

Abstract
BACKGROUND AND PURPOSE
Muscarinic and adrenergic G protein-coupled receptors (GPCRs) are the targets of rare peptide toxins isolated from snake or cone snail venoms. We used a screen to identify novel toxins from Dendroaspis angusticeps targeting aminergic GPCRs. These toxins may offer new candidates for the development of new tools and drugs.
EXPERIMENTAL APPROACH
In binding experiments with 3H-rauwolscine, we studied the interactions of green mamba venom fractions with α2-adrenoceptors from rat brain synaptosomes. We isolated, sequenced and chemically synthesized a novel peptide, ρ-Da1b. This peptide was pharmacologically characterized using binding experiments and functional tests on human α2-adrenoceptors expressed in mammalian cells.
KEY RESULTS
ρ-Da1b, a 66-amino acid peptide stabilized by four disulphide bridges, belongs to the three-finger-fold peptide family. Its synthetic homologue inhibited 80% of 3H-rauwolscine binding to the three α2-adrenoceptor subtypes, with an affinity between 14 and 73 nM and Hill slopes close to unity. Functional experiments on α2A-adrenoceptor demonstrated that ρ-Da1b is an antagonist, shifting adrenaline activation curves to the right. Schild regression revealed slopes of 0.97 and 0.67 and pA2 values of 5.93 and 5.32 for yohimbine and ρ-Da1b, respectively.
CONCLUSIONS AND IMPLICATIONS
ρ-Da1b is the first toxin identified to specifically interact with α2-adrenoceptors, extending the list of class A GPCRs sensitive to toxins. Additionally, its affinity and atypical mode of interaction open up the possibility of its use as a new pharmacological tool, in the study of the physiological roles of α2-adrenoceptor subtypes.
Keywords: three-finger-fold toxins, binding experiments, α2-adrenoceptor antagonists, venom fractionation, mass fragmentation, snake venoms
Introduction
Animal venoms represent a vast library of peptide toxins. With 1400 species of scorpions, 400 species of venomous snakes, 600 species of sea cone snails and 35 000 species of spiders, each with venoms containing more than 300 different toxins, this natural venom bank consists of more than 10 million peptide toxins, biochemically stable and with particular pharmacological properties. Currently, almost 1600 of these peptides have been sequenced (representing less than 0.02% of the natural bank), and this number is growing exponentially (King et al., 2008). The ability to sequence toxins directly from mass spectrometry analysis of venoms, together with sequencing the genomes of venomous animals (Putnam et al., 2007), will further accelerate the rate of peptide toxin discovery over the next few years. The pharmacological activity of toxins mainly arises from their action at ion channels, such as voltage-gated and ligand-gated ion channels, which are important in controlling the mobility of the venomous animal's prey (Terlau and Olivera, 2004). Thus, toxins targeting ion channels became valuable tools to study neurotransmission or neuromuscular junction and for labelling specific ion channels. Due to their high affinity and selectivity for various ion channels, some animal toxins are being used as drugs or are being tested in several preclinical trials (Becker and Terlau, 2008; Han et al., 2008; Halai and Craik, 2009). For example, Prialt is a ω-conotoxin, isolated from the venom of the cone snail species Conus magus, which selectively blocks N-type voltage-gated calcium channels and is the only approved non-opioid therapy for severe chronic pain (Williams et al., 2008). Because venomous animals are predators that can quickly immobilize and kill their prey, it has been logical to consider only ion channels as the primary targets of toxins. Toxins are thus usually identified and characterized according to their toxicity (Terlau and Olivera, 2004). Consequently, very few alternative targets for toxins have been identified, and this includes the G protein-coupled receptors (GPCR) for which no extensive study has been performed. Of the 1600 known toxins, less than 30 are active at GPCRs and these can be divided into two families. Members of the first family mimic the natural agonist of the receptor target and include the snake sarafotoxins, functional analogs of the endogenous endothelins (Ducancel, 2005), the cone snail toxin conopressin, similar to the arginine-vasopressin peptide (Cruz et al., 1987), or the cone snail toxin contulakin-G, similar to the neurotensin peptide (Craig et al., 1999). The second family includes only 12 different toxins which are all highly reticulated peptides with folds unrelated to natural ligands. Nine are active at muscarinic receptors and have been isolated from mamba venoms (Servent and Fruchart-Gaillard, 2009) and three are active at adrenergic receptors: ρ-TIA, from the conus tulipa, specific to the α1B-adrenoceptor (Sharpe et al., 2001), β-cardiotoxin, from the snake Ophiophagus Hannah, active at β-adrenoceptors (Rajagopalan et al., 2007) and ρ-Da1a (previously called AdTx1), from the snake Dendroaspis angusticeps, specific to the α1A-adrenoceptor (Quinton et al., 2010). Due to their size and peptide nature, toxins acting on aminergic receptors display a range of atypical modes of action, including competitive and non-competitive interactions, insurmountable antagonism and negative allosterism. These toxins can be used to investigate the pharmacological and functional properties of their receptor targets and could also open new avenues for drug development (Harvey et al., 2002; Chen et al., 2004; Kukkonen et al., 2004; Ducancel, 2005; Fruchart-Gaillard et al., 2006; Antosova et al., 2009). GPCRs are the molecular targets for almost half of currently used therapeutic drugs.
We have been studying green mamba venom to discover new toxins acting on aminergic GPCRs. Among the 300 different toxins detected by mass analysis in this venom (N. Gilles, unpubl. data), less than 10% are pharmacologically characterized. The first one, isolated more than 20 years ago (Adem et al., 1988) and the last one identified 10 years later (Carsi et al., 1999), interact with various subtypes of muscarinic acetylcholine receptors. We hypothesized that this venom contains more toxins active on GPCRs and recently discovered the first toxin specific for the α1A-adrenoceptor, which was initially called AdTx1 and then renamed according to a rational nomenclature ρ-Da1a (King et al., 2008; Quinton et al., 2010). Our strategy was based on the successive purification of venom associated with binding experiments using specific radiolabelled ligands and approximately 20 different receptors preparations. In this study, we described the identification of a novel peptide, called ρ-Da1b, which displays nanomolar affinities for the three α2-adrenoceptors subtypes and which antagonizes α2A-adrenoceptor activation non-competitively.
Methods
Venom fractionation
One gram of Dendroaspis angusticeps venom (Latoxan, Valence, France) was separated into 13 fractions by ion exchange (2 × 15 cm) on Source 15S using a protocol as previously described (Quinton et al., 2010). Fraction H was purified by reverse-phase chromatography (Waters 600, Milford, MA, USA) on a preparative column (C18, 15 µm, 20 cm, Vydac, Sigma-Aldrich, Saint Quentin Fallavier, France, 10 mL·min−1), using a linear gradient from 0 to 100% acetonitrile and 0.1% trifluoroacetic acid in 100 min. Fraction D was further purified on an analytical HPLC C18 Vydac column (4.6 mm, 5 µm, 15 cm 1 mL·min−1) using a gradient of 0.5% acetonitrile·min−1.
Protein quantification
Fraction A protein concentration and membrane protein concentration were determined using the Bio-Rad (Hercules, CA, USA) protein assay, with bovine serum albumin (BSA) as standard.
Edman sequencing
N-terminal sequencing of fraction A (100 pmol loaded on a Biobrene-coated filter) was carried out using Edman chemistry on a Procise Model 492 automatic sequencer from Applied Biosystems (Foster City, CA, USA).
Mass spectrometry analysis
Mass spectrometry was carried out with a 7-T APEX III FT-ICR mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with a 7 Tesla magnet. A voltage of −700 V was applied between the nano-electrospray needles (Proxeon, Odense, Denmark) and the entrance of the glass capillary, for ion transfer at a temperature of 50°C. Mass spectra were acquired from m/z 200 to 3000 with 512 k data points. Monoisotopic peaks were labelled using XMASS 6.1.4 software (Bruker Daltonics). Peptides were sequenced by MALDI-‘in-source decay’, peptide mass fingerprinting and MS/MS experiments on an ULTRAFLEX II MALDI-TOF/TOF (Bruker Daltonics) mass spectrometer equipped with a Nd-YAG Smartbeam laser (MLN 202, LTB). 1,5-Diaminonaphthalene saturated in acetronitrile/formic acid 0.1% 50/50 (v/v) was used for in-source decay and 2,5-dihydroxybenzoic acid at 20 mg·mL−1 in acetronitrile/formic acid 0.1% 50/50 (v/v) was used for peptide mass fingerprinting and MS/MS experiments. ρ-Da1b (about 70 ng) was reduced by 5 µL of 100 mM dithiothreitol in 100 mM NH4HCO3 for 1 h at 50°C and then alkylated by 1 µL of 500 mM iodoacetamide for 45 min in the dark before hydrolysis with 1 ng of trypsin (sequencing grade, from bovine pancreas), overnight at 37°C. The resulting peptides were desalted using a Zip-Tip C18 microcolumn (Millipore, Billarica, MA, USA), spotted on the DHB matrix and analyzed by MALDI-TOF/TOF.
