

Abstract
Different primary lysosomal trafficking defects lead to common alterations in lipid trafficking, suggesting cooperative interactions among lysosomal lipids. However, cellular analysis of the functional consequences of this phenomenon is lacking. As a test case, we studied cells with defective Niemann-Pick C1 (NPC1) protein, a cholesterol trafficking protein whose defect gives rise to lysosomal accumulation of cholesterol and other lipids, leading to NPC disease. NPC1 cells also develop a secondary defect in acid sphingomyelinase (SMase) activity despite a normal acid SMase gene (SMPD1). When acid SMase activity was restored to normal levels in NPC1-deficient CHO cells through SMPD1 transfection, there was a dramatic reduction in lysosomal cholesterol. Two other defects, excess lysosomal bis-(monoacylglycerol) phosphate (BMP) and defective transferrin receptor (TfR) recycling, were also markedly improved. To test its relevance in human cells, the acid SMase activity defect in fibroblasts from NPC1 patients was corrected by SMPD1 transfection or acid SMase enzyme replacement. Both treatments resulted in a dramatic reduction in lysosomal cholesterol. These data show that correcting one aspect of a complex lysosomal lipid storage disease can reduce the cellular consequences even if the primary genetic defect is not corrected.
Keywords: acid sphingomyelinase, cholesterol, lipid trafficking, lysosomal storage disease, lysosomes, Niemann-Pick C
Lysosomal lipid storage diseases (LSDs) are caused by mutations in specific lysosomal hydrolases, trafficking proteins or their co-factors, leading to the accumulation of substrate compounds in late endosome-derived structures called lysosomal storage organelles (LSOs) (1,2). The primary cellular abnormality often perturbs the trafficking of multiple lipids and proteins, which probably contributes to the overall pathophysiology of the disease (3,4). These trafficking abnormalities and other secondary defects may, in turn, amplify the cellular pathophysiology triggered by the primary mutation. Identification and functional assessment of these secondary defects may therefore offer new therapeutic opportunities even if the primary genetic defect is not corrected.
To test this concept, we studied cells lacking the late endosomal protein Niemann-Pick C1 (NPC1). NPC1, a membrane protein, co-operates in some manner with NPC2, a cholesterol-binding protein in the lumen of endosomes, to transfer endocytosed cholesterol from the lumen to the membrane of late endosomes (5,6). The cholesterol is then transferred from the endosomal membrane to peripheral cellular sites through one or more processes that are still under intense investigation [cf. (7)]. Cells lacking functional NPC1 and NPC2 show defects in the transport of cholesterol and other lipids from late endosomes to peripheral sites in the cell (8-10). Mutations in NPC1 and NPC2 give rise to Niemann-Pick C disease, which is characterized by hepatosplenomegaly, liver disease and potentially devastating neurological disease (9,11,12).
At the cellular level, cells with defective NPC1 accumulate cholesterol along with excess sphingomyelin (SM), glycosphingolipids and bis- (monoacylglycerol) phosphate (BMP) (3,4,13). It remains unclear whether the primary defect in NPC1 mutants is directly associated with cholesterol transport or whether the cholesterol accumulation is secondary to accumulation of other lipids, which associate with cholesterol in membranes (4,10). In this regard, it is noteworthy that cholesterol-enriched NPC cells and tissues from NPC1-mutant mice and humans have a secondary, post-translational defect in the activity of a lysosomal enzyme, acid sphingomyelinase (SMase) (14-19). This alteration in acid SMase activity can be observed in wild-type (WT) cells with increased levels of late endosomal cholesterol resulting from incubation with low-density lipoprotein (LDL) and progesterone (16). Thus, elevated cholesterol and elevated SM appear to be synergistically linked in a positive feedback loop.
Although the mechanism of suppression of acid SMase activity by cholesterol is not known, we reasoned that this defect might have functional significance. In particular, primary deficiency of acid SMase (types A and B Niemann-Pick disease) leads to cellular defects and disease characteristics that share certain features with NPC disease. For example, both types A/B and type C Niemann-Pick disease patients have hepatosplenomegaly and neurological abnormalities (12,20), and we showed that macrophages lacking acid SMase have defective late endosomal cholesterol trafficking (21). We therefore wondered whether we could correct some of the trafficking defects in cells with NPC disease by correcting the secondary enzymatic activity defect (i.e. acid SMase). In this study, we present cell culture data that provide support for this concept.
