

Abstract
BACKGROUND AND PURPOSE
Mutations of SCN1A, the gene encoding the pore-forming subunit of the voltage-gated sodium channel NaV1.1, have been associated with a spectrum of genetic epilepsies and a familial form of migraine. Several mutant NaV1.1 channels exhibit increased persistent current due to incomplete inactivation and this biophysical defect may contribute to altered neuronal excitability in these disorders. Here, we investigated the ability of ranolazine to preferentially inhibit increased persistent current evoked by mutant NaV1.1 channels.
EXPERIMENTAL APPROACH
Human wild-type (WT) and mutant NaV1.1 channels were expressed heterologously in human tsA201 cells and whole-cell patch clamp recording was used to assess tonic and use-dependent ranolazine block.
KEY RESULTS
Ranolazine (30 µM) did not affect WT NaV1.1 channel current density, activation or steady-state fast inactivation but did produce mild slowing of recovery from inactivation. Ranolazine blocked persistent current with 16-fold selectivity over tonic block of peak current and 3.6-fold selectivity over use-dependent block of peak current. Similar selectivity was observed for ranolazine block of increased persistent current exhibited by NaV1.1 channel mutations representing three distinct clinical syndromes, generalized epilepsy with febrile seizures plus (R1648H, T875M), severe myoclonic epilepsy of infancy (R1648C, F1661S) and familial hemiplegic migraine type 3 (L263V, Q1489K). In vitro application of achievable brain concentrations (1, 3 µM) to cells expressing R1648H channels was sufficient to suppress channel activation during slow voltage ramps, consistent with inhibition of persistent current.
CONCLUSIONS AND IMPLICATIONS
Our findings support the feasibility of using selective suppression of increased persistent current as a potential new therapeutic strategy for familial neurological disorders associated with certain sodium channel mutations.
Keywords: sodium channel, epilepsy, antiepileptic drugs, ranolazine, migraine
Introduction
Mutation of SCN1A, which encodes the pore-forming α-subunit of the brain voltage-gated sodium channel NaV1.1 (nomenclature follows Alexander et al., 2009), is a cause of genetic epilepsies ranging in severity from the typically mild generalized epilepsy with febrile seizures plus (GEFS+) to the debilitating severe myoclonic epilepsy of infancy (SMEI) (George, 2005; Meisler et al., 2005; Mulley et al., 2005; Lossin, 2009). Mutation of SCN1A has also been linked to familial hemiplegic migraine type 3 (FHM3) (Dichgans et al., 2005; Vanmolkot et al., 2007; Kahlig et al., 2008). A common feature observed for several NaV1.1 channel mutants is a significantly increased persistent current (Lossin et al., 2002; Rhodes et al., 2004; Spampanato et al., 2004; Kahlig et al., 2006; 2008;), which can be predicted to affect neuronal excitability. Therapeutic strategies capable of selectively inhibiting increased persistent current might have clinical benefits in this setting.
Ranolazine, a novel anti-anginal drug that chemically contains a piperazine ring flanked by lidocaine and catecholamine-like moieties, exerts its clinical action in part by inhibiting persistent sodium current carried by cardiac sodium channels (Abrams et al., 2006; Nash et al., 2008). Persistent sodium current in myocytes is predicted to alter ionic homeostasis by raising intracellular sodium ion concentration that in turn blunts activity of the sodium/calcium exchanger and elevates the level of intracellular calcium. The resulting ionic imbalance is thought to precipitate myocardial dysfunction leading to electrical instability, arrhythmia and reduced contractility (Shryock et al., 2008). Ranolazine also inhibits sodium currents recorded from cultured pituitary tumour GH3 neurons as well as the neuron-like NG108-15 mouse/rat hybridoma cell line (Chen et al., 2009; Wu et al., 2009), but the target of ranolazine in these in vitro models is unclear as the cell lines may express a low level of NaV1.5 channels.
Although ranolazine exhibits activity against other molecular targets (Allely et al., 1993; Letienne et al., 2001; Chen et al., 2009), the primary therapeutic mechanism of action is thought to involve block of persistent sodium current due to the lidocaine moiety. This effect was first demonstrated in a guinea pig ventricular myocyte model of long-QT syndrome (LQTS) in which persistent current was induced by the sea anemone toxin ATX-II (Song et al., 2004; Wu et al., 2004). Subsequently, ranolazine was shown to preferentially block the increased persistent current evoked by LQTS mutations in NaV1.5 channels (Fredj et al., 2006; Rajamani et al., 2009), as well as toxin- or mutation-induced persistent current carried by muscle (NaV1.4; (Wang et al., 2008) and peripheral nerve (NaV1.7 and NaV1.8; (Rajamani et al., 2008a; Wang et al., 2008) sodium channels. However, the ability of ranolazine to inhibit brain sodium channel isoforms such as NaV1.1 has not been reported.
In this study, we have demonstrated the ability of ranolazine to preferentially block the increased persistent current generated by mutant NaV1.1 channels. Our findings illustrate a proof of principle that selective suppression of persistent current may provide a useful new therapeutic strategy for SCN1A-associated epilepsy and migraine syndromes.
Methods
Expression of human NaV1.1 channel cDNA
All wild-type (WT) and mutant plasmid constructs have been studied previously by our laboratory and cDNA expression was performed as previously described (Lossin et al., 2002; Rhodes et al., 2004; Kahlig et al., 2008). Briefly, heterologous transient expression of NaV1.1 α1 channels in combination with human β1 and β2 accessory subunit cDNAs was achieved by transfection of tsA201 cells using Qiagen Superfect reagent (5.5 µg of DNA was transfected at a plasmid mass ratio of 10:1:1 for α1: β1: β2 channel subunits).
Electrophysiology
Whole-cell voltage-clamp recordings were used to measure the biophysical properties of WT and mutant NaV1.1 channels, as described previously (Kahlig et al., 2008). Briefly, the pipette solution consisted of (in mM) 110 CsF, 10 NaF, 20 CsCl, 2 EGTA, 10 HEPES, with a pH of 7.35 and osmolarity of 300 mOsmol/kg. The bath (control) solution contained in (mM): 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 dextrose, 10 HEPES, with a pH of 7.35 and osmolarity of 310 mOsmol/kg. Cells were allowed to stabilize for 10 min after establishment of the whole-cell configuration before current was measured. Series resistance was compensated 90% to assure that the command potential was reached within microseconds with a voltage error <2 mV. Leak currents were subtracted by using an online P/4 procedure and all currents were low-pass Bessel filtered at 5 kHz and digitized at 50 kHz. For clarity, representative ramp currents were low pass filtered off-line at 50 Hz. Specific voltage-clamp protocols assessing channel activation, fast inactivation and availability during repetitive stimulation were used as depicted in figure insets.