Synthesis of ρ-Da1b
ρ-Da1b was synthesized on an Applied Biosystems 433A peptide synthesizer, purified and folded according to the method described for the muscarinic toxin MT1 (Mourier et al., 2003). Briefly, this involved solid-phase synthesis using a Fmoc strategy, peptide cleavage and purification on a reverse-phase column. The linear peptide was then folded at 4°C for 48 h in the presence of glycerol (25%) and oxidized and reduced glutathione (1 mM) in Tris buffer at pH 8.
Animals
All animal care and experimental procedures were in compliance with French legislation, following the European Council Directive 86/609/EEC regarding the protection of animals used for experimental purposes.
Toxins and molecular targets nomenclature
Receptor nomenclature follows Alexander et al. (2009). Toxin nomenclature follows the recent suggestions from the International Society on Toxinology (Tytgat et al., 1999; King et al., 2008).
Binding analysis
Rat brain synaptosomes (RBS) were prepared from adult albino Sprague Dawley rats (Favreau et al., 2001). After dissection, the entire brain was homogenized in ice-cold 0.3 M mannitol buffer containing 10 mM EDTA and 10 mM HEPES-Tris, pH 7.4. After centrifugation at 1000×g for 10 min, the supernatant was removed and centrifuged at 27 000×g for 30 min (P2 pellet). All buffers contained a cocktail of protease inhibitors composed of: phenylmethylsulphonyl fluoride (50 mg·mL−1), pepstatin A (1 mM), iodoacetamide (1 mM) and 1 mM f 1,10-phenanthroline. Human α2-adrenoceptors and α1-adrenoceptors subtypes were stably expressed in mammalian CHO cells kindly provide by Dr Paris, INSERM 858, Toulouse, France and Dr Cottechia, Lausanne University, Switzerland, respectively. Membranes from CHO cells were prepared as described previously (Fruchart-Gaillard et al., 2006). Membranes from cells expressing β-adrenoceptors were purchased from PerkinElmer (Courtaboeuf, France). Binding experiments were performed in 96-well plates. Reaction mixtures contained 50 mM Tris–HCl, pH 7.4, 10 mM MgCl2 and 1 g·L−1 BSA in a final volume of 100 µL. Plates were incubated for 16 h (except for the kinetic experiments) at room temperature. Binding reactions were stopped by filtration through a GF/C filter pre-soaked in 0.5% polyethyleneimine on a cell harvester (PerkinElmer) and plates were dried. Ultimagold O (25 µL; PerkinElmer) was added to each well and samples were counted using a TopCount counter (PerkinElmer) (Counting yield of 55%). Non-specific binding was measured in the presence of 1 µM prazosin for 3H-prazosin binding, 1 µM yohimbine for 3H-rauwolscine, 3 µM propanolol for 3H-CGP-12177 and was subtracted from the total binding. A one-site inhibition mass action curve was fitted to inhibition binding data using Kaleidagraph (Synergy software, Reading, PA, USA). IC50 values were converted to Ki for competition experiments using the Cheng-Prusoff equation (Cheng and Prusoff, 1973). Equilibrium saturation experiments were performed with various concentrations of radioactive ligand and a constant quantity of membrane. Kinetic experiments were carried out using a constant amount of radioactive ligand and membrane, with various incubation times. The dissociation kinetics of 3H-rauwolscine were determined after addition of 1 µM of rauwolscine and 10 µM of ρ-Da1b. The dissociation rate constant (koff) was determined directly from a first-order plot of ligand dissociation versus time. Binding results are presented as mean ± SD (standard deviation) with n, number of independent experiments.
Bioluminescence resonance energy transfer measurement
We used bioluminescence resonance energy transfer (BRET) to follow the structural changes of the G trimeric protein under receptor activation, as previously described (Gales et al., 2006). BRET signals were measured in HEK293T cells coexpressing Gαi1-Renilla luciferase (RLuc) (RLuc energy donor inserted at position 91 of amino acid sequence of Gβi1), GFP10-Gγ2 (GFP10 energy acceptor inserted at the N-terminus of Gγ2) and in the presence of the complementary Gβ1 subunits and α2A-adrenoceptor. Measurements were conducted in resuspended cells in 96-well microplates (white optiplate; PerkinElmer). BRET readings were collected 1 min after addition of 5 µM coelenterazine 400a (Optima), using Infinite F500 (Tecan group Ltd, Männedorf, Switzerland) that allows sequential integration of signals emitted by GFP10 and RLuc. Results are expressed as the difference in BRET signals measured between the BRET partners in the presence and absence of the α2A-adrenoceptor-agonist UK14304 (10 µM, UK-modulated BRET). Results are expressed as mean ± SD with n, number of independent experiments.
Functional tests on human-α2A-adrenoceptor
Functional tests were carried out in 96-well plates using COS cells transfected with DNA encoding the human α2A-adrenoceptor (33 ng/well) and the ubiquitous G protein GqTop [16.6 ng/well, kindly provided by Dr J.P. Pin, Montpellier, France (Selvam et al., 2010)], using the lipofectamine reagent (according to the manufacture protocol). Forty-eight hours after transfection, cells were incubated for 1 h at 37°C with 10 µL of 10× solutions of the various compounds tested and 100 µL of dye solution (FLIPR Calcium Kit buffer, Molecular Devices, Sunnyvale, CA, USA) supplemented with 2.5 mM probenecid. Changes in fluorescence induced by adrenaline were measured at 37°C using a Flex station I (Molecular Devices), with an excitation wavelength of 485 nm and emission wavelength of 525 nm. The resulting activation curves obtained were analyzed using Softmax Pro software (Molecular Devices). Receptor activation for the different experimental conditions was evaluated using maximum and minimum values and analyzed by Kaleidagraph (Synergy software). Results are expressed as mean ± SD with n, number of independent experiments.
Materials
Radioactive components were from PerkinElmer. All chemical products and cell culture media were from Sigma-Aldrich. Protected amino acid derivatives and resins were from Novabiochem (France Biochem, Meudon, France).
Results
Isolation and characterization of ρ-Da1b
Green mamba venom was separated into 13 fractions by cation exchange chromatography (Figure 1A). We investigated the capacity of each fraction to inhibit 3H-rauwolscine binding to RBS. Fraction H, which significantly reduced 3H-rauwolscine binding, was sub-fractionated by reverse-phase chromatography (Figure 1B). Pharmacological activity was recovered in peak D. This fraction, composed of one major (average mass of 6826 Da) and one minor (average mass of 7512 Da) toxin, was further fractionated by reverse phase chromatography (Figure 1C). The minor fraction A, determined to be around 200 µM concentration, inhibited 3H-rauwolscine binding in a concentration-dependent manner, giving a binding curve with a Hill slope of around 0.75 and with an affinity in the nanomolar range (Figure 1D). The monoisotopic molecular mass of the toxin was, after deconvolution of 7507.507 Da (Figure 2A) and 7515.582 Da, before and after reduction, respectively, indicating the presence of four disulphide bonds. Its sequence was determined by a combination of Edman's degradation (for the 54 first residues) and mass analysis. After reduction, the toxin was fragmented by In-source Decay (Quinton et al., 2007). A c-ion series was generated, allowing characterization of 48 central amino acids out of a total of 66 (73% sequence coverage, results not shown), but leaving the N- and C-terminal ends undetermined. To determine the C-terminal part of the toxin sequence, ρ-Da1b was hydrolyzed by trypsin and the peptide mixtures were analyzed by MALDI-TOF/TOF (Figure 2B). The ion at m/z 1680.68, corresponding to the C-terminus part (red arrow), was selected and fragmented by LIFT-TOF/TOF (Suckau et al., 2003). The fragmentation spectrum (Figure 2C) revealed y and b-ion fragments, allowing the C-terminus sequence to be determined without any ambiguity since the accuracy of mass measurement is less than 0.5 m/z. Determination of the whole sequence thus gave LTCVTKDTIFGITTQNCPAGQNLCFIRRHYINHRYTEITRGCTATCPKPTNVRETIHCCNTDKCNE. This sequence has a theoretical mass (7507.4869 Da) differing by 2.7 ppm from the experimental value. We chemically synthesized, purified and refolded the corresponding peptide, as previously described (Mourier et al., 2003). Comparative analytical HPLC showed that the synthetic linear-reduced peptide (Figure 3, line a) was more hydrophobic than the folded and oxidized form (Figure 3, line b), which was eluted at the same position as the native peptide (Figure 3, line c), as confirmed by co-injection of both peptides (Figure 3, line d). We named this new peptide ρ-Da1b. The Greek letter signifies its activity at adrenoceptors, Da corresponds to the animal genus (Dendroaspis angusticeps), 1 relates to the three-finger fold of the peptide and b to the second homologue toxin, the first one being ρ-Da1a, specific to the α1A-adrenoceptor, previously called AdTx1 (McIntosh et al., 1999; Tytgat et al., 1999; King et al., 2008; Quinton et al., 2010). Protein sequence data reported here have been deposited in the UniProt database under accession number P86419.
