co-incubation experiment. Such inhibitory effects were clearly present and, when expressed as a function of the initial clozapine concentration in the pre-incubation step, it exhibited almost the same potency as the shifted curve in the TSC experiment (Figure 5F). Similar control washout experiments were also carried out for spiperone (+)-butaclamol and haloperidol (Figure 5C–E). The washout media were all found to contain D2L receptor binding activity but, contrary to the picture seen with clozapine, the corresponding IC50 values were =10-fold higher when compared with the (unshifted) TSC curve of each of these antagonists. This implies that, although the cells seem act as a sink for these antagonists, their subsequent release into the medium is not sufficient to account for their competition behaviour in the TSC experiment.
Taranabant and rimonabant dissociation from CB1 receptors
The dissociation rate of the antagonists taranabant and rimonabant from the CB1 cannabinoid receptors was also indirectly estimated by the TSC approach. To this end, plated HEK293-CB1r cells were pre-incubated for 30 min at 37°C with increasing concentrations of rimonabant or taranabant, rapidly washed or not and further incubated with 5 nM [3H]-rimonabant for 60 min after which the amount of CB1 receptor-associated (i.e. specifically bound) radioligand was determined. Figure 6A and B compares the binding under both conditions as a function of the initial competitor concentration. Compared with the control curve in unwashed cells, the taranabant TSC curve underwent a 33% upward shift in the washed cells (Figure 6A). Based on the kinetic parameters of [3H]-rimonabant for this receptor isoform (i.e. k1= 1 107 M−1·min−1 and k2= 0.11 min−1, Wennerberg et al., 2010) and Equation 5, the calculated k4 of taranabant amounted 0.0077 (0.0063–0.0092) min−1[t1/2= 90 (75–109) min]. In contrast to taranabant, the TSC curve of rimonabant was only shifted 25-fold to the right of the reference curve (Figure 6B). As for clozapine (Figure 5B) this behaviour could be ascribed to the release of a fast-dissociating competitor during the radioligand incubation-step. Indeed, the corresponding medium decreased [3H]-rimonabant binding to fresh HEK293-CB1r cells with almost the same potency (expressed as a function of the initial rimonabant concentration in the pre-incubation step) as the shifted curve in the TSC experiment (Figure 6B).
For direct [3H]-rimonabant and [3H]-taranabant dissociation measurements, plated HEK293-CB1r cells were incubated with 2 nM [3H]-taranabant or 5 nM [3H]-rimonabant for 60 min, rapidly washed and further incubated for different periods of time with 5 nM unlabelled rimonabant (for conditions to match the TSC approach). As depicted in Figure 6C and D, binding of both radioligands decreased exponentially. [3H]-taranabant dissociated with a t1/2 of 122 min. [3H]-Rimonabant dissociated with a t1/2 of 6.4 min and the vast majority of specific binding sites were free at the end of the experiment, i.e. after 60 min.
Discussion
Several radioligand binding strategies can be adopted to quantitate the dissociation rate (k4) of a receptor-bound ligand. Besides direct radioligand dissociation experiments, competitor k4 values can also be estimated indirectly in co-incubation experiments (Motulsky and Mahan, 1984; Dowling and Charlton, 2006). Fewer data need to be generated by alternative approaches that include a rapid intermediate wash-step. Among them, the ‘delayed association’ approach is based on the ability of bound competitors to delay the association of a subsequently added radioligand molecules (Hara et al., 1995; Packeu et al., 2008). The presently explored TSC approach is based on the same principle but consists of pre-incubating the receptors with a range of ligand concentrations (instead of a single one), washing off ligand molecules in solution and monitoring the binding of fixed concentration of radioligand after a fixed time. In principle, and with prior knowledge of the radioligand's kinetic parameters, analysis of the TSC binding data should be straightforward as, at a receptor near-saturating concentration of competitor (as determined from concurrent reference competition binding curves without an intermediate wash step), the appropriate kinetic rate equation (Malany et al., 2009) is simplified so that k4 can be calculated from the upward shift of the TSC curve (Equation 5). As a first approach, visual inspection of the upward shift will already provide an approximate k4, at least if the radioligand associates swiftly (Figure 3).