In vitro pharmacology
A stock solution of 20 mM ranolazine (Gilead Sciences Inc., Palo Alto, CA, USA) was prepared in 0.1 M HCl. A fresh dilution of ranolazine in the bath solution was prepared every experimental day and the pH was readjusted to 7.35. Direct application of the test solution to the clamped cell was achieved using the Perfusion Pencil system (Automate, Berkeley, CA, USA). Direct cell superfusion was driven by gravity at a flow rate of 350 µL/min using a 250 micron tip. This system sequesters the clamped cell within a stream and enables complete solution exchange within 1 s. The clamped cell was superfused continuously starting immediately after establishing the whole-cell configuration. Control currents were measured in drug-free solution. Ranolazine containing solutions were superfused for 3 min prior to current recordings to allow equilibrium (tonic) drug block. Tetrodotoxin (TTX) was then applied in the presence of ranolazine to enable offline digital subtraction of TTX-insensitive current.
Tonic block of peak and persistent currents were measured from this steady-state condition and three sequential current traces (0.1 Hz pulsing frequency) were averaged to obtain a mean current for each recording condition (control, ranolazine and TTX) that were utilized for offline analysis. Steady-state persistent current was measured during the final 10 ms of a 200 ms voltage step to −10 mV with an inter-pulse duration of 10 s.
Use-dependent block of peak current was measured during pulse number 300 of a pulse train (−10 mV, 5 ms, 300 pulses) at frequencies between 10 and 135 Hz from a holding potential of −120 mV. Two sequential pulse train stimulations were averaged to obtain mean current traces for each recording condition, which were then used for offline subtraction and analysis. Use-dependent block of persistent current was measured by first conditioning cells with a 10 Hz pulse train (−10 mV, 5 ms, 300 pulses) followed immediately by a 200 ms voltage step to −10 mV (effectively the 301st pulse of the train). TTX-sensitive persistent current was measured during the final 10 ms of the 200 ms step.
Block of ramp current was assessed by voltage ramps to +20 mV from a holding potential of −120 mV at 20 mV/s stimulated every 30 s. To minimize time-dependent current drift, only one trace recorded during control, ranolazine or TTX superfusion was analyzed.
Concentration inhibition curves were fitted with the Hill equation: I/Imax= 1/[1+10^(log IC50− I)*k], where IC50 is the concentration that produces half inhibition and k is the Hill slope factor. The upper and lower limits were set to 1 and 0.
In vivo pharmacology
All animal care and experimental use were approved by the Institutional Animal Care and Use Committee, Gilead Sciences. Male Sprague Dawley rats (250–350 g, Charles River Laboratories, Hollister, CA, USA) were cannulated in the jugular vein and used to study brain penetration of ranolazine in vivo. Three rats per group were infused intravenously with ranolazine in saline at 85.5 µg/kg/min. After 1, 2.5 or 5 h, animals were killed for plasma and brain collection, and ranolazine concentrations were measured by liquid chromatography coupled with tandem mass spectrometry. Brain tissue was homogenized in 5% sodium fluoride solution containing 1% 2N HCl (final homogenate was diluted threefold). Plasma and brain homogenate samples (50 µL) were precipitated, with 300 µL of deuterated D3-ranolazine (150 ng/mL; synthesized by PPD Discovery, Morrisville, NC, USA) as an internal standard, in 9:1 acetonitrile: methanol, vortexed, then centrifuged at 3273× g for 10 min at 22°C. The supernatant (50 µL) was transferred and diluted with water (450 µL) prior to injection (10 µL). High-performance liquid chromatography was performed using a Shimadzu LC-10AD liquid chromatograph and a Luna C18(2) (Shimadzu Scientific Instruments, Inc., Columbia, MD, USA), 3 µm, 20 × 2.0 mm column with a mobile phase consisting of water containing 0.1% formic acid (solution A) and acetonitrile (solution B) carried out under isocratic conditions (75% solution A, 25% solution B; flow rate 0.300 mL/min). Mass spectrometric analyses were performed using an API3000 mass spectrometer (Applied Biosystems, Foster City, CA, USA) operating in positive ion mode with multiple reaction monitoring for ion transition 428.1 > 98. Brain-to-plasma ranolazine ratios were calculated for each sample as ng ranolazine/g brain divided by ng ranolazine/mL plasma.
Data analysis
Results from the electrophysiological experiments are presented as mean ± SEM, and unless otherwise noted, statistical comparisons were made using one-way anova followed by a Tukey post hoc test in reference to WT NaV1.1 channels.
Materials
Unless otherwise noted, all reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Results
In this study, we determined the ability of ranolazine to inhibit WT NaV1.1 channels and a panel of NaV1.1 channel mutants associated with GEFS+, SMEI and FHM3 to evaluate the ability of ranolazine to preferentially block the abnormal increased persistent current carried by these mutant channels.
Ranolazine effects on activation and inactivation of WT NaV1.1 channels
We first determined the effects of ranolazine on activation and inactivation properties of WT NaV1.1 channels expressed heterologously in tsA201 cells. Figure 1A illustrates representative whole-cell sodium currents recorded from a cell expressing WT NaV1.1 channels in control solution (drug-free) and the same cell during superfusion with 30 µM ranolazine. Application of the drug had no overt effects on WT NaV1.1 channel function even at this high concentration. Similarly, there was no significant effect of the drug on peak current density recorded during sequential application of control solution and 30 µM ranolazine (Figure 1B). Furthermore, 30 µM ranolazine did not significantly shift the voltage dependence of the activation or inactivation of WT NaV1.1 channels (Figure 1C, Table 1). These results indicate that ranolazine does not interfere with activation of the channels. However, 30 µM ranolazine did cause a slight but significant slowing of recovery from inactivation (Figure 1D, Table 1) consistent with increased stability of the inactivated state. These results indicate that 30 µM ranolazine has minimal effects on the function of WT NaV1.1 channels.
Figure 1.