Figure 1.
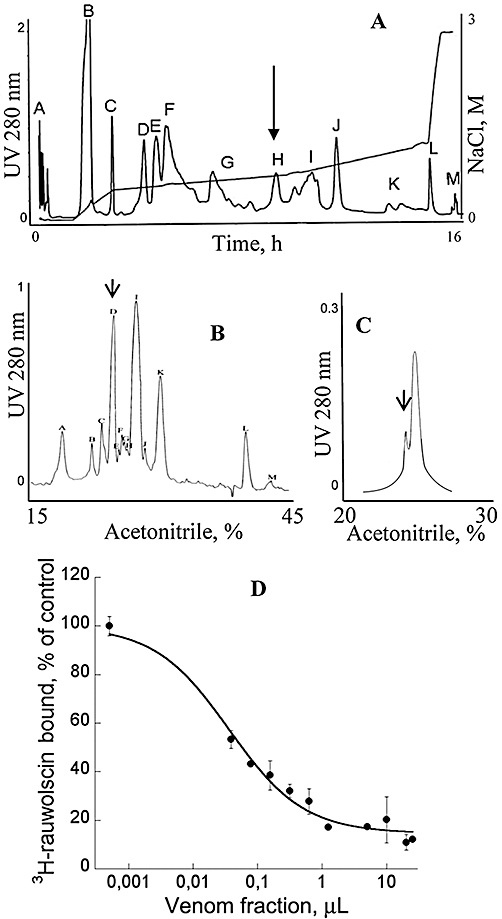
Purification and preliminary pharmacological characterization of ρ-Da1b. (A) Ion-exchange chromatography of Dendroaspis angusticeps crude venom. Labelled peaks were collected (13 fractions). (B) Reverse-phase chromatography of fraction H on a Vydac C18 preparative column. Labelled peaks were collected (20 fractions). Fraction D was eluted at around 27% acetonitrile. (C) Reverse-phase chromatography of fraction D on a Vydac C18 analytical column. Arrows show active peaks. (D) Inhibition of 3H-rauwolscine (1 nM) binding on rat brain synaptosomes by the minor peak purified in C.
Figure 2.
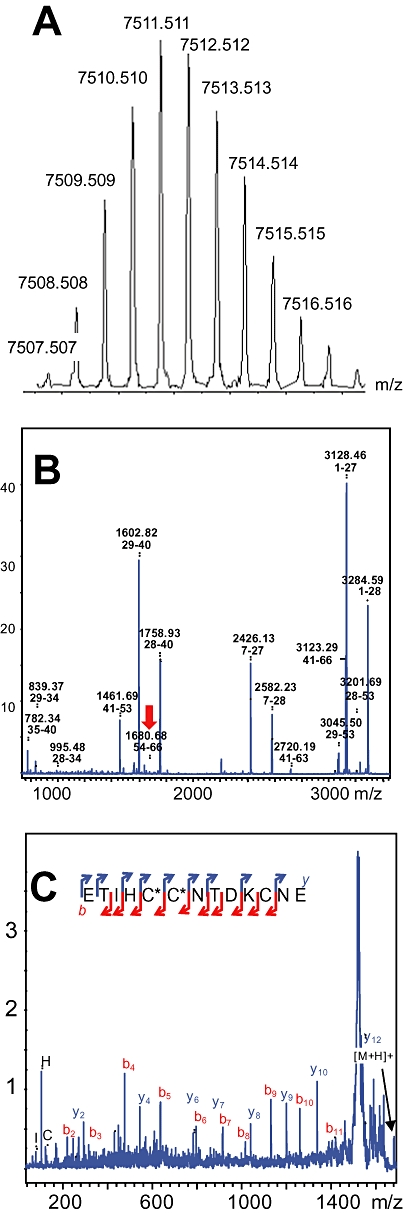
Mass and sequence analysis of ρ-Da1b. (A) Isotopic profile of ρ-Da1b. (B) Peptide mass fingerprint of ρ-Da1b after trypsin treatment. (C) Fragmentation by MALDI-LIFT-TOF/TOF of the m/z 1680.68 ion (red arrow) obtained after trypsin digestion. b- and y-ion types used for the sequencing are indicated in red and blue, respectively.
Figure 3.
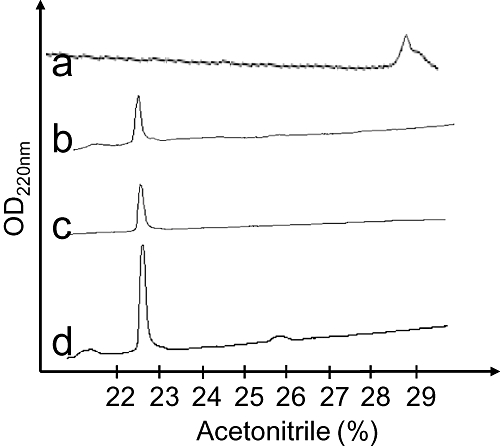
Analytical reverse-phase chromatography of the reduced synthetic ρ-Da1b (line a), oxidized synthetic ρ-Da1b (line b) and natural ρ-Da1b (line c). Line d shows the co-elution of the natural and synthetic oxidized ρ-Da1b.
ρ-Da1b is specific for α2-adrenoceptors
We first characterized the binding of 3H-rauwolscine to the three α2-adrenoceptor subtypes stably expressed in CHO cells. Saturation binding experiments performed with increasing concentrations of 3H-rauwolscine (0.1 to 7 nM) were used to obtain affinities of the tritiated ligand and the number of ligand-binding sites present on the cells expressing the receptor. 3H-rauwolscine had an affinity of 0.83 nM, with a Bmax (in pmol /mg membrane protein) of 1.6 pmol·mg−1, for the hα2A-adrenoceptor; an affinity of 0.97 nM and Bmax of 2.1 pmol·mg−1 for the hα2B subtype; and an affinity of 0.15 nM with a Bmax of 27 pmol·mg−1 for hα2C-adrenoceptors (data not shown). These rauwolscine Kd values were used to calculate Ki values for ρ-Da1b using the Cheng-Prusoff equation. ρ-Da1b inhibited 3H-rauwolscine binding with an IC50 of 28.1 ± 4.1 nM, a Ki value of 14 ± 2 nM and a Hill slope of 1.2 for hα2A-adrenoceptors; an IC50 of 144 ± 12 nM, a Ki value of 73 ± 6 nM and a Hill slope of 0.80 for hα2B-adrenoceptors; and an IC50 of 260 ± 40 nM, a Ki value of 38 ± 6 nM and a Hill slope of 1.0 for hα2C-adrenoceptors (n= 3, Figure 4). To confirm the selectivity of ρ-Da1b for adrenoceptor type, we evaluated its inhibition of 3H-prazosin binding to the three α1-adrenoceptor subtypes and 3H-CGP12177 binding to the three β-adrenoceptors. The toxin inhibited 3H-prazosin binding to the h-α1A-adrenoceptor with an IC50 of 6.4 ± 2.2 µM, corresponding to a Ki of 2.1 ± 0.7 µM and a Hill slope of 0.84 (n= 2, Figure 4) but did not affect binding, at a concentration of 10 µM, for any of the other subtypes tested (data not shown).