In practice, a number of issues need special attention. First, the TSC approach avoids the measurement of radioligand binding at different time intervals and incubations can be kept relatively short. While this issue is, of course, a matter of personal appreciation, shortening the pre-incubation and incubation steps in intact cell experiments would help to limit cell death and other potential interfering phenomena, linked to alteration of the medium and the secretion of metabolites. Another particular feature of the TSC approach is that it provides at the same time, at least preliminary indications about the competitor's affinity (from the reference competition binding curves), its dissociation rate and its ability to partition between the medium and ‘non-effector sinks’, such as cell membranes. More refined estimations can then be envisioned by performing measurements after longer pre-incubation periods, after different incubation periods, with different radioligand concentrations and by testing the washout media for the presence of free competitor molecules. In this respect, small k4 values could point to the need for longer pre-incubation periods to achieve equilibrium binding of the competitor. While this issue matters for estimating the competitor's affinity, it is not supposed to affect its k4 estimate as reference competition curves are only required to find out a suitable receptor-saturating concentration of competitor and to estimate the ‘rightward shift’ of the TSC curves. Also, as illustrated in Figure 3B, k4 estimates could become more inaccurate in case of very small or large upward shifts of the TSC curve and, in this situation, more reliable estimates could be obtained by increasing and decreasing the incubation time with radioligand respectively. It is also preferable for the radioligand to be present at a sufficiently high concentration. While this does not matter for the calculation of k4 according to Equation 5 (Figure 3B), the resulting decrease in the radioligand kobs should yield more reliable estimations of k4 by visual inspection of the TSC curve, as in case of pronounced partitioning of the competitor (see below). At a high concentration, the radioligand should also be more effective at preventing rebinding of freshly dissociated competitor molecules to the same or adjacent receptor molecules (Fierens et al., 1999; Vauquelin and Szczuka, 2007) and this obviously also applies to delayed association-based experiments. If allowed to take place, such as in traditional ‘organ bath’-based kinetic experiments in where the washout step takes place in medium only, this rebinding phenomenon could be responsible for an apparent delay in the competitor's dissociation. Finally, particular to the ‘delayed association’ and TSC methods is that the intermediate wash-step should be fast enough to prevent significant dissociation of the competitor-receptor complexes and yet effective enough to remove the great majority of competitor molecules in solution. To meet this requirement, it is convenient to perform binding studies on intact cells firmly attached to microwell plates, so that washing merely requires a change of medium. Incomplete removal of competitor in solution should result in a rightward shift (on top of a potential upward shift, see simulations in Figure 7A) of the TSC curve when compared with the reference curve.
Intact cell binding studies were performed to evaluate the merits and limits of the TSC approach. The k4 values, estimated here, of the fast-dissociating D2 dopamine receptor antagonists supiride and raclopride (Figures 4A, 5 and 6) and the slow dissociating CB1 cannabinoid receptor antagonist taranabant were in good agreement with those obtained by the alternative indirect ‘delayed association’ and direct radiolabelled antagonist dissociation approaches (Table 2). The pronounced (≫100-fold) rightward shift of their TSC curves (if even perceptible at the competitor concentrations used) indicates that a succession of rapid washes is highly effective in removing competitor molecules from the solution. In agreement, non-specific binding of [3H]-raclopride and [3H]-taranabant (which in the intact cell binding paradigm also accounts for incomplete removal of radioligand from the solution) was also very small after an equivalent rapid wash procedure (at 2 nM: <10% of specific/receptor binding for both radioligands).
The TSC curves of the D2 receptor antagonist clozapine and of the CB1 receptor antagonist rimonabant only experienced limited rightward shifts of 4- and 25-fold respectively (Figures 5F and 6B). To find out whether this was related to the presence of competitor molecules in solution during the radioligand binding step, an equivalent washout medium was checked for its ability to decrease radioligand binding to fresh cells. Such effect was clearly present for both competitors and, when expressed as a function of their initial concentration in the pre-incubation step, the curves exhibited IC50 values similar to those from the TSC curves (Figures 5F and 6B). Hence, the TSC curves of these competitors account for two independent phenomena. The almost complete recovery of the radioligand binding at low initial rimonabant and clozapine concentrations should point to their relatively fast dissociation from their receptors. For rimonabant, this was confirmed by the direct dissociation experiments with its tritiated equivalent (Figure 6D). For clozapine, this is in tune with its classification as an atypical antipsychotic (Kapur and Seeman, 2000; 2001;). At higher initial concentrations, enough free rimonabant and clozapine molecules are present in solution to produce, mass-action equilibrium-based inhibition of the radioligand binding. As the resulting shifts of the TSC curves are less pronounced as in case of supiride, raclopride and taranabant, the free rimonabant and clozapine molecules are likely to originate from their release from cellular stores.