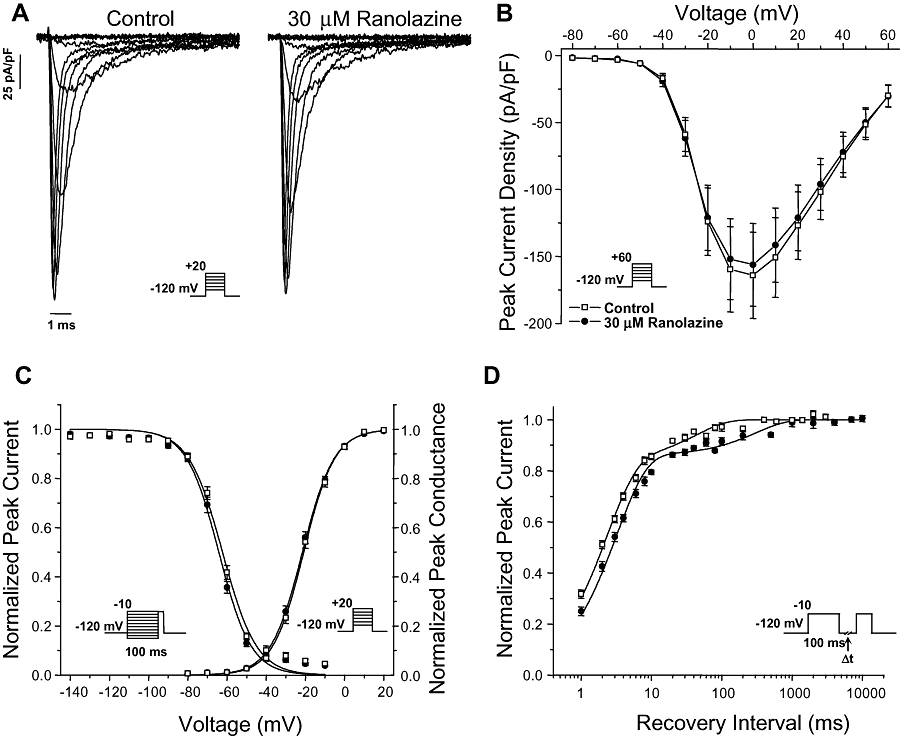
Ranolazine effects on wild-type (WT) NaV1.1 channels. (A) Representative whole-cell sodium currents recorded during sequential superfusion of control solution followed by 30 µM ranolazine. Currents were activated by voltage steps to between −80 and +20 mV from a holding potential of −120 mV. (B) Peak current density elicited by test pulses to various potentials and normalized to cell capacitance recorded during sequential superfusion of control solution followed by 30 µM ranolazine. (C) Voltage dependence of activation measured during voltage steps to between −80 and +20 mV plotted together with voltage dependence of fast inactivation determined with 100 ms pre-pulses to between −140 and −10 mV (symbols as defined in B). Pulse protocols are shown as panel insets and fit parameters are provided in Table 1. (D) Time-dependent recovery from fast inactivation following an inactivating pre-pulse of 100 ms to −10 mV (symbols as defined in B). Pulse protocols are shown as panel insets and fit parameters are provided in Table 1.
Table 1.
Biophysical parameters for activation and fast inactivation of WT NaV1.1 channels
| Activation | Inactivation | Recovery from Inactivation§ | |||||||
|---|---|---|---|---|---|---|---|---|---|
| V1/2(mV) | k (mV) | n | V1/2(mV) | k (mV) | n | τf(ms) | τs(ms) | n | |
| Control | −20.9 ± 0.9 | 7.7 ± 0.2 | 10 | −62.3 ± 0.8 | −8.6 ± 0.6 | 10 | 2.2 ± 0.2 [82 ± 4%] | 63.5 ± 12.1 [18 ± 4%] | 10 |
| 30 µM Ranolazine | −21.6 ± 0.8 | 8.0 ± 0.2 | 10 | −64.2 ± 0.8 | −8.0 ± 0.5 | 10 | 3.2 ± 0.2**[85 ± 1%] | 412.4 ± 86.1**[15 ± 1%] | 10 |
Values in brackets represent fractional amplitudes. Values significantly different from Control are indicated as follows
P < 0.01.
Preferential ranolazine block of persistent current
We examined the concentration-dependent tonic inhibition of peak and persistent current carried by WT NaV1.1 and a mutant NaV1.1 channel (R1648H) associated with GEFS+ that we previously demonstrated to exhibit significantly increased persistent current as the only apparent biophysical defect (Lossin et al., 2002; Kahlig et al., 2006). Figure 2A illustrates whole-cell sodium currents recorded from WT NaV1.1 channels during sequential application of control solution (black trace) followed by 30 µM ranolazine (red trace). Tonic block of the peak current in WT NaV1.1 channels was minimal as illustrated by the figure inset where the data were plotted on an expanded time scale. In Figure 2B, which illustrates the same experimental sequence for the R1648H mutant channels, persistent current was substantially reduced during superfusion of ranolazine as compared to the drug-free condition. As observed for WT NaV1.1, 30 µM ranolazine exerted minimal tonic block of peak current in R1648H channels (Figure 2B inset).
Figure 2.
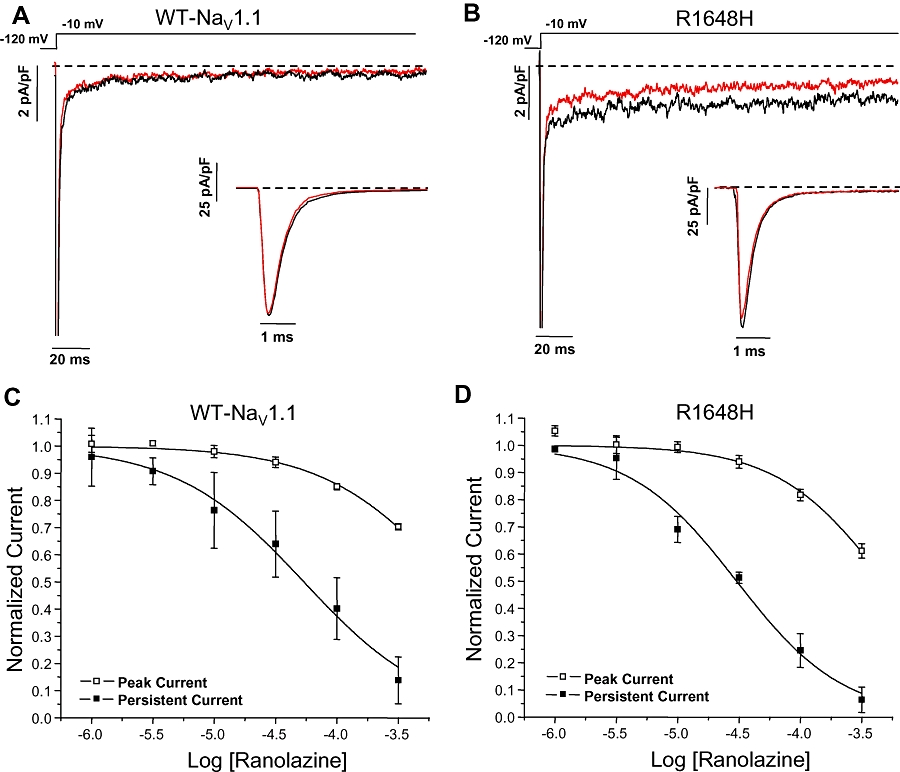
Ranolazine preferentially inhibits the persistent current in NaV1.1 channels. Tonic inhibition of NaV1.1 channel peak and persistent current measured using a 200 ms voltage step to −10 mV from a holding potential of −120 mV. Representative tetrodotoxin-subtracted whole-cell sodium currents recorded for wild-type (WT) NaV1.1 (A) and R1648H channels (B) during sequential superfusion of control solution (black trace) followed by 30 µM ranolazine (red trace). The dashed line indicates zero current level. (C, D) Ranolazine exhibits a concentration dependent tonic block of WT NaV1.1 and R1648H channels peak and persistent currents. The peak and persistent current measured during ranolazine superfusion was normalized to the current measured in control solution. Derived parameters are shown in Tables 2 and n= 5–6 for both WT NaV1.1 and R1648H channels.
Ranolazine exhibited greater degrees of tonic inhibition of persistent current as compared with peak current for both WT NaV1.1 and R1648H channels (Figure 2C and D, respectively). Fits of concentration–inhibition curves with the Hill equation provided IC50 values of 871 µM for WT NaV1.1 and 490 µM for R1648H channels for tonic peak current block (Table 2), whereas ranolazine block of persistent current carried by WT NaV1.1 and R1648H channels exhibited IC50 values of 53.7 µM and 30.2 µM, respectively. These results demonstrate that ranolazine has approximately 16-fold selectivity for tonic block of persistent current carried by either WT NaV1.1 or R1648H channels.