Figure 4.
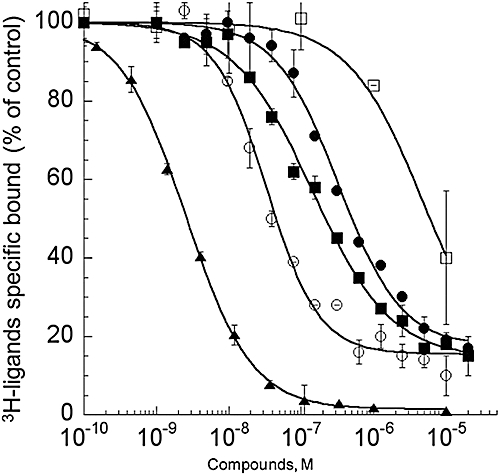
Inhibition binding curves for human adrenoceptors of 3H-rauwolscine (1 nM) in CHO membranes expressing, hα2A-adrenoceptor (12 µg) with yohimbine (▴) and ρ-Da1b (○) and membranes expressing hα2B (11 µg,  ) or hα2C-adrenoceptor (3.1 µg, •) with ρ-Da1b. Inhibition of 3H-prazosin (1 nM) by ρ-Da1b in CHO membranes expressing hα1A-adrenoceptor (3.8 µg).
) or hα2C-adrenoceptor (3.1 µg, •) with ρ-Da1b. Inhibition of 3H-prazosin (1 nM) by ρ-Da1b in CHO membranes expressing hα1A-adrenoceptor (3.8 µg).
Regardless of the α2-adrenoceptor subtype used, ρ-Da1b could not totally abolish 3H-rauwolscine binding, leaving residual binding between 15 and 20%. This contrasts with yohimbine, which is able to fully displace 3H-rauwolscine binding, with a Ki of 0.85 ± 0.01 nM and a Hill slope of 0.96 (Figure 4).
ρ-Da1b prevents α2A-adrenoceptor-induced activation of Gi
Activation of GPCR induces conformational changes in the associated trimeric G protein. This structural reorganization can be studied using a system in which Gαi is fused to the RLuc (Gαi1-RLuc) and Gγ2 is fused to the fluorescent protein GFP (GFP10-Gγ2, Figure 5A). Addition of coelenterazine induces emission of light by RLuc, which leads to the activation of GFP if the two protein constructs are in close proximity, that is, in this case, if the constituents of the trimeric G protein remain associated (Figure 5A). We tested the capacity of yohimbine and ρ-Da1b to activate or antagonize the α2A-adrenoceptor. Receptor activation was not observed upon addition of 10 µM of yohimbine or ρ-Da1b (Figure 5B), whereas 10 µM UK14304 significantly reduced the BRET signal, consistent with its action as a specific α2-adrenoceptor agonist. Cells were then pretreated for 1 h with 10 µM yohimbine or ρ-Da1b and then stimulated with UK14304. Both yohimbine and ρ-Da1b significantly reduced the agonist-induced activation of the α2A-adrenoceptor, demonstrating their antagonist activity at this receptor (n= 3, Figure 5B).
Figure 5.
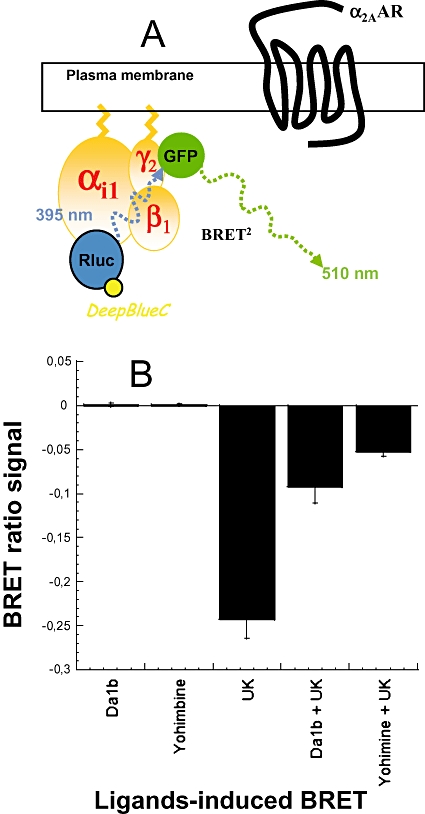
ρ-Da1b prevented α2A-adrenoceptor mediated Gi-inhibition. (A) Construction of the trimeric G protein with Renilla luciferase (RLuc) as a donor and GFP as acceptor link to the αi1 and γ2 subunits, respectively. (B) Cells were first treated directly with 10 µM ρ-Da1b or yohimbine to evaluate their agonistic property. Results are expressed as the difference in bioluminescence resonance energy transfer (BRET) signals measured before and after application of the compounds. Then, cells were stimulated with 10 µM UK14304 alone or after a pre-incubation with 10 µM of ρ-Da1b or yohimbine. Results are expressed as the difference in BRET signals measured before and after the α2-agonist UK14304 stimulation. Data represent the mean ± SD of three independent experiments.
Characterization of the ρ-Da1b antagonist potency on α2A-adrenoceptors
The ρ-Da1b antagonist properties were further evaluated in functional experiments on COS mammalian cells co-expressing the hα2A-adrenoceptor and the chimeric G protein GqTop (Selvam et al., 2010), allowing the cell signal response to be measured by calcium release. Receptor activation was followed with a fluorophore sensitive to the intracellular Ca2+ concentration. Cells were pre-incubated with antagonists (from 0 to 100 µM) for 2 h before addition of adrenaline (1 nM to 300 µM). The EC50 of adrenaline without pre-incubation with antagonist was 0.16 ± 0.03 µM (n= 8). Activation curves shifted to the right with increased antagonist concentration, without changing adrenaline efficacy. The shifts were significantly greater in the presence of yohimbine (EC50 between 0.16 and 12.0 µM, Figure 6A) than in the presence of ρ-Da1b (EC50 between 0.16 and 2.21 µM, Figure 6B). Hill slopes of the activation curves remained close to 1 for yohimbine and ρ-Da1b (Table 1). Schild regressions (Figure 6C) were linear for both antagonists, with a slope close of 0.97 for yohimbine and 0.67 for ρ-Da1b. The pA2 values calculated were 5.93 for yohimbine and 5.32 for ρ-Da1b.
Figure 6.
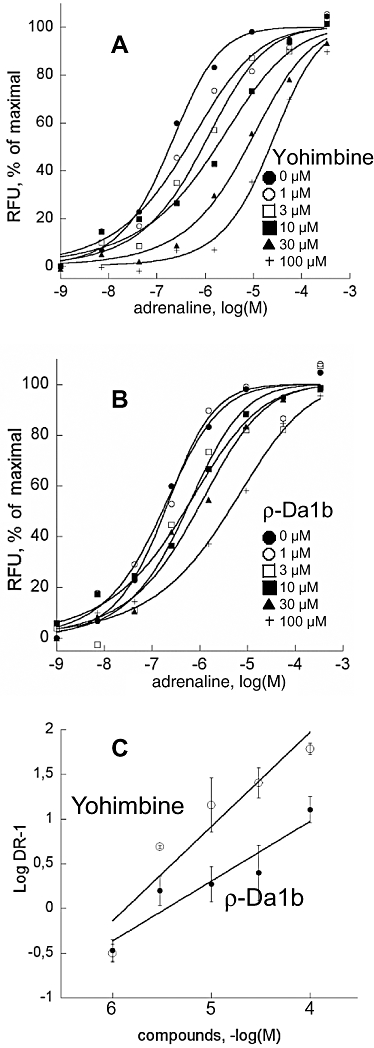
Functional characterization of yohimbine and ρ-Da1b on COS cells co-expressing the hα2A-adrenoceptor and the chimeric G protein GqTop. Concentration–response curves for epinephrine were obtained in the presence of increasing concentrations of yohimbine (A) or ρ-Da1b (B). Error bars have been omitted for clarity. (C) Schild plot representations of the evolution of epinephrine EC50 in the presence of yohimbine (○) or ρ-Da1b (•) were fitted by a linear regression.
Table 1.