Finally, the TSC curves of the D2 receptor antagonists spiperone, haloperidol and (+)-butaclamol overlapped with the reference curves (Figure 5C–E). Although the equivalent washout media were able to decline radioligand binding to fresh cells, the inhibitory activity therein was too weak to explain the overlap. As no upward shift was perceived either, the simplest explanation is that these antagonists dissociate very slowly from their receptors. For spiperone, this explanation is corroborated by the results obtained from the ‘delayed association’ and direct [3H]-spiperone dissociation experiments (Table 2).
Very similar modes of behaviour were recently also reported to take place for β2 adrenoceptor agonists in related functional experiments on intact cells (Summerhill et al., 2008). In that study, β2 adrenoceptor-expressing CHO cells were also pre-incubated with increasing concentrations of agonist, briefly washed and (instead of challenging with radioligand such as in the present studies) further incubated with fresh medium to monitor post-wash cyclic AMP production. Compared with the reference concentration–response curves in unwashed conditions, the curves for the washed cells experienced a pronounced rightward shift for the hydrophilic agonist salbutamol, a moderate shift for formoterol and no shift along with a nearly unchanged maximal response for salmeterol.
Formoterol possesses adequate lipophilic properties to permit its reversible partitioning between the cell plasma membrane and the surrounding aqueous phase (Rhodes et al., 1992; Bergendal et al., 1996). Hence, when formoterol-pretreated cells or tissues are briefly washed, membrane-associated formoterol will progressively leak into the aqueous medium from where it can reach and stimulate its receptor (Johnson and Coleman, 1995; Johnson, 2001). According to the dedicated ‘diffusion microkinetic’ model, the membrane thus acts as a reservoir whose size depends on the concentration of formoterol initially added (Anderson, 1993; Anderson et al., 1994). Additionally, formoterol-mediated responses are rapidly reversed upon addition of antagonist molecules (Anderson, 1993). This points at rapid dissociation of the formoterol–receptor complexes. In view of the marked lipophilicity of rimonabant and clozapine (Norman et al., 1979; Härtter et al., 2003; Fan et al., 2006), the limited shift of their TSC curves could also be attributed to their reversible partitioning between the cell plasma membrane and the surrounding aqueous phase. In this respect, TSC binding experiments similar to the ones examined here have recently been performed for xanomeline-related M1 muscarinic receptor agonists both on intact CHO cells and membrane preparations thereof (Jakubik et al., 2003; Kane et al., 2007). Attention was focused on the rightward shift of the curves and the important influence of the cell membrane, including ionic interactions with the polar phospholipid headgroups, was clearly demonstrated.
Release of membrane-associated competitor molecules into the medium will result in a new mass-action equilibrium binding between these molecules and the receptor. This phenomenon is hinted at when the TSC curves are only moderately shifted to the right of the reference curve. In the ‘delayed association’ experiments, this new equilibrium may cause a pronounced, long-lasting blunting of the radioligand binding (see simulations in Figure 7B). The resulting radioligand association curves are no longer adequately analysed by the non-linear regression fitting procedure developed by Malany et al. (2009) and, as illustrated by recent ‘delayed association’ studies with clozapine (Packeu et al., 2010a) and AT1 angiotensin II receptor antagonists (Kakuta et al., 2005), this could even lead to the erroneous conclusion that the competitor undergoes long-lasting binding to the receptor.