Table 2.
Tonic block of WT NaV1.1 and R1648H channels
| Tonic block of peak current | Tonic block of persistent current | |||
|---|---|---|---|---|
| Log IC50 | k | Log IC50 | k | |
| WT NaV1.1 | −3.06 ± 0.13 (871 µM) | −0.84 ± 0.15 | −4.27 ± 0.04 (53.7 µM) | −0.83 ± 0.06 |
| R1648H | −3.31 ± 0.09 (490 µM) | −1.00 ± 0.19 | −4.52 ± 0.04 (30.2 µM) | −1.00 ± 0.09 |
We also assessed use-dependent block of the peak current in WT NaV1.1 and R1648H channels by ranolazine. Figure 3A illustrates the whole-cell sodium currents recorded from WT NaV1.1 channels in response to a repetitive depolarization protocol (5 ms, −10 mV, 300 pulses, 10 Hz) during superfusion of control solution. In the drug-free control condition, the availability of WT NaV1.1 channels was unchanged during repetitive depolarization. In contrast, application of 30 µM ranolazine to the same cell caused a reduction in peak current during repetitive pulsing consistent with use-dependent block of the channel (Figure 3B). The concentration dependence of ranolazine use-dependent block of the peak current in WT NaV1.1 and R1648H channels was characterized by IC50 values of 195 µM and 138 µM, respectively (Figure 3C and D, Table 2). The use-dependent block of persistent current was measured for R1648H channels using a repetitive depolarization protocol (5 ms, −10 mV, 300 pulses, 10 Hz) followed immediately by the 200 ms voltage step necessary to elicit steady-state persistent current (Figure 4A). Use-dependent block of persistent current by ranolazine exhibited an IC50 value of 30.1 µM for R1648H channels (Figure 4B, Table 3), which is indistinguishable from tonic block of persistent current. These results demonstrated that ranolazine was 3.6-fold and 4.6-fold more potent at inhibiting the persistent current carried by WT NaV1.1 and R1648H channels, respectively, as compared to use-dependent block of peak current.
Figure 3.
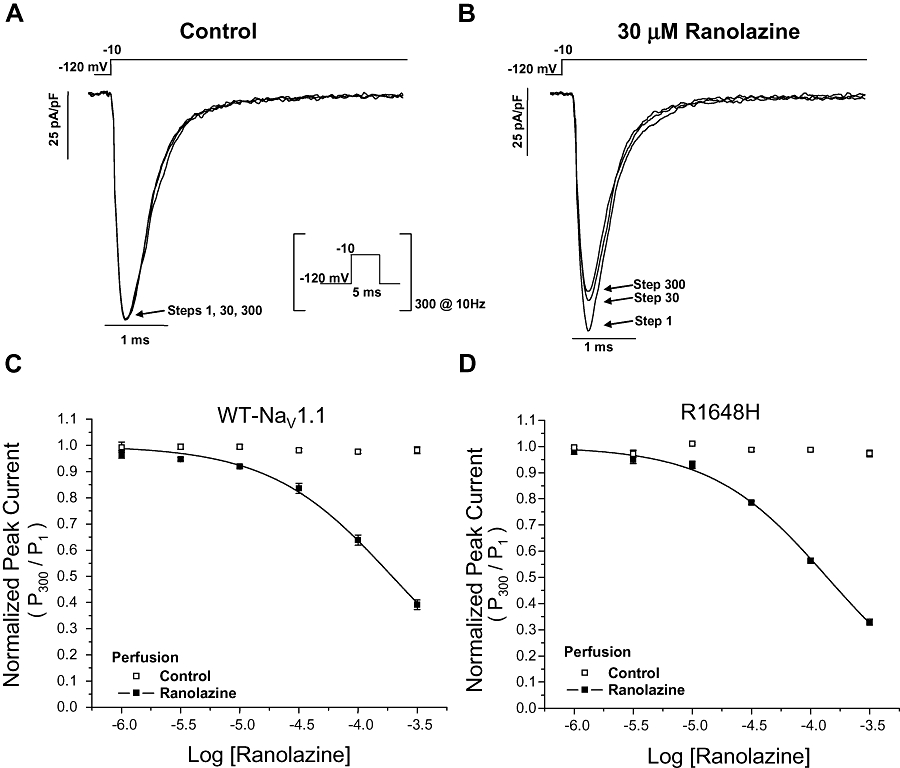
Use-dependent block of NaV1.1 channel peak current by ranolazine. NaV1.1 channel availability during repetitive stimulation was assessed with a depolarizing pulse train (−10 mV, 5 ms, 300 pulses, 10 Hz) from a holding potential of −120 mV. Representative whole-cell sodium currents recorded from wild-type (WT) NaV1.1 channels during sequential superfusion of control solution (A) followed by 30 µM ranolazine (B). Only the current traces from pulses 1, 30 and 300 are shown for clarity. (C,D) Ranolazine exhibits concentration-dependent and use-dependent block of the peak current in WT NaV1.1 and R1648H channels (n= 5–6). Neither WT NaV1.1 nor R1648H channels exhibited use-dependent reduction in peak current when exposed to drug-free control solution. Derived parameters are shown in Table 3.
Figure 4.
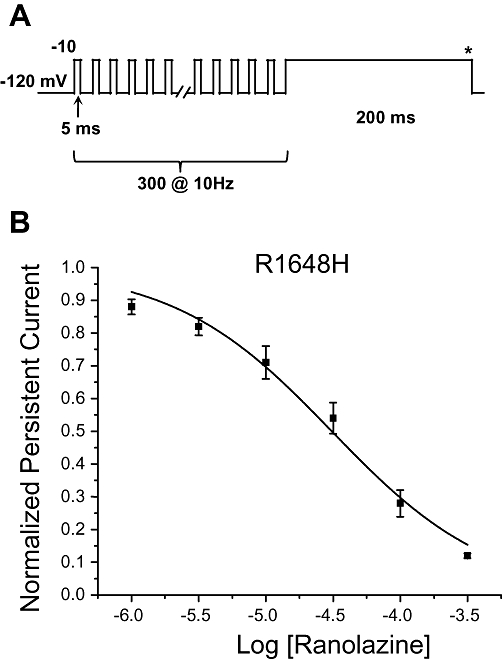
Use-dependent block of persistent current in R1648H channels by ranolazine. (A) Use-dependent inhibition of the persistent current in R1648H channels was measured using a conditioning pulse train (−10 mV, 5 ms, 300 pulses, 10 Hz) followed by a 200 ms step to −10 mV. The steady-state persistent current was measured during the final 10 ms of the pulse (asterisk). (B) Ranolazine exhibits a concentration dependent block of R1648H channel persistent current (n= 5–6). The persistent current measured during ranolazine superfusion was normalized to the current measured in control solution. Derived parameters are shown in Table 3.