Results
Restoration of acid SMase activity in NPC1-defective CT60 cells by transfection with WT and/or C629S acid SMase (SMPD1) cDNA
As a model of NPC1 cells, we studied CT60 cells, which are NPC1-deficient CHO cells derived from a parental line called 25RA (22,23). Consistent with previous data (16), we found that CT60 cells had much lower acid SMase activity than the 25RA cells (Figure 1A) despite no decrease in acid SMase protein. In preparation for studies addressing the role of defective acid SMase activity in CT60 cells, we sought to restore enzymatic activity in these cells through human acid SMase gene (SMPD1) transfection. For this purpose, we used two cDNA constructs: WT SMPD1 cDNA and a site-directed mutant cDNA (C629S SMPD1), which encodes acid SMase in which C-terminal Cys-629 of the enzyme is replaced with Ser. Qiu et al. (24)proposed that C629S acid SMase mimics a naturally occurring processed form of the enzyme that has increased enzymatic activity. As shown in Figure 1A, cells transfected with either form of the cDNA demonstrated restoration of acid SMase activity to a level similar to that in the parental 25RA cells, whereas those transfected with a construct not containing the SMPD1 cDNA (VEC) had a level of acid SMase activity similar to that in non-transfected CT60 cells. Despite our hypothesis on the potential functional consequences of acid SMase in NPC1 cells, we predicted that the overall quantitative effect of the NPC1 mutation and subsequent acid SMase correction on total cellular SM mass would be relatively modest, because only the pool of SM in late endosomes/lysosomes should be accessible to this enzyme. We found that total cellular SM mass was 20% higher in CT60 and CT60 cells transfected with empty vector (CT60-VEC) compared with that in 25RA cells (p < 0.05), and transfection of CT60 cells with WT or C629S SMPD1 cDNA lowered SM mass close to the value in 25RA cells (data not shown). In summary, WT levels of acid SMase activity can be functionally restored in CT60 cells through transfection with either WT or C629S SMPD1 cDNA, thus allowing an assessment of whether this restoration can correct trafficking defects in these cells.
Figure 1.
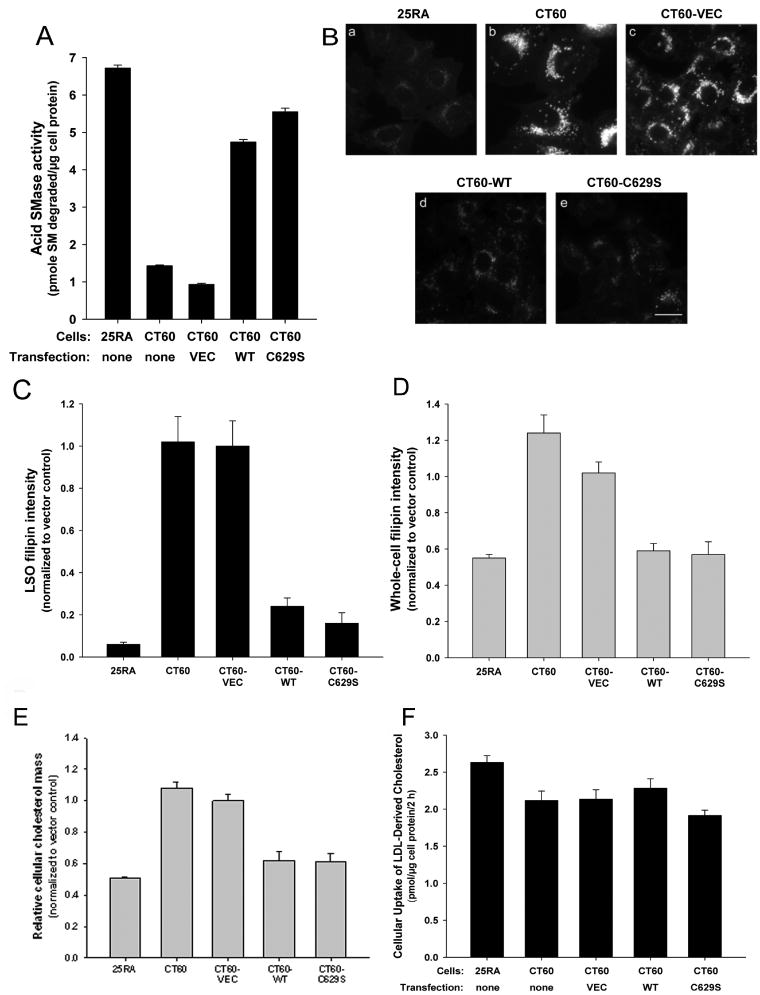
Decrease in cholesterol accumulation in LSOs by restoration of acid SMase activity in CT60 cells. A) CT60 cells were transfected with empty vector (VEC) or with vector containing either the WT or the C629S SMPD1 cDNA constructs. Two days later, extracts of these cells and non-transfected 25RA and CT60 cells were assayed for acid SMase activity. The activity levels in CT60-WT and CT60-C629S cells were significantly different from those in non-transfected CT60 cells and CT60-VEC cells (p < 0.0001). B) The five cell groups described in (A) were fixed and stained with filipin. The images are displayed with the same gray scale range. Scale bar, 20 μm. C) Quantification of filipin fluorescence in the LSOs and D) in whole cells. Each bar in the data quantification represents the average of 30 images from two independent experiments ±SEM. The CT60-WT and CT60-C629S values were significantly different from the CT60 and CT60-VEC values (p < 0.0001). NB: The 25RA and CT60 cells used in this experiment are the same ones used in Figure 4, i.e. they express the hTfR. However, they have the same level of cholesterol accumulation in LSOs and the same response to acid SMase restoration as cells not expressing the TfR (data not shown). E) Free cholesterol levels in each of the five cell groups described in (A) were assayed by gas chromatography. Each bar represents an average of four samples from two independent experiments and is normalized to CT60-VEC value, which was 45.7 ± 1.7 μg cholesterol/mg cell protein. The CT60-WT and CT60-C629S values were significantly different from the CT60 and CT60-VEC values (p < 0.0001). F) Monolayers of the five groups of cells described above were incubated for 2 h with 5 μg/mL LDL reconstituted with [14C]cholesteryl ester (CE). Lipid extracts of the cells were then fractionated by thin-layer chromatography, and the areas of the plate corresponding to cholesterol and CE, which accounted for all of the radioactivity, were scraped and counted for [14C]cpm. [14C]cholesterol represents hydrolyzed LDL–CE in the cells, and [14C]CE represents either unhydrolyzed LDL–CE or hydrolyzed LDL-cholesterol that was re-esterified in the cells to CE. The data shown are derived from the total LDL-derived cholesterol in the cells (free cholesterol + CE) and are mean of 5 values ±SEM. The values for cellular-free cholesterol derived from LDL were similar among all the five cell types: 1.92, 1.88, 1.93, 2.05, 1.72 pmol/μg cell protein, respectively. None of the differences in total or free LDL-derived cellular cholesterol among the five groups of cells reached statistical significance.
Restoration of acid SMase activity in CT60 cells leads to a decrease in cholesterol accumulation in LSOs
Previous mechanistic studies have shown that enrichment of membranes with SM can disturb cholesterol trafficking because of SM–cholesterol interactions, perturbations in membrane biophysical properties and defective interaction of cholesterol transport proteins with SM-rich membranes (5,21,25,26). We therefore wondered whether we could correct some of the trafficking defects in NPC cells by restoring normal levels of acid SMase activity. To test this hypothesis, we quantified LSO filipin fluorescence in CT60 cells stably transfected with the cDNA constructs described in Materials and Methods. Shown in Figure 1B, a–c, is the expected increase in LSO filipin fluorescence in CT60 or CT60-VEC cells compared with that in 25RA cells. Most importantly, the data show a striking loss of LSO filipin fluorescence in the cells expressing WT or C629S acid SMase (Figure 1B, d and e). Quantification of LSO and whole-cell fluorescence confirmed these observations (Figure 1C,D). We and others have shown previously that estimation of cholesterol levels using filipin assay is comparable with free cholesterol levels quantified by biochemical methods (27,28). To confirm our filipin results, we also estimated the free cholesterol using gas chromatography (Figure 1E). A previous report showed that the free cholesterol in control 25RA cells is only ~40% compared with that of CT60 cells (29). We obtained similar data as can be seen by the ~45–50% decrease in free cholesterol content in control 25RA cells compared with that in CT60 or CT60-VEC cells. In agreement with our whole-cell filipin results, we found that the level of free cholesterol was markedly decreased in CT60-WT and CT60-C629S cells and approached the level found in 25RA cells. Note that the loss of LSO filipin fluorescence in the SMPD1-transfected cells could not be explained by a decreased uptake of LDL-cholesterol, which was similar among all five cell types (Figure 1E). Thus, correcting the acid SMase activity defect in NPC1-mutant CT60 cells has a dramatic effect on improving a fundamental characteristic of these cells, namely, cholesterol accumulation in LSOs.