The unshifted TSC curves for spiperone, haloperidol and (+)-butaclamol in the present study are comparable with the behaviour of salmeterol (Summerhill et al., 2008). This β2 adrenoceptor agonist is highly lipophilic and, as its activity is rapidly reversed in presence of an excess of hydrophilic antagonists, its post-wash long-lasting activity in organ bath experiments and its ability to re-activate the receptors after antagonist removal has been attributed to its long-lasting presence in the cell membrane along with its ability to reach the receptor's active site through lateral diffusion across its transmembrane-spanning α-helices. Whether this long-lasting effect is only governed by the ‘diffusion microkinetic’ model (Austin et al., 2003) or also requires additional mechanisms like exosite binding (Coleman et al., 1996) or rebinding (Vauquelin and Szczuka, 2007; Szczuka et al., 2009) is still matter of debate. In contrast, [3H]-spiperone-D2 receptor binding is long-lasting in intact cells, even in the presence of a receptor-saturating concentration of raclopride (Packeu et al., 2010b) and this provides the simplest explanation for its unshifted TSC curve. Similar kinetic properties can provisionally also be attributed to haloperidol and (+)-butaclamol.
In conclusion, the presently explored TSC approach constitutes a practical alternative to the ‘delayed association’ approach for estimating the dissociation rate of unlabelled competitive ligands in radioligand binding experiments on plated cells. It provides concurrently, at least preliminary indications about the competitor's affinity, its dissociation rate and, above all, its ability to partition between the medium and ‘sinks’ like cell membranes. Competitor k4 values are conveniently calculated from the upward shift of the TSC curve, in case of minor partitioning or, otherwise, already approximated by visual inspection of the TSC curve.
Acknowledgments
Chinese hamster ovary cells (CHO-K1) stably transfected with the cDNA for apo-aquorin of Aquoria victoria, the GTP-binding protein Gα16 (CHO-AEQ cells) were kindly donated by Dr M. Detheux (Euroscreen s.a., Gosselies, Belgium). We are very much indebted to the radiolabelling group at AstraZeneca Mölndal for providing us the radiolabelled cannabinoid receptor antagonists and to Dr Niklas Larsson and his group for developing the HEK293-CB1r cell line.
Glossary
Abbreviations
- CHO-D2S cells
recombinant Chinese hamster ovary cells stably expressing the human D2S dopamine receptor
- HEK293-CB1r cells
recombinant human embryonic kidney cells stably expressing human Cannabinoid CB1 receptor
- IC50
concentration at which the competitor produces half-maximal inhibition of specific radioligand binding
- k4
dissociation rate constant of an unlabelled competitor
- TSC
two-step competition
Conflicts of interest
None to declare.
References
- Aiyar N, Baker E, Vickery-Clark L, Ohlstein EH, Gellai M, Fredrickson TA, et al. Pharmacology of a potent long-acting imidazole-5-acrylic acid angiotensin AT1 receptor antagonist. Eur J Pharmacol. 1995;283:63–72. doi: 10.1016/0014-2999(95)00287-u. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC) Br J Pharmacol. 2009;158(Suppl. 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson GP. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective β2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–2160. doi: 10.1016/0024-3205(93)90729-m. [DOI] [PubMed] [Google Scholar]
- Anderson GP, Lindén A, Rabe KF. Why are long-acting beta-adrenoceptor agonists long-acting? Eur Respir J. 1994;7:569–578. doi: 10.1183/09031936.94.07030569. [DOI] [PubMed] [Google Scholar]