Table 3.
Use-dependent block of WT NaV1.1 and R1648H channels
| Use-dependent block of peak current | Use-dependent block of persistent current | |||
|---|---|---|---|---|
| Log IC50 | k | Log IC50 | k | |
| WT NaV1.1 | −3.71 ± 0.02 (195 µM) | −0.84 ± 0.05 | ||
| R1648H | −3.86 ± 0.02 (138 µM) | −0.88 ± 0.03 | −4.51 ± 0.04 (30.9 µM) | −0.73 ± 0.05 |
Ranolazine block of mutant NaV1.1 channels
We compared the degree of ranolazine block among six NaV1.1 mutant channels representing three clinical syndromes: GEFS+ (R1648H, T875M), SMEI (R1648C, F1661S) and FHM3 (L263V, Q1489K). Figure 5A illustrates tonic block of peak and persistent current by 30 µM ranolazine for this panel of mutant channels normalized to current amplitudes recorded in drug-free control solution. For WT and all mutant channels, we observed a much greater degree of ranolazine block of persistent current as compared to peak current. We also assessed the ability of ranolazine to reduce the magnitude of persistent current exhibited by mutant channels to the level conducted by WT NaV1.1 channels. In Figure 5B, persistent current was expressed as a percent of peak current and was not normalized to the drug-free condition. In general, the level of persistent current carried by mutant channels was reduced by approximately 50% (range 44–60%), and for some mutant channels (R1648H, T875M, L263V), the level in the presence of ranolazine was not significantly different from that observed in WT NaV1.1 channels in the absence of drug.
Figure 5.
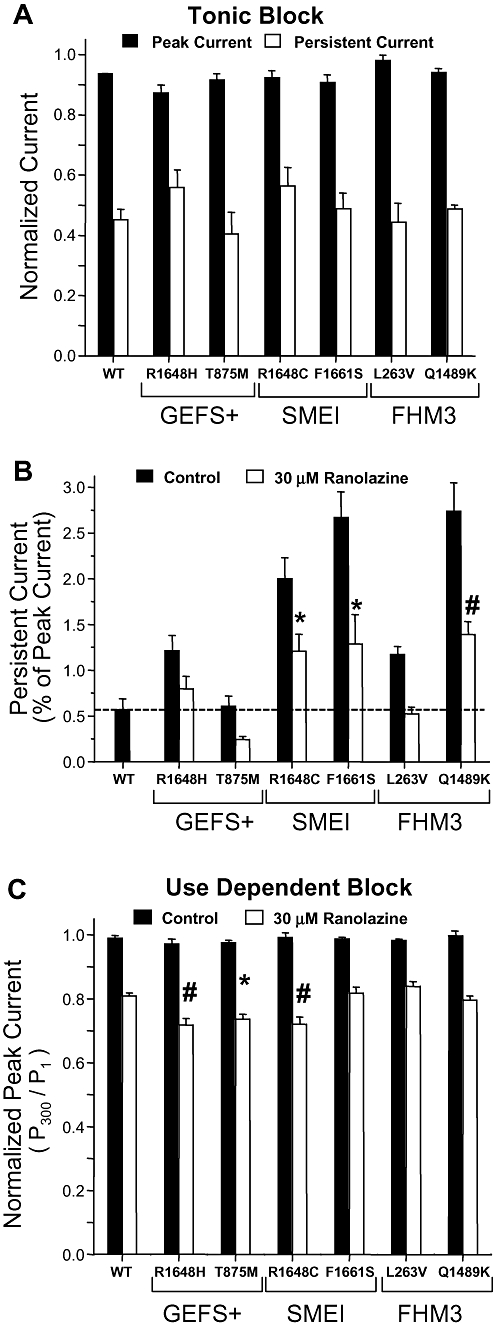
Preferential block of persistent current by ranolazine. Tonic block of peak and persistent current measured using a 200 ms voltage step to −10 mV during application of 30 µM ranolazine for wild-type (WT) NaV1.1 and mutant NaV1.1 channels. (A) Peak and persistent current amplitudes were normalized to values recorded in drug-free control solution for each cell (n= 5–7). (B) Persistent current expressed as a percentage of peak current recorded during the same voltage protocol for 30 µM ranolazine. Significant differences from WT NaV1.1 channels in drug-free solution are indicated by *P < 0.05 and #P < 0.01. (C) Use-dependent block of WT NaV1.1 and mutant channels during superfusion of 30 µM ranolazine (n= 5–7). Neither WT NaV1.1 nor mutant NaV1.1 channels exhibited use-dependent reduction in availability when exposed to drug-free control solution. Significant differences from WT channels are indicated by *P < 0.05 and # P < 0.01. GEFS+, generalized epilepsy with febrile seizures plus; SMEI, severe myoclonic epilepsy of infancy; FHM3, familial hemiplegic migraine type 3.
We also assessed use-dependent block of the peak and residual currents in mutant NaV1.1 channels by ranolazine. Figure 5C illustrates use-dependent block of peak current for WT NaV1.1 and mutant channels by 30 µM ranolazine. Neither WT NaV1.1 nor any mutant channel exhibited significant loss of channel availability in control solution by the 300th pulse, but there was significant loss of channel availability during ranolazine application for both WT NaV1.1 and mutant channels. However, the mutant channels R1648H, T875M and R1648C exhibited a significantly greater reduction in channel availability in the presence of 30 µM ranolazine as compared to WT NaV1.1 channels.
By dividing the degree of persistent current block by the extent of use-dependent block of peak current, we calculated a selectivity index for the effect of ranolazine on mutant NaV1.1 channels. Ranolazine exhibited the most selective block of persistent current on L263V and F1661S, and least selective block on R1648H and R1648C channels with an overall rank order of L263V > F1661S > Q1489K > T875M > R1648H = R1648C. Although the physiological impact remains to be investigated, these relationships may help predict molecular subsets of NaV1.1 channel mutations that might be more amenable to selective suppression of increased persistent current.
Brain penetration of ranolazine
The ability of ranolazine to cross the blood–brain barrier has not been reported previously. We measured the degree of brain penetration of ranolazine in rats following continuous intravenous infusion of the drug (85.5 µg/kg/min) for 1, 2.5 and 5 h. Ranolazine exhibited significant brain penetration at all time points peaking after 5 h at 470 ng ranolazine/g brain (approximately 1.1 µM, Table 4). Throughout the time course studied, the mean brain levels of ranolazine were approximately one-third of the corresponding plasma levels. Given that the therapeutic plasma concentration of ranolazine is 2–10 µM, brain concentrations up to 3.3 µM should be possible to attain with systemic dosing at therapeutic levels.
Table 4.