Maximal signal (Max, arbitrary units), EC50 (µM) and Hill slope (nH), for activation by adrenaline in the presence of various concentrations of yohimbine and ρ-Da1b
| Antagonists | |||||||
|---|---|---|---|---|---|---|---|
| 0 µM | 1 µM | 3 µM | 10 µM | 30 µM | 100 µM | ||
| Yohimbine | EC50 | 0,16 ± 0,03 | 0,508 ± 0,087 | 0,681 ± 0,37 | 1,61 ± 1,2 | 4,11 ± 1,1 | 12,0 ± 5,8 |
| nH | 1.11 | 0.93 | 0.96 | 0.87 | 0.89 | 0.96 | |
| Max | 6060 ± 1010 | 4306 ± 585 | 6536 ± 150 | 6299 ± 776 | 5151 ± 165 | 5787 ± 660 | |
| n | 8 | 3 | 3 | 6 | 4 | 6 | |
| ρ-Da1b | EC50 | 0.16 ± 0.03 | 0.173 ± 0.038 | 0.419 ± 0.098 | 0.611 ± 0.48 | 0.745 ± 0.56 | 2.21 ± 0.68 |
| nH | 1.11 | 1.04 | 1.01 | 0.89 | 0.92 | 0.81 | |
| Max | 6060 ± 1010 | 6766 ± 186 | 6125 ± 762 | 6414 ± 701 | 6680 ± 1243 | 7232 ± 126 | |
| n | 8 | 3 | 4 | 6 | 6 | 3 | |
n, number of experiments.
Characterization of the mode of action of ρ-Da1b on α2A-adrenoceptors
As described above, ρ-Da1b, in contrast to yohimbine, could not fully inhibit 3H-rauwolscine binding to α2-adrenoceptors and antagonized activation by adrenaline, with a non-competitive mechanism. To further characterize the mode of interaction of ρ-Da1b with α2A-adrenoceptor, we performed additional equilibrium and kinetic binding experiments.
Saturation binding experiments were carried out with COS cells transiently expressing the human α2A-adrenoceptor. We first checked that, using this expression system, ρ-Da1b inhibits 3H-rauwolscine with the same affinity and induces the same amount of residual binding as observed for stable CHO cell lines (data not shown). Saturation binding experiments with 3H-rauwolscine gave an affinity of 2.5 ± 0.2 nM with a membrane capacity of 27 ± 1 pmol of receptor (mg protein)−1. With a large excess of ρ-Da1b (10 µM), 3H-rauwolscine affinity was 4.0 ± 1.1 nM, with a membrane capacity of 7.0 ± 0.5 pmol of receptor (mg protein)−1 (Figure 7A). Thus, the presence of ρ-Da1b did not significantly modify 3H-rauwolscine affinity constant but reduced the number of accessible ligand binding sites at the membrane by a factor of four.
Figure 7.
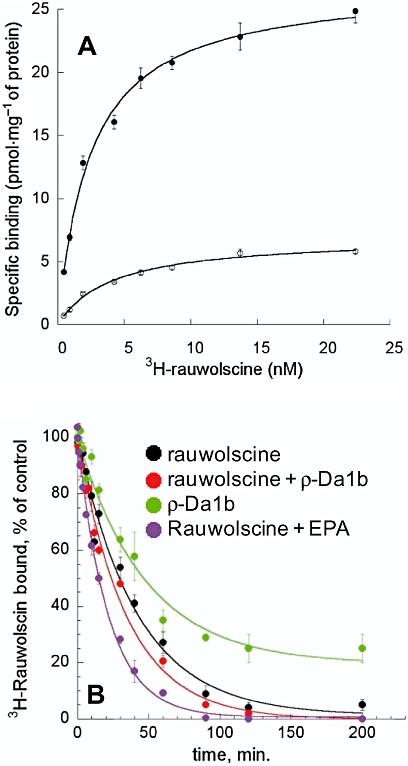
Characterization of the mode of action of ρ-Da1b on α2A-adrenoceptor expressed on CHO cells. (A) Saturation binding experimentof 3H-rauwolscine in the absence (•) or presence of ρ-Da1b (○, 10 µM). (B) Dissociation kinetic rate of 3H-rauwolscine in the presence of rauwolscine (black), rauwolscine plus ρ-Da1b (red), ρ-Da1b (green) and rauwolscine + 5-(N-ethyl-N-isopropyl)-amiloride (EPA) (purple).
Most allosteric modulators affect the orthosteric ligand dissociation rate. We measured the Koff for 3H-rauwolscine (Figure 7B) in the presence of 1 µM of rauwolscine alone (Koff rauwolscine= 0.024 ± 0.005 min−1, n= 4) and in the presence of rauwolscine with ρ-Da1b (10 µM) (Koff rauwolscine+ρ-Da1b= 0.029 ± 0.008 min-1, n= 3) or with 1 mM of 5-N-ethyl-N-isopropyl-amiloride (EPA, Koff rauwolscine+EPA= 0.058 ± 0.004 min-1, n= 3). Whereas the rauwolscine dissociation rate was doubled in the presence of EPA, consistent with previous observations (Leppik et al., 2000), it was not significantly affected by ρ-Da1b. Finally, we measured the 3H-rauwolscine dissociation rate in the presence of 10 µM of ρ-Da1b alone. The kinetic rate obtained under these conditions (Koff ρ-Da1b= 0.021 ± 0.007 min−1, n= 3) was similar to that obtained for rauwolscine alone, suggesting a competitive mechanism of action for the toxin. Additionally, only 80% of 3H-rauwolscine binding was reversed by ρ-Da1b, consistent with equilibrium binding experiments.
Discussion
Screening green mamba venom against GPCR targets enabled us to isolate ρ-Da1b, the second original toxin active on adrenoceptors. Analysis of the ρ-Da1b sequence demonstrated that this toxin belongs to the three-finger-fold toxin family, one of the most common folds found in snake venoms. This fold consists of 61 to 74 residues, reticulated by four conserved disulphide bridges. The tips of the fingers usually constitute the active site, with the palm of the ‘hand’ formed by the four bridges. Some three-finger-fold toxins have an additional disulphide bridge located at the tip of the first or second finger, affecting their pharmacological profiles (Agrawal et al., 1984; Servent et al., 1997). The three-finger-fold toxin was initially described for its activity at nicotinic acetylcholine receptors (Changeux et al., 1970). Since this finding, many other pharmacological activities have been attributed to this toxin family. Most three-finger-fold toxins interact with cholinergic systems, including nicotinic receptors, muscarinic acetylcholine receptors and acetylcholinesterases (Servent and Menez, 2001). However, toxins from this family also interact with coagulation factors (Banerjee et al., 2005), calcium channels (de Weille et al., 1991), phospholipids (Kumar et al., 1997) and integrin receptors (Wu et al., 2006). Recent findings have described two new three-finger-fold toxins, one is active on β-adrenoceptors (Rajagopalan et al., 2007) and the other on α1A-adrenoceptors (Quinton et al., 2010). Indeed, the three-finger-fold toxin family is probably associated with the most diverse range of characterized pharmacological activities for toxins to date.
In this study, the four peptides sharing the highest level of sequence identity (74–77%) with ρ-Da1b were MTα, from Dendroaspis polylepis, and MT3, MT4 and MT1 from Dendroaspis angusticeps (Table 2). These toxins are all active at muscarinic receptors (Joubert, 1985; Jolkkonen et al., 1995, 2001), although MT1 also binds to α1-adrenoceptors (Harvey et al., 2002). The second most similar group of toxins, showing 64 to 67% identity with ρ-Da1b, included ρ-Da1a, MTβ and Cm3. Nothing is known about the pharmacological characteristics of Cm-3, except that it is not toxic at the high dose of 50 mg·kg−1 (Joubert, 1985). MTβ is weakly active at all muscarinic receptor subtypes (Joubert, 1985; Jolkkonen et al., 1995, 2001) and may thus interact with other molecular targets such as the adrenoceptors. ρ-Da1a is specific to the α1A-adrenoceptor (Quinton et al., 2010). The third group, with 54 to 55% identity, contains MT2, MTLP-2 and MT7. MT7 is the only highly specific toxin for the M1-muscarinic receptor subtype (Servent and Fruchart-Gaillard, 2009), whereas MTLP-2 is not active at this receptor. The only other snake toxin described with adrenergic activity is β-cardiotoxin, which antagonizes β1- and β2-adrenoceptors at micromolar concentrations (Rajagopalan et al., 2007). However, this toxin displays only 35% sequence identity with ρ-Da1b. Some toxins acting on muscarinic receptors have a higher sequence identity with ρ-Da1b than those acting on adrenoceptors. This suggests a relationship between these two pharmacological profiles with a potential cross-reactivity of three-finger-fold toxins for muscarinic receptors and adrenoceptors receptor families, as previously shown for the MT1 toxin (Harvey et al., 2002). A detailed analysis of the few residues which interact these toxins with their respective targets initially requires a characterization of their precise pharmacological effects on the various subtypes of muscarinic receptors and adrenoceptors.