- Anthes JC, Gilchrest H, Richard C, Ecke lS, Hesk D, West RE, Jr, et al. Biochemical characterization of desloratadine, a potent antagonist of the human histamine H(1) receptor. Eur J Pharmacol. 2002;449:229–237. doi: 10.1016/s0014-2999(02)02049-6. [DOI] [PubMed] [Google Scholar]
- Austin RP, Barton P, Bonnert RV, Brown RC, Cage PA, Cheshire DR, et al. QSAR and the rational design of long-acting dual D2-receptor/beta2-adrenoceptor agonists. J Med Chem. 2003;46:3210–3320. doi: 10.1021/jm020886c. [DOI] [PubMed] [Google Scholar]
- Bergendal A, Lindén A, Skoogh B-E, Gerspacher M, Anderson GP, Lolfdahl C-G. Salmeterol mediated reassertion of relaxation persists in guinea-pig trachea pretreated with aliphatic side chain structural analogues. Br J Pharmacol. 1996;117:1009–1015. doi: 10.1111/j.1476-5381.1996.tb16690.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el Bizri H, Clarke PB. Blockade of nicotinic receptor-mediated release of dopamine from striatal synaptosomes by chlorisondamine administered in vivo. Br J Pharmacol. 1994;111:414–418. doi: 10.1111/j.1476-5381.1994.tb14750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower PR. Granisetron: relating pharmacology to clinical efficacy. Support Care Cancer. 2003;11:93–100. doi: 10.1007/s00520-002-0410-z. [DOI] [PubMed] [Google Scholar]
- Coleman RA, Johnson M, Nials AT, Vardey CJ. Exosites: their current status, and their relevance to the duration of action of long-acting beta 2-adrenoceptor agonists. Trends Pharmacol Sci. 1996;17:324–330. [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug-target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- De Arriba AF, Gomez-Casajus LA, Cavalcanti F, Almansa C, Garcia-Rafanel J, Forn J. In vitro pharmacological characterization of a new selective angiotensin AT1 receptor antagonist, UR 7280. Eur J Pharmacol. 1996;318:341–347. doi: 10.1016/s0014-2999(96)00794-7. [DOI] [PubMed] [Google Scholar]
- Dowling MR, Charlton SJ. Quantifying the association and dissociation rates of unlabelledantagonists at the muscarinic M3 receptor. Br J Pharmacol. 2006;148:927–937. doi: 10.1038/sj.bjp.0706819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan H, Raver HT, Holt DP, Dannals R, Horti AG. Synthesis of 1-(2,4 dichlorophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide ([11C]JHU75528) and 1-(2-bromophenyl)-4-cyano-5-(4-[11C]methoxyphenyl)-N-(piperidin-1-yl)-1H-pyrazole-3-carboxamide([11C]JHU75575) as potential radioligands for PET imaging ofα cerebral cannabinoid receptor. J Label Compd Radiopharm. 2006;49:1021–1036. [Google Scholar]
- Fierens FLP, Vanderheyden PML, De Backer J-P, Vauquelin G. Binding of the antagonist [3H]candesartan to angiotensin II AT1 receptor-transfected Chinese hamster ovary cells. Eur J Pharmacol. 1999;367:413–422. doi: 10.1016/s0014-2999(98)00965-0. [DOI] [PubMed] [Google Scholar]
- Hara M, Kiyama R, Nakajima S, Kawabata T, Kawakami M, Ohtani K, et al. Kinetic studies on the interaction of nonlabelled antagonists with the angiotensin II receptor. Eur J Pharmacol. 1995;289:267–273. doi: 10.1016/0922-4106(95)90103-5. [DOI] [PubMed] [Google Scholar]
- Härtter S, Hüwel S, Lohmann T, Abou El Ela A, Langguth P, Hiemke C. How does the benzamide antipsychotic amisulpride get into the brain? An in vitro approach comparing amisulpride with clozapine. Neuropsychopharmacology. 2003;28:1916–1922. doi: 10.1038/sj.npp.1300244. [DOI] [PubMed] [Google Scholar]
- Heise CE, Sullivan S, Crowe PD. Scintillation proximity assay as high-throughput method to identify slowly dissociating nonpeptide ligand binding to the GnRH receptor. J Biomol Screen. 2007;12:235–239. doi: 10.1177/1087057106297362. [DOI] [PubMed] [Google Scholar]
- Jakubik J, Tucek S, El-Fakahany EE. Role of receptor ptoein and membrane lipids in xanomeline wash-resistent binding to muscarinic M1 receptors. J Pharmacol Exp Ther. 2003;308:105–110. doi: 10.1124/jpet.103.058594. [DOI] [PubMed] [Google Scholar]