Penetration of ranolazine into brain
| Time (h) | Brain (ng/g) | Plasma (ng/mL) | Brain/Plasma (%) |
|---|---|---|---|
| 1 | 298 ± 88.6 (0.70 µM) | 777 ± 255 (1.82 µM) | 38 |
| 2.5 | 446 ± 302 (1.04 µM) | 1180 ± 456 (2.76 µM) | 38 |
| 5 | 470 ± 300 (1.10 µM) | 1590 ± 488 (3.72 µM) | 30 |
Suppression of persistent current by therapeutic ranolazine concentrations
We next examined the ability of achievable brain concentrations of ranolazine (1 and 3 µM) to suppress activation of R1648H channels during slow depolarizing voltage ramps, a phenomenon attributed to increased persistent current. Figure 6A illustrates representative inward currents produced in response to a slow depolarizing voltage ramp. Cells expressing R1648H channels exhibited an increased depolarizing current (compared to WT; red vs. black traces) that was blocked by 3 µM ranolazine (blue trace). The average inward charge (pC) was calculated for multiple cells as the area under the current trace between −40 and 0 mV and normalized to the corresponding peak current (nA) generated by a voltage step to −10 mV to account for variation in channel expression. Figure 6B demonstrates that sequential superfusion of control solution followed by either 1 µM or 3 µM ranolazine reduced the charge conducted by R1648H channels. Neither 1 µM nor 3 µM ranolazine produced significantly different peak current amplitude compared to that observed in cells expressing WT channels recorded in the absence of drug.
Figure 6.
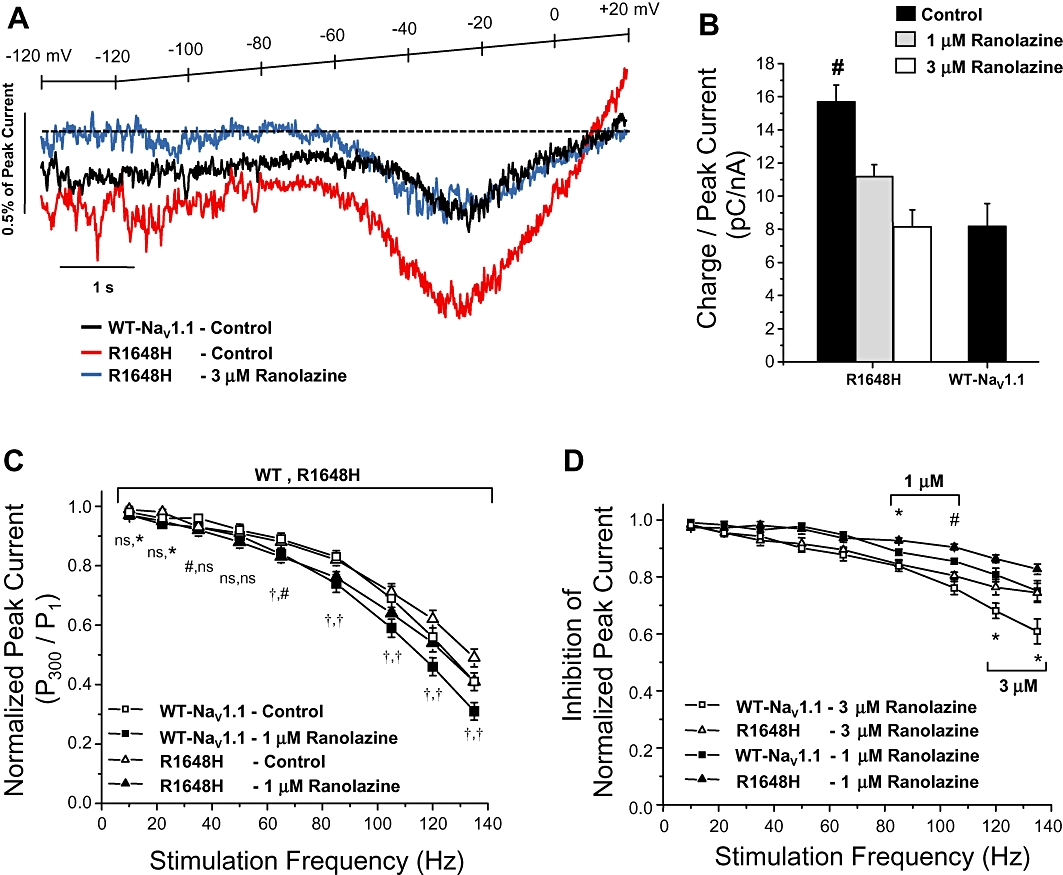
Ranolazine inhibits ramp and use-dependent currents. (A) Representative tetrodotoxin-subtracted ramp currents measured during a 20 mV/s voltage ramp from a holding potential of −120 mV during sequential superfusion of control solution followed by 3 µM ranolazine. The dotted line indicates zero current. (B) R1648H channels conducted significantly more charge between −40 and 0 mV of the ramp, which was inhibited in a concentration dependent manner to the level of wild-type (WT) NaV1.1 channels by 1 µM or 3 µM ranolazine (n= 9–10). (C) WT NaV1.1 and R1648H channel availability during repetitive stimulation was assessed with a depolarizing pulse train (−10 mV, 5 ms, 300 pulses) at frequencies between 10 and 135 Hz during sequential superfusion of control solution followed by 1 µM ranolazine. Normalized peak current (pulse 300/pulse 1) was plotted versus frequency for each pulse train (n= 9 and 8, respectively). (D) Inhibition of normalized peak current calculated as the ratio of channel availability during either 1 µM or 3 µM ranolazine and control conditions. Significant differences are indicated by *P < 0.05, #P < 0.01 and †P < 0.001.
Finally, we assessed use-dependent block of WT and mutant NaV1.1 by ranolazine. Figure 6C illustrates use-dependent block of WT NaV1.1 and R1648H channels at pulsing frequencies between 10 and 135 Hz. In control solution, both WT and R1648H channels exhibited an expected degree of frequency-dependent loss of channel availability. Application of 1 µM ranolazine exaggerated loss of availability at all frequencies greater than 60 Hz. Application of 3 µM ranolazine exaggerated loss of availability at all frequencies greater than 22 Hz (Supporting Information Figure S1). These results are consistent with significant use-dependent block by ranolazine. Figure 6D shows that 1 µM and 3 µM ranolazine produced a similar degree of block of WT and R1648H channels up to 60 Hz.
Discussion and conclusions
Voltage-gated sodium channels are important targets for several widely used anti-epileptic drugs such as phenytoin and lamotrigine. These drugs act in part by stabilizing the inactivated state, thereby reducing sodium channel availability and limiting the ability of neurons to fire repetitively. In addition to reducing sodium channel availability during repetitive neuronal activity, another potentially important effect of these drugs may be the suppression of persistent sodium current (Stafstrom, 2007). Several types of neurons throughout the brain exhibit low amplitude depolarizing persistent current resulting from incomplete sodium channel inactivation. Persistent sodium current is active at subthreshold potentials and contributes to amplification of synaptic inputs and subthreshold oscillations, thereby enabling action potential generation and propagation, as well as supporting repetitive neuronal firing. Also, the persistent sodium current, although small in magnitude, can influence neuronal firing behaviour substantially and may be critical to enabling spread of epileptic activity (Stafstrom, 2007).