Table 2.
Alignment of sequences for ρ-Da1b and various other toxins acting on adrenoceptors and muscarinic receptors
| Edman's degradation | LTXVTKDTIFGITTQNXPAGQNLXFIRRHYINHRYTEITRGXTATXPKPTNVRE------------ | ||
|---|---|---|---|
| Mass sequencing | -----------------–-----------------------------------ETIHCCNTDKCNE | ||
| Loops | -------1----- ---------2------- -----3----- | % | |
| ρ-Da1b | Da | LTCVTKDTIFGITTQNCPAGQNLCFIRRHYINHRYTEITRGCTATCPKPTNVRETIHCCNTDKCNE | |
| MT-α | Dpp | LTCVTSKSIFGITTENCPDGQNLCFKKWYYLNHRYSDITWGCAATCPKPTNVRETIHCCETDKCNE | 77 |
| MT3 | Da | LTCVTKNTIFGITTENCPAGQNLCFKRWHYVIPRYTEITRGCAATCPIPENY-DSIHCCKTDKCNE | 76 |
| MT4 | Da | LTCVTSKSIFGITTENCPDGQNLCFKKWYYIVPRYSDITWGCAATCPKPTNVRETIHCCETDKCNE | 76 |
| MT1 | Da | LTCVTSKSIFGITTENCPDGQNLCFKKWYYIVPRYSDITWGCAATCPKPTNVRETIRCCETDKCNE | 74 |
| MT-β | Dpp | LTCVTSKSIFGITTEDCPDGQNLCFKRRHYVVPKIYDITRGCVATCPIPENY-DSIHCCKTDKCNE | 67 |
| ρ-Da1a | Da | LTCVTSKSIFGITTEDCPDGQNLCFKRRHYVVPKIYDSTRGCAATCPIPENY-DSIHCCKTDKCNE | 66 |
| Cm3 | Dpp | LTCVTSKSIFGITTEDCPDGQNLCFKRRHYVVPKIYDITRGCVATCPIPENY-DSIHCCKTEKCNN | 64 |
| MT2 | Da | LTCVTTKSIGGVTTEDCPAGQNVCFKRWHYVTPKNYDIIKGCAATCPKVDNN-DPIRCCGTDKCND | 55 |
| MT7 | Da | LTCVKSNSIWFPTSEDCPPGQNLCFKRWQYISPRMYDFTRGCAATCPKAEYR-DVINCCGTDKCNK | 54 |
| β-ca | Oh | RKCLNTPLPLIYTT--CPIGQDKCVKMTIKKLPSKYDVIRGCIDICPKSSA-DVEVLCCDTNKCNK | 36 |
Percentage identity, relative to ρ-Da1b, is indicated for each sequence. Residues that differ from ρ-Da1b are in bold. Da, Dendroaspis angusticeps; Dpp, Dendroaspis polylepis polylepis; Nak, Naja kaouthia; β-Ca, β-cardiotoxin; Oh, Ophiophagus hannah.
Very little is known about the molecular interactions between these toxins and their receptor targets, except for MT7, which exploits the tip of its three fingers (Fruchart-Gaillard et al., 2008) to strongly interact with the second external loop of the M1 muscarinic acetylcholine receptor (Kukkonen et al., 2004). In the last decade, few structures of class A GPCRs have been solved (Palczewski et al., 2000; Cherezov et al., 2007; Rasmussen et al., 2007; Jaakola et al., 2008). If structural similarities between GPCRs reside mainly in their transmembrane domains, it is not the case for their external loops that are highly diverse in sequence and spatial organization (Palczewski et al., 2000; Cherezov et al., 2007; Rasmussen et al., 2007; Jaakola et al., 2008). Assuming that all three-finger-fold toxins interact with the external part of their molecular targets (as MT7 does), the structural organization of the external domains of each GPCR should account for the selectivity of the interacting toxin.
ρ-Da1b interacts with hα2-adrenoceptors with an affinity of 14 nM for the α2A-adrenoceptor, showing a weak selectivity, five and three times, for α2B- and α2C-adrenoceptors, respectively. It is the first peptide ligand found to be specific for this receptor subfamily, showing almost no activity at the other adrenoceptors. ρ-Da1b did not fully displace rauwolscine binding, leaving the same level of residual binding on each of the three α2-subtypes. To further investigate this residual binding, we performed 3H-rauwolscine saturation binding experiments in the presence of an excess of the toxin. Rauwolscine was still able to bind to the α2A-adrenoceptor with high affinity in these experiments, but the total number of sites available for binding was reduced by a factor of four. That rauwolscine could still bind receptors saturated with ρ-Da1b and with similar affinity suggests that two populations of receptor are present. Approximately 75% of the total receptor population is sensitive to ρ-Da1b; the remaining 25% is thus resistant to the toxin. We also examined the effect of ρ-Da1b on the dissociation rate of rauwolscine. We did not find a significant effect of the toxin, ruling out a potential allosteric mode of interaction. Additionally, ρ-Da1b added alone resulted in rauwolscine dissociation at the same rate as observed upon addition of yohimbine, confirming the competitive mode of action of the toxin. Moreover, only 80% of its binding was reversible, consistent with our findings from equilibrium binding experiments. Similar results had been previously obtained for ρ-Da1a, which does not fully inhibit 3H-prazosin binding on α1A-adrenoceptor and does not affect 3H-prazosin dissociation rate, suggesting a competitive mode of interaction of ρ-Da1a (Quinton et al., 2010). By contrast, the MT7 toxin, which leaves residual binding at the M1 muscarinic receptor, substantially reduces the dissociation rate for 3H-N-methyl-scopolamine and was described as a potent negative allosteric modulator (Olianas et al., 2000; Mourier et al., 2003).
We used two different strategies for the functional characterization of the ρ-Da1b interaction. The first strategy involved monitoring conformational changes of the trimeric G protein induced upon receptor activation. We monitored this conformational rearrangement between G protein subunits by measuring BRET signal between a donor, the Gα-RLuc and an acceptor, the Gγ-GFP (Gales et al., 2006). This strategy allowed us to evaluate the effects of ρ-Da1b on one of the initial events in activation, leading to the characterization of this peptide toxin as an antagonist. The α2A-adrenoceptor is normally coupled to the Gi and Gs proteins and plays a role in a number of physiological functions. This receptor has been implicated in several disorders of the CNS [epilepsy, depression (Bekker and Sturaitis, 2005)] and others expressed in the periphery, such as diabetes, intestinal motility, cardiac function and pain (Rosengren et al., 2009). We shifted its natural activation pathway to one mediated by a chimeric G protein, Gq-Top (Selvam et al., 2010). This allowed α2A-adrenoceptor activation to be detected and quantified through measurement of calcium release. Yohimbine and ρ-Da1a induced rightward shifts of the activation curves without changing adrenaline efficacy. Schild regressions were clearly linear with a slope that deviated from unity for ρ-Da1b, suggesting that this ligand does not employ a simple competitive mode of action. The pA2 values for yohimbine determined using this system (5.93) were significantly lower than values found in the literature: 6.88 in the pineal gland (Pratt and Takahashi, 1987), 7.25 in the uterine artery (Ribeiro and Macedo, 1986) or 8.4 in the mesenteric artery (Agrawal et al., 1984). This disparity could be due to the use of a chimeric G protein to funnel GPCR signal transduction to a common pathway involving Ca2+ release from intracellular stores. Indeed, shifts in agonist potency and efficacy have previously been shown, for example, upon co-transfection of receptors with Gα16 or with other chimeric G proteins (Kostenis, 2001). Our system may therefore have underestimated the potency of both yohimbine and ρ-Da1b, with pA2 values differing substantially from the affinity constants determined in binding experiments.
Further experiments are needed to check whether ρ-Da1b has similar pharmacological properties on the two other subtypes of α2-adrenoceptor and to determine the origin of the residual binding and non-competitive antagonist properties observed in our competition experiments. Studies are also needed, particularly at the molecular level, to determine more precisely the interaction of ρ-Da1b with α2-adrenoceptors.