- Johnson M. Beta2-adrenoceptors: mechanisms of action of beta2-agonists. Paediatr Respir Rev. 2001;2:57–62. doi: 10.1053/prrv.2000.0102. [DOI] [PubMed] [Google Scholar]
- Johnson M, Coleman RA. Mechanisms of action of β2-adrenoceptor agonists. In: Busse WW, Holgate ST, editors. Asthma & Rhinitis. Cambridge: Blackwell; 1995. pp. 1278–1295. [Google Scholar]
- Kakuta H, Sudoh K, Sasamata M, Yamagashi S. Telmisartan has the strongest binding affinity to angiotensin II type 1 receptor: comparison with other angiotensin II type 1 receptor blockers. Int J Clin Pharmacol. 2005;25:41–46. [PubMed] [Google Scholar]
- Kane BE, Gramt MKO, El-Fakahany EE, Ferguson DM. Synthesis and evaluation of xanomeline analogs-probing the wash-resistant phenomenon at M1 muscarinic acdetylcholine receptor. Bioorg Med Chem. 2007;16:1376–1392. doi: 10.1016/j.bmc.2007.10.058. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. Antipsychotic agents differ in how fast they come off the dopamine D2 receptors. Implications for atypical antipsychotic action. J Psychiatry Neurosci. 2000;25:161–166. [PMC free article] [PubMed] [Google Scholar]
- Kapur S, Seeman P. Does fast dissociation from the dopamine d(2) receptor explain the action of atypical antipsychotics?: a new hypothesis. Am J Psychiatry. 2001;158:360–369. doi: 10.1176/appi.ajp.158.3.360. [DOI] [PubMed] [Google Scholar]
- Kukkonen JP, Huifang G, Jansson JC, Wurster S, Cockroft V, Salvola JM, et al. Different apparent modes of inhibition of alpha2A-adrenoceptor by alpha2-adrenoceptor antagonists. Eur J Pharmacol. 1997;335:99–105. doi: 10.1016/s0014-2999(97)01180-1. [DOI] [PubMed] [Google Scholar]
- Malany S, Hernandez LM, Smith WF, Crowe PD, Hoare SRJ. Analytical method for simultaneously measuring ex vivo drug receptor occupancy and dissociation rate: application to (R)-dimethindene occupancy of central histamine H1 receptors. J Recept Signal Transduct Res. 2009;29:84–93. doi: 10.1080/10799890902721339. [DOI] [PubMed] [Google Scholar]
- Morsing P, Adler G, Brandt-Eliasson U, Karp L, Ohlson K, Renberg L, et al. Mechanistic differences of various AT1-receptor blockers in isolated vessels of different origin. Hypertension. 1999;33:1406–1413. doi: 10.1161/01.hyp.33.6.1406. [DOI] [PubMed] [Google Scholar]
- Motulsky HJ, Mahan LC. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol Pharmacol. 1984;25:1–9. [PubMed] [Google Scholar]
- Norman JA, Drummond AH, Moser P. Inhibition of calcium-dependent regulator-stimulated phosphodiesterase activity by neuroleptic drugs is unrelated to their clinical efficacy. Mol Pharmacol. 1979;16:1089–1094. [PubMed] [Google Scholar]
- Ojima M, Inada Y, Shibouta Y, Wada T, Sanada T, Kubo K, et al. Candesartan (CV-11974) dissociates slowly from the angiotensin AT1 receptor. Eur J Pharmacol. 1997;319:137–146. doi: 10.1016/s0014-2999(96)00837-0. [DOI] [PubMed] [Google Scholar]
- Packeu A, De Backer J-P, Van Liefde I, Vanderheyden P, Vauquelin G. Antagonist- dopamine D2L- receptor interactions in intact cells. Biochem Pharmacol. 2008;75:2192–2203. doi: 10.1016/j.bcp.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Packeu A, Béghin T, De Backer J-P, Vauquelin G. Antagonist-D2S-dopamine receptor interactions in intact Chinese recombinant ovary cells. Fund Clin Pharmacol. 2010a;24:293–303. doi: 10.1111/j.1472-8206.2009.00777.x. [DOI] [PubMed] [Google Scholar]
- Packeu A, De Backer J-P, Vauquelin G. Non-competitive interaction between raclopride and spiperone on human D2L-receptors in intact recombinant Chinese Hamster Ovary cells. Fund Clin Pharmacol. 2010b;24:283–291. doi: 10.1111/j.1472-8206.2009.00793.x. [DOI] [PubMed] [Google Scholar]