The importance of persistent sodium current in the pathogenesis of epilepsy received additional attention when the functional consequences of neuronal sodium channel mutations discovered in various epilepsies were revealed. Several mutations in SCN1A associated with GEFS+ and other epilepsies result in increased persistent sodium current, sometimes as the predominant biophysical abnormality (Lossin et al., 2002; Rhodes et al., 2004; Spampanato et al., 2004; Kahlig et al., 2006; 2008;). An abnormally large persistent current would be predicted to accentuate subthreshold depolarizations, thereby facilitating action potential generation and inducing neuronal hyperexcitability. These findings highlighted increased persistent current as a plausible pathophysiological factor in epileptogenesis and stimulated the idea that selective suppression of persistent current may offer a therapeutic strategy for rare familial epilepsies associated with mutations that promote this type of sodium channel dysfunction. Our findings in this study suggest that therapeutic levels of ranolazine can suppress abnormal increased persistent current and may re-establish normal subthreshold sodium conductance and hence normalize neuronal excitability.
The goal of our study was to test the hypothesis that ranolazine, a drug approved for the treatment of chronic stable angina pectoris, was capable of selectively suppressing increased persistent current evoked by NaV1.1 mutant channels. This hypothesis was inspired in part by the observation that pharmacological suppression of increased persistent current caused by cardiac sodium channel mutations is therapeutically beneficial in subjects with congenital LQTS (Schwartz et al., 1995; Wang et al., 1997). We observed that ranolazine exhibited 16-fold and fivefold greater inhibition of persistent current as compared to tonic block and use-dependent block of peak current, respectively. This inhibition was concentration dependent with greatest selectivity in the low micromolar concentration range, which matches the usual therapeutic plasma concentration of 2–10 µM (Chaitman, 2006; Sicouri et al., 2008). Ranolazine did not have significant effects on current density, activation and voltage dependence of inactivation, but we did observe slowing of recovery from inactivation that may indicate some degree of inactivated state stabilization. Ranolazine also exerts use-dependent block of WT and mutant NaV1.1 channels providing further evidence of inactivation stabilization, but the concentrations required for these effects are much higher than the usual therapeutic plasma levels of the drug.
The binding of ranolazine to NaV1.1 channels may involve drug–receptor site interactions reported for other sodium isoforms. In a previous report investigating block of NaV1.4 and NaV1.7 channels, Wang et al. (2008) determined that ranolazine selectively binds open states with minimal binding to either closed or inactivated states. Their study utilized voltage-train protocols with increasing step durations to correlate ranolazine use-dependent inhibition with the presentation of open conformations. The authors also reported a moderately rapid association rate (kon= 8.2 µM−1 s−1) for NaV1.4 channels, which they suggested would allow drug binding only after channels respond normally to membrane depolarization. Unfortunately, to control current magnitude, this study employed an inverse sodium gradient (65 mM external and 130 mM internal), and the resultant non-physiological efflux of sodium ions may have affected drug binding kinetics, especially if ranolazine binds near the ion conduction pathway in open conformations. A second study by Rajamani et al. (2008a) also investigated the state-dependent binding of ranolazine to NaV1.7 and NaV1.8 channels. This study used a similar voltage-train protocol to demonstrate ranolazine binding to open channels. The authors also report significant shifts in the voltage dependence of channel activation and inactivation that were interpreted as evidence for drug binding to inactivated states. Our findings demonstrate that NaV1.1 channels respond differently to ranolazine with no observable effects on voltage dependence of activation or inactivation.
Our results combined with earlier data highlight the diverse actions of ranolazine among sodium channel isoforms. Nevertheless, each study investigating the inhibition of sodium channels by ranolazine has reported preferential block of persistent current with a selectivity of between 9 and 17-fold (Fredj et al., 2006; Wang et al., 2008; Rajamani et al., 2009). Further investigations are needed to determine the mechanism of persistent current block by ranolazine. Among the possible mechanisms of action are: (i) binding to open states and occluding the pore; (ii) binding to open states and providing secondary inactivation stabilization; (iii) binding to inactivated states to directly stabilization inactivation; or (iv) a combination of each. Evidence for involvement of the intracellular local anaesthetic binding site was supported by the observation that mutating the binding site in NaV1.5 and NaV1.4 channels reduces the efficacy of ranolazine (Fredj et al., 2006; Wang et al., 2008).
At usual clinical dosages, ranolazine is well tolerated with a minority of patients experiencing mild adverse effects such as dizziness, nausea, headache and constipation (Nash et al., 2008). Ranolazine also blocks the cardiac voltage-gated potassium channel HERG (Rajamani et al., 2008b) and this accounts for the mild degree of QT interval prolongation observed in some subjects. We demonstrated that ranolazine is able to cross the blood–brain barrier, which may explain certain adverse effects such as dizziness and headache reported by subjects receiving the drug. Furthermore, demonstration of ranolazine brain penetration prompts us to speculate that this drug could exert an anti-epileptic effect in persons carrying certain sodium channel mutations such as those we have tested in this study.
The strategy of selective suppression of persistent sodium current might not be appropriate for all subjects with sodium channel mutations. In particular, we predict that this approach could be counterproductive in SMEI caused by non-functional alleles (e.g. nonsense, frame shift mutations) based on anecdotal evidence that sodium channel blocking anti-epileptic drugs may aggravate this condition (Kassai et al., 2008). However, some cases of borderline SMEI and related syndromes carry missense mutations in SCN1A that promote increased persistent sodium current (Rhodes et al., 2005). We speculate that selective suppression of persistent current would have greatest value in epilepsy prophylaxis rather than in aborting active seizures based on the somewhat limited degree of use-dependent block exerted by the drug. Some degree of sodium channel use-dependent inhibition is likely to be important for an anticonvulsant effect and the therapeutic value of drugs selective for persistent current such as ranolazine might depend on the right balance of these two pharmacological actions. Perhaps, a combination of a highly selective persistent current blocker with a more conventional anti-epileptic drug would offer synergistic benefit. Testing these concepts in appropriate animal models is an important and logical next step.
Acknowledgments
The authors would like to thank Melissa Daniels for her valuable technical assistance and Christopher Thompson for critical review of this manuscript. This article is supported by NIH grant NS032387 (A.L.G.) and funds from Gilead Sciences Palo Alto Inc.