Nevertheless, the high affinity and selectivity of ρ-Da1b for this receptor subfamily make it a useful tool in the study of α2-adrenoceptors physiology and in the development of novel drug candidates. Thus, no α2-adrenoceptors antagonist are currently used clinically, although these receptors are involved in several pathologies such as intestinal motility (Blandizzi, 2007), orthostatic hypotension (Pang, 2001) or Parkinson's disease (Brotchie, 2005). This is largely because none of the compounds so far tested are sufficiently selective for α2-adrenoceptors (Crassous et al., 2007). By itself, ρ-Da1b has no activity on any other adrenoreceptor families. In addition, the natural venom fraction H, from which ρ-Da1b was isolated, shows no activity on 20 different GPCRs (N. Gilles, unpubl. data), suggesting that the ρ-Da1b selectivity covers a large number of aminergic GPCR. Finally, the peptide nature of the toxin should not allow it to pass the blood brain barrier, avoiding central side effects. For these reasons, ρ-Da1b has been patented and is under evaluation for treatments of several peripheral diseases.
Acknowledgments
This work was supported by the French National Research Agency (L’Agence Nationale de la Recherche), as part of the programs adrenergicpeptides and gpcrcanotox.
We would like to thank Amelie Goudet for technical assistance and Jean-Philippe Pin (IGF, Montpellier, France) for the generous gift of the GqTop plasmid expression.
Glossary
Abbreviations
- BRET
bioluminescence resonance energy transfer
- EPA
5-(N-ethyl-N-isopropyl)-amiloride
- GPCR
G protein-coupled receptor
- RBS
rat brain synaptosomes
Conflicts of interest
None.
References
- Adem A, Asblom A, Johansson G, Mbugua PM, Karlsson E. Toxins from the venom of the green mamba Dendroaspis angusticeps that inhibit the binding of quinuclidinyl benzilate to muscarinic acetylcholine receptors. Biochim Biophys Acta. 1988;968:340–345. doi: 10.1016/0167-4889(88)90025-0. [DOI] [PubMed] [Google Scholar]
- Agrawal DK, Triggle CR, Daniel EE. Pharmacological characterization of the postsynaptic alpha adrenoceptors in vascular smooth muscle from canine and rat mesenteric vascular beds. J Pharmacol Exp Ther. 1984;229:831–838. [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC). 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antosova Z, Mackova M, Kral V, Macek T. Therapeutic application of peptides and proteins: parenteral forever? Trends Biotechnol. 2009;27:628–635. doi: 10.1016/j.tibtech.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Banerjee Y, Mizuguchi J, Iwanaga S, Kini RM. Hemextin AB complex, a unique anticoagulant protein complex from Hemachatus haemachatus (African Ringhals cobra) venom that inhibits clot initiation and factor VIIa activity. J Biol Chem. 2005;280:42601–42611. doi: 10.1074/jbc.M508987200. [DOI] [PubMed] [Google Scholar]
- Becker S, Terlau H. Toxins from cone snails: properties, applications and biotechnological production. Appl Microbiol Biotechnol. 2008;79:1–9. doi: 10.1007/s00253-008-1385-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekker A, Sturaitis MK. Dexmedetomidine for neurological surgery. Neurosurgery. 2005;57:1–10. doi: 10.1227/01.neu.0000163476.42034.a1. discussion 1–10. [DOI] [PubMed] [Google Scholar]
- Blandizzi C. Enteric alpha-2 adrenoceptors: pathophysiological implications in functional and inflammatory bowel disorders. Neurochem Int. 2007;51:282–288. doi: 10.1016/j.neuint.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Brotchie JM. Nondopaminergic mechanisms in levodopa-induced dyskinesia. Mov Disord. 2005;20:919–931. doi: 10.1002/mds.20612. [DOI] [PubMed] [Google Scholar]
- Carsi JM, Valentine HH, Potter LT. m2-toxin: a selective ligand for M2 muscarinic receptors. Mol Pharmacol. 1999;56:933–937. doi: 10.1124/mol.56.5.933. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Kasai M, Lee CY. Use of a snake venom toxin to characterize the cholinergic receptor protein. Proc Natl Acad Sci U S A. 1970;67:1241–1247. doi: 10.1073/pnas.67.3.1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Rogge G, Hague C, Alewood D, Colless B, Lewis RJ, et al. Subtype-selective noncompetitive or competitive inhibition of human alpha1-adrenergic receptors by rho-TIA. J Biol Chem. 2004;279:35326–35333. doi: 10.1074/jbc.M403703200. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human 2-adrenergic G protein coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AG, Norberg T, Griffin D, Hoeger C, Akhtar M, Schmidt K, et al. Contulakin-G, an O-glycosylated invertebrate neurotensin. J Biol Chem. 1999;274:13752–13759. doi: 10.1074/jbc.274.20.13752. [DOI] [PubMed] [Google Scholar]
- Crassous PA, Denis C, Paris H, Senard JM. Interest of alpha2-adrenergic agonists and antagonists in clinical practice: background, facts and perspectives. Curr Top Med Chem. 2007;7:187–194. doi: 10.2174/156802607779318190. [DOI] [PubMed] [Google Scholar]
- Cruz LJ, de Santos V, Zafaralla GC, Ramilo CA, Zeikus R, Gray WR, et al. Invertebrate vasopressin/oxytocin homologs. Characterization of peptides from Conus geographus and Conus straitus venoms. J Biol Chem. 1987;262:15821–15824. [PubMed] [Google Scholar]
- Ducancel F. Endothelin-like peptides. Cell Mol Life Sci. 2005;62:2828–2839. doi: 10.1007/s00018-005-5286-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favreau P, Gilles N, Lamthanh H, Bournaud R, Shimahara T, Bouet F, et al. A new omega-conotoxin that targets N-type voltage-sensitive calcium channels with unusual specificity. Biochemistry. 2001;40:14567–14575. doi: 10.1021/bi002871r. [DOI] [PubMed] [Google Scholar]
- Fruchart-Gaillard C, Mourier G, Marquer C, Menez A, Servent D. Identification of various allosteric interaction sites on M1 muscarinic receptor using 125I-Met35-oxidized muscarinic toxin 7. Mol Pharmacol. 2006;69:1641–1651. doi: 10.1124/mol.105.020883. [DOI] [PubMed] [Google Scholar]
- Fruchart-Gaillard C, Mourier G, Marquer C, Stura E, Birdsall NJ, Servent D. Different interactions between MT7 toxin and the human muscarinic M1 receptor in its free and N-methylscopolamine-occupied states. Mol Pharmacol. 2008;74:1554–1563. doi: 10.1124/mol.108.050773. [DOI] [PubMed] [Google Scholar]
- Gales C, Van Durm JJ, Schaak S, Pontier S, Percherancier Y, Audet M, et al. Probing the activation-promoted structural rearrangements in preassembled receptor-G protein complexes. Nat Struct Mol Biol. 2006;13:778–786. doi: 10.1038/nsmb1134. [DOI] [PubMed] [Google Scholar]
- Halai R, Craik DJ. Conotoxins: natural product drug leads. Nat Prod Rep. 2009;26:526–536. doi: 10.1039/b819311h. [DOI] [PubMed] [Google Scholar]
- Han TS, Teichert RW, Olivera BM, Bulaj G. Conus venoms – a rich source of peptide-based therapeutics. Curr Pharm Des. 2008;14:2462–2479. doi: 10.2174/138161208785777469. [DOI] [PubMed] [Google Scholar]
- Harvey AL, Kornisiuk E, Bradley KN, Cervenansky C, Duran R, Adrover M, et al. Effects of muscarinic toxins MT1 and MT2 from green mamba on different muscarinic cholinoceptors. Neurochem Res. 2002;27:1543–1554. doi: 10.1023/a:1021660708187. [DOI] [PubMed] [Google Scholar]
- Jaakola VP, Griffith MT, Hanson MA, Cherezov V, Chien EY, Lane JR, et al. The 2.6 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolkkonen M, Adem A, Hellman U, Wernstedt C, Karlsson E. A snake toxin against muscarinic acetylcholine receptors: amino acid sequence, subtype specificity and effect on guinea-pig ileum. Toxicon. 1995;33:399–410. doi: 10.1016/0041-0101(94)00102-e. [DOI] [PubMed] [Google Scholar]