- Rhodes DG, Newton R, Butler R, Herbette L. Equilibrium and kinetic studies of the interactions of salmeterol with membrane bilayers. Mol Pharmacol. 1992;42:596–602. [PubMed] [Google Scholar]
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebocontrolled trials. Am J Geriatr Psychiatry. 2006;14:191–210. doi: 10.1097/01.JGP.0000200589.01396.6d. [DOI] [PubMed] [Google Scholar]
- Seeman P, Chau-Wong M, Tedesco J, Wong K. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C, Rahman T, Toohey N, Mazurkiewicz J, Herrick-Davis K, Teitler M. Risperidone Irreversibly Binds to and Inactivates the h5-HT7 Serotonin Receptor. Mol Pharmacol. 2006;70:1264–1270. doi: 10.1124/mol.106.024612. [DOI] [PubMed] [Google Scholar]
- Summerhill S, Stroud T, Nagendra R, Perros-Huguet C, Trevethik M. A cell-based asay to asess the persistence of action of agonists acting at recombinant human β2 adrenoceptors. J Pharmacol Toxicol Methods. 2008;58:189–197. doi: 10.1016/j.vascn.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nat Rev Drug Discov. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- Swinney DC. Can Binding Kinetics Translate to a Clinically Differentiated Drug? From Theory to Practice. Lett Drug Des Discov. 2006;3:569–574. [Google Scholar]
- Szczuka A, Packeu A, Wennerberg M, Vauquelin G. Molecular mechanism of the persistent bronchodilatory effect of the partial β2-adrenoceptor agonist salmeterol. Br J Pharmacol. 2009;158:183–194. doi: 10.1111/j.1476-5381.2009.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans PBMWM. Pharmacological properties of angiotensin II receptor antagonists. Can J Cardiol. 1999;15:16F–28F. [PubMed] [Google Scholar]
- Tummino PJ, Copeland RA. Residence time of receptor-ligand complexes and its effect on biological function. Biochemistry. 2008;47:5481–5492. doi: 10.1021/bi8002023. [DOI] [PubMed] [Google Scholar]
- Unger T. Significance of angiotensin type 1 receptor blockade: why are angiotensin II receptor blockers different? Am J Cardiol. 1999;84:9S–15S. doi: 10.1016/s0002-9149(99)00728-6. [DOI] [PubMed] [Google Scholar]
- Vanderheyden PML, Fierens FLP, De Backer J-P, Vauquelin G. Reversible and syntopic interaction between angiotensin II AT1 receptor antagonists and human AT1 receptors expressed in CHO-K1 cells. Biochem Pharmacol. 2000;59:927–935. doi: 10.1016/s0006-2952(99)00403-7. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Szczuka A. Kinetic versus allosteric mechanisms to explain insurmountable antagonism and delayed ligand dissociation. Neurochem Int. 2007;51:254–260. doi: 10.1016/j.neuint.2007.05.005. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I. Slow antagonist dissociation and long-lasting in vivo receptor protection. Trends Pharmacol Sci. 2006;27:356–359. doi: 10.1016/j.tips.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Fierens F, Verheyen I, Vanderheyden P. Insurmountable AT1 receptor antagonism: the need for different antagonist binding states of the receptor. Trends Pharmacol Sci. 2001;22:343–344. doi: 10.1016/s0165-6147(00)01739-9. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, Van Liefde I, Vanderheyden P. Models and methods for studying insurmountable antagonism. Trends Pharmacol Sci. 2002;23:514–518. doi: 10.1016/s0165-6147(02)02081-3. [DOI] [PubMed] [Google Scholar]
- Wennerberg M, Ballendran A, Clapham J, Vauquelin G. Unravelling the complex dissociation of [3H]-rimonabant from plated CB1 cannabinoid receptor-expressing cells. Fund Clin Pharmacol. 2010;24:181–187. doi: 10.1111/j.1472-8206.2009.00756.x. [DOI] [PubMed] [Google Scholar]
- Wienen W, Hauel N, Van Meel JCA, Narr B, Ries U, Entzeroth M. Pharmacological characterization of the novel nonpeptide angiotensin II receptor antagonist, BIBR 277. Br J Pharmacol. 1993;110:245–252. doi: 10.1111/j.1476-5381.1993.tb13800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]