Glossary
Abbreviations
- FHM3
familial hemiplegic migraine type 3
- GEFS+
generalized epilepsy with febrile seizures plus
- LQTS
long-QT syndrome
- SMEI
severe myoclonic epilepsy of infancy
- TTX
tetrodotoxin
- WT
wild type
Statement of conflicts of interest
Drs Lepist, Leung and Rajamani are employees of Gilead Sciences, Inc. that sells ranolazine. Dr George received grant funding from Gilead Sciences, Inc. to support this study.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 Ranolazine (3 μM) inhibits usedependent current. WT NaV1.1 and R1648H availability during repetitive stimulation was assessed with a depolarizing pulse train (−10 mV, 5 ms, 300 pulses) at frequencies between 10 and 135 Hz during sequential superfusion of control solution followed by 3 μM ranolazine. Normalized peak current (pulse 300/pulse 1) was plotted versus frequency for each pulse train (n = 9 and 8, respectively).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Abrams J, Jones CA, Kirkpatrick P. Ranolazine. Nat Rev Drug Discov. 2006;5:453–454. doi: 10.1038/nrd2069. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allely MC, Brown CM, Kenny BA, Kilpatrick AT, Martin A, Spedding M. Modulation of α1-adrenoceptors in rat left ventricle by ischaemia and acyl carnitines: protection by ranolazine. J Cardiovasc Pharmacol. 1993;21:869–873. doi: 10.1097/00005344-199306000-00004. [DOI] [PubMed] [Google Scholar]
- Chaitman BR. Ranolazine for the treatment of chronic angina and potential use in other cardiovascular conditions. Circulation. 2006;113:2462–2472. doi: 10.1161/CIRCULATIONAHA.105.597500. [DOI] [PubMed] [Google Scholar]
- Chen BS, Lo YC, Peng H, Hsu TI, Wu SN. Effects of ranolazine, a novel anti-anginal drug, on ion currents and membrane potential in pituitary tumor GH3 cells and NG108-15 neuronal cells. J Pharmacol Sci. 2009;110:295–305. doi: 10.1254/jphs.09018fp. [DOI] [PubMed] [Google Scholar]
- Dichgans M, Freilinger T, Eckstein G, Babini E, Lorenz-Depiereux B, Biskup S, et al. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of LQT-3 mutant sodium channels: evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AL., Jr Inherited disorders of voltage-gated sodium channels. J Clin Invest. 2005;115:1990–1999. doi: 10.1172/JCI25505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Misra SN, George AL., Jr Impaired inactivation gate stabilization predicts increased persistent current for an epilepsy-associated SCN1A mutation. J Neurosci. 2006;26:10958–10966. doi: 10.1523/JNEUROSCI.3378-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Rhodes TH, Pusch M, Freilinger T, Pereira-Monteiro JM, Ferrari MD, et al. Divergent sodium channel defects in familial hemiplegic migraine. Proc Natl Acad Sci USA. 2008;105:9799–9804. doi: 10.1073/pnas.0711717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai B, Chiron C, Augier S, Cucherat M, Rey E, Gueyffier F, et al. Severe myoclonic epilepsy in infancy: a systematic review and a meta-analysis of individual patient data. Epilepsia. 2008;49:343–348. doi: 10.1111/j.1528-1167.2007.01423.x. [DOI] [PubMed] [Google Scholar]
- Letienne R, Vie B, Puech A, Vieu S, Le GB, John GW. Evidence that ranolazine behaves as a weak β1- and β2-adrenoceptor antagonist in the cat cardiovascular system. Naunyn Schmiedebergs Arch Pharmacol. 2001;363:464–471. doi: 10.1007/s002100000378. [DOI] [PubMed] [Google Scholar]
- Lossin C. A catalog of SCN1A variants. Brain Dev. 2009;31:114–130. doi: 10.1016/j.braindev.2008.07.011. [DOI] [PubMed] [Google Scholar]
- Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34:877–884. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA. Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest. 2005;115:2010–2017. doi: 10.1172/JCI25466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulley JC, Scheffer IE, Petrou S, Dibbens LM, Berkovic SF, Harkin LA. SCN1A mutations and epilepsy. Hum Mutat. 2005;25:535–542. doi: 10.1002/humu.20178. [DOI] [PubMed] [Google Scholar]
- Nash DT, Nash SD. Ranolazine for chronic stable angina. Lancet. 2008;372:1335–1341. doi: 10.1016/S0140-6736(08)61554-8. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Shryock JC, Belardinelli L. Block of tetrodotoxin-sensitive, NaV1.7 and tetrodotoxin-resistant, NaV1.8, Na+ channels by ranolazine. Channels. 2008a;2:449–460. doi: 10.4161/chan.2.6.7362. [DOI] [PubMed] [Google Scholar]
- Rajamani S, Shryock JC, Belardinelli L. Rapid kinetic interactions of ranolazine with HERG K+ current. J Cardiovasc Pharmacol. 2008b;51:581–589. doi: 10.1097/FJC.0b013e3181799690. [DOI] [PubMed] [Google Scholar]
- Rajamani S, El-Bizri N, Shryock JC, Makiyama T, Belardinelli L. Use-dependent block of cardiac late Na+ current by ranolazine. Heart Rhythm. 2009;6:1625–1631. doi: 10.1016/j.hrthm.2009.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes TH, Lossin C, Vanoye CG, Wang DW, George AL., Jr Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc Natl Acad Sci USA. 2004;101:11147–11152. doi: 10.1073/pnas.0402482101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes TH, Vanoye CG, Ohmori I, Ogiwara I, Yamakawa K, George AL., Jr Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures. J Physiol. 2005;569:433–445. doi: 10.1113/jphysiol.2005.094326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Priori SG, Locati EH, Napolitano C, Cantù F, Towbin JA, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate – Implications for gene-specific therapy. Circulation. 1995;92:3381–3386. doi: 10.1161/01.cir.92.12.3381. [DOI] [PubMed] [Google Scholar]
- Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. doi: 10.1038/sj.bjp.0707522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicouri S, Glass A, Belardinelli L, Antzelevitch C. Antiarrhythmic effects of ranolazine in canine pulmonary vein sleeve preparations. Heart Rhythm. 2008;5:1019–1026. doi: 10.1016/j.hrthm.2008.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the pro-arrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- Spampanato J, Kearney JA, de Haan G, McEwen DP, Escayg A, Aradi I, et al. A novel epilepsy mutation in the sodium channel SCN1A identifies a cytoplasmic domain for beta subunit interaction. J Neurosci. 2004;24:10022–10034. doi: 10.1523/JNEUROSCI.2034-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanmolkot KR, Babini E, de VB, Stam AH, Freilinger T, Terwindt GM, et al. The novel p.L1649Q mutation in the SCN1A epilepsy gene is associated with familial hemiplegic migraine: genetic and functional studies. Hum Mutat. 2007;28:522. doi: 10.1002/humu.9486. [DOI] [PubMed] [Google Scholar]
- Wang DW, Yazawa K, Makita N, George AL, Jr, Bennett PB. Pharmacological targeting of long QT mutant sodium channels. J Clin Invest. 1997;99:1714–1720. doi: 10.1172/JCI119335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GK, Calderon J, Wang SY. State- and use-dependent block of muscle NaV1.4 and neuronal NaV1.7 voltage-gated Na+ channel isoforms by ranolazine. Mol Pharmacol. 2008;73:940–948. doi: 10.1124/mol.107.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Shryock JC, Song Y, Li Y, Antzelevitch C, Belardinelli L. Antiarrhythmic effects of ranolazine in a guinea pig in vitro model of long-QT syndrome. J Pharmacol Exp Ther. 2004;310:599–605. doi: 10.1124/jpet.104.066100. [DOI] [PubMed] [Google Scholar]
- Wu SN, Chen BS, Hsu TI, Peng H, Wu YH, Lo YC. Analytical studies of rapidly inactivating and non-inactivating sodium currents in differentiated NG108-15 neuronal cells. J Theor Biol. 2009;259:828–836. doi: 10.1016/j.jtbi.2009.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.