- Jolkkonen M, Oras A, Toomela T, Karlsson E, Jarv J, Akerman KE. Kinetic evidence for different mechanisms of interaction of black mamba toxins MT alpha and MT beta with muscarinic receptors. Toxicon. 2001;39:377–382. doi: 10.1016/s0041-0101(00)00141-0. [DOI] [PubMed] [Google Scholar]
- Joubert FJ. The amino acid sequence of protein CM-3 from Dendroaspis polylepis polylepis (black mamba) venom. Int J Biochem. 1985;17:695–699. doi: 10.1016/0020-711x(85)90367-2. [DOI] [PubMed] [Google Scholar]
- King GF, Gentz MC, Escoubas P, Nicholson GM. A rational nomenclature for naming peptide toxins from spiders and other venomous animals. Toxicon. 2008;52:264–276. doi: 10.1016/j.toxicon.2008.05.020. [DOI] [PubMed] [Google Scholar]
- Kostenis E. Is Galpha16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors? Trends Pharmacol Sci. 2001;22:560–564. doi: 10.1016/s0165-6147(00)01810-1. [DOI] [PubMed] [Google Scholar]
- Kukkonen A, Perakyla M, Akerman KE, Nasman J. Muscarinic toxin 7 selectivity is dictated by extracellular receptor loops. J Biol Chem. 2004;279:50923–50929. doi: 10.1074/jbc.M406424200. [DOI] [PubMed] [Google Scholar]
- Kumar TK, Jayaraman G, Lee CS, Arunkumar AI, Sivaraman T, Samuel D, et al. Snake venom cardiotoxins-structure, dynamics, function and folding. J Biomol Struct Dyn. 1997;15:431–463. doi: 10.1080/07391102.1997.10508957. [DOI] [PubMed] [Google Scholar]
- Leppik RA, Mynett A, Lazareno S, Birdsall NJ. Allosteric interactions between the antagonist prazosin and amiloride analogs at the human alpha(1A)-adrenergic receptor. Mol Pharmacol. 2000;57:436–445. doi: 10.1124/mol.57.3.436. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Olivera BM, Cruz LJ. Conus peptides as probes for ion channels. Methods Enzymol. 1999;294:605–624. doi: 10.1016/s0076-6879(99)94034-x. [DOI] [PubMed] [Google Scholar]
- Mourier G, Dutertre S, Fruchart-Gaillard C, Menez A, Servent D. Chemical synthesis of MT1 and MT7 muscarinic toxins: critical role of Arg-34 in their interaction with M1 muscarinic receptor. Mol Pharmacol. 2003;63:26–35. doi: 10.1124/mol.63.1.26. [DOI] [PubMed] [Google Scholar]
- Olianas MC, Maullu C, Adem A, Mulugeta E, Karlsson E, Onali P. Inhibition of acetylcholine muscarinic M(1) receptor function by the M(1)-selective ligand muscarinic toxin 7 (MT-7) Br J Pharmacol. 2000;131:447–452. doi: 10.1038/sj.bjp.0703606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: a G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Pang CC. Autonomic control of the venous system in health and disease: effects of drugs. Pharmacol Ther. 2001;90:179–230. doi: 10.1016/s0163-7258(01)00138-3. [DOI] [PubMed] [Google Scholar]
- Pratt BL, Takahashi JS. Alpha-2 adrenergic regulation of melatonin release in chick pineal cell cultures. J Neurosci. 1987;7:3665–3674. doi: 10.1523/JNEUROSCI.07-11-03665.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, et al. Sea anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007;317:86–94. doi: 10.1126/science.1139158. [DOI] [PubMed] [Google Scholar]
- Quinton L, Demeure K, Dobson R, Gilles N, Gabelica V, De Pauw E. New method for characterizing highly disulfide-bridged peptides in complex mixtures: application to toxin identification from crude venoms. J Proteome Res. 2007;6:3216–3223. doi: 10.1021/pr070142t. [DOI] [PubMed] [Google Scholar]
- Quinton L, Girard E, Maiga A, Rekik M, Lluel P, Masuyer G, et al. Isolation and pharmacological characterization of AdTx1, a natural peptide displaying specific insurmountable antagonism of the alpha(1A)-adrenoceptor. Br J Pharmacol. 2010;159:316–325. doi: 10.1111/j.1476-5381.2009.00532.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopalan N, Pung YF, Zhu YZ, Wong PT, Kumar PP, Kini RM. Beta-cardiotoxin: a new three-finger toxin from Ophiophagus hannah (king cobra) venom with beta-blocker activity. FASEB J. 2007;21:3685–3695. doi: 10.1096/fj.07-8658com. [DOI] [PubMed] [Google Scholar]
- Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- Ribeiro CA, Macedo TA. Pharmacological characterization of the postsynaptic alpha-adrenoceptors in human uterine artery. J Pharm Pharmacol. 1986;38:600–605. doi: 10.1111/j.2042-7158.1986.tb03088.x. [DOI] [PubMed] [Google Scholar]
- Rosengren AH, Jokubka R, Tojjar D, Granhall C, Hansson O, Li DQ, et al. Overexpression of alpha2A-adrenergic receptors contributes to type 2 diabetes. Science. 2009;327:217–220. doi: 10.1126/science.1176827. [DOI] [PubMed] [Google Scholar]
- Selvam C, Oueslati N, Lemasson IA, Brabet I, Rigault D, Courtiol T, et al. A virtual screening hit reveals new possibilities for developing group III metabotropic glutamate receptor agonists. J Med Chem. 2010;53:2797–2813. doi: 10.1021/jm901523t. [DOI] [PubMed] [Google Scholar]
- Servent D, Fruchart-Gaillard C. Muscarinic toxins: tools for the study of the pharmacological and functional properties of muscarinic receptors. J Neurochem. 2009;109:1193–1202. doi: 10.1111/j.1471-4159.2009.06092.x. [DOI] [PubMed] [Google Scholar]
- Servent D, Menez A. Snake neurotoxins that interact with nicotinic acetylcholine receptors. In: Massaro EJ, editor. Handbook of Neurotoxicology. Totowa, NJ: Humana Press; 2001. pp. 385–425. [Google Scholar]
- Servent D, Winckler-Dietrich V, Hu HY, Kessler P, Drevet P, Bertrand D, et al. Only snake curaremimetic toxins with a fifth disulfide bond have high affinity for the neuronal alpha7 nicotinic receptor. J Biol Chem. 1997;272:24279–24286. doi: 10.1074/jbc.272.39.24279. [DOI] [PubMed] [Google Scholar]
- Sharpe IA, Gehrmann J, Loughnan ML, Thomas L, Adams DA, Atkins A, et al. Two new classes of conopeptides inhibit the alpha1-adrenoceptor and noradrenaline transporter. Nat Neurosci. 2001;4:902–907. doi: 10.1038/nn0901-902. [DOI] [PubMed] [Google Scholar]
- Suckau D, Resemann A, Schuerenberg M, Hufnagel P, Franzen J, Holle A. A novel MALDI LIFT-TOF/TOF mass spectrometer for proteomics. Anal Bioanal Chem. 2003;376:952–965. doi: 10.1007/s00216-003-2057-0. [DOI] [PubMed] [Google Scholar]
- Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol Rev. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- Tytgat J, Chandy KG, Garcia ML, Gutman GA, Martin-Eauclaire MF, van der Walt JJ, et al. A unified nomenclature for short-chain peptides isolated from scorpion venoms: alpha-KTx molecular subfamilies. Trends Pharmacol Sci. 1999;20:444–447. doi: 10.1016/s0165-6147(99)01398-x. [DOI] [PubMed] [Google Scholar]
- de Weille JR, Schweitz H, Maes P, Tartar A, Lazdunski M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the L-type calcium channel. Proc Natl Acad Sci U S A. 1991;88:2437–2440. doi: 10.1073/pnas.88.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Day M, Heavner JE. Ziconotide: an update and review. Expert Opin Pharmacother. 2008;9:1575–1583. doi: 10.1517/14656566.9.9.1575. [DOI] [PubMed] [Google Scholar]
- Wu PL, Lee SC, Chuang CC, Mori S, Akakura N, Wu WG, et al. Non-cytotoxic cobra cardiotoxin A5 binds to alpha(v)beta3 integrin and inhibits bone resorption. Identification of cardiotoxins as non-RGD integrin-binding proteins of the Ly-6 family. J Biol Chem. 2006;281:7937–7945. doi: 10.1074/jbc.M513035200. [DOI] [PubMed] [Google Scholar]