Cholesterol leaving LSOs in acid SMase-transfected CT60 cells might accumulate in other cellular membranes, become esterified by acyl-CoA:cholesterol acyltransferase and/or get effluxed from the cells. The fact that whole-cell filipin fluorescence is lower in the SMPD1-transfected cells and becomes comparable with that in the parental 25RA cells suggests that accumulation of high concentrations of unesterified cholesterol in non-LSO sites is not a major fate of the cholesterol. Moreover, when cells were incubated with [3H]cholesterol-labeled LDL for 4 h and then chased for up to 24 h, we did not observe an increase in esterification in CT60 cells transfected with SMPD1 versus CT60-VEC cells (data not shown), suggesting that trafficking to and esterification by ACAT in the endoplasmic reticulum (ER) are also not a major fate of the acid SMase-mediated released LSO-derived cholesterol. However, efflux of the LDL-derived [3H]cholesterol, which is markedly decreased in CT60 cells, was restored by ~20% after 24 h in SMPD1-transfected cells (Figure 2A). The corresponding acid SMase activity measurements are shown in Figure 2B. These data suggest that a portion of the cholesterol exiting the LSO in acid SMase-restored CT60 cells is effluxed, with the rest probably being distributed diffusely to other cellular membranes.
Figure 2.
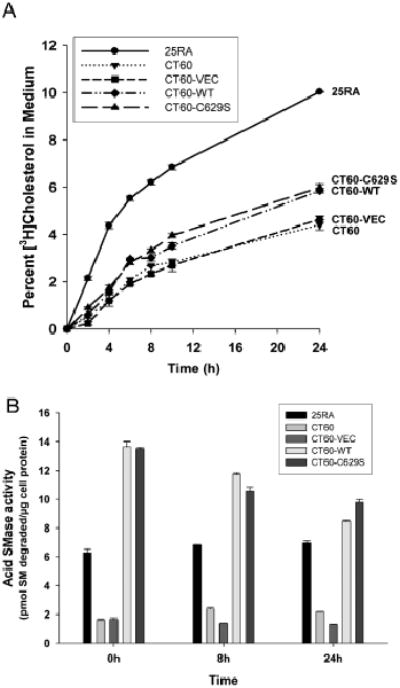
Partial correction of efflux of LDL-derived [3H]cholesterol from CT60 cells by restoration of acid SMase activity. A) Monolayers of 25RA, CT60, CT60-VEC, CT60-WT and CT60-C629S were incubated for 4 h in serum-free medium containing 10 μg/mL [3H]CE-labeled LDL. The cells were then rinsed and incubated with fresh medium containing 50 μg/mL HDL3 for the indicated times. Tritium radioactivity in the media and cells was measured to calculate the percent [3H]cholesterol in the medium. The values for CT60-WT and CT60-C629S cells were significantly different from the other three values at 24 h (p < 0.005). B) The bottom graph shows acid SMase activity in the five groups of cells at 0, 8 and 24 h after incubation in conditions nearly identical to those in (A), except that cells were incubated with unlabeled LDL. The values for CT60-WT and CT60-C629S cells were significantly different from that of CT60 and CT60-VEC (p <0.005).
Restoration of acid SMase activity in CT60 cells leads to a decrease in BMP accumulation and an increase in the half-life of transferrin receptor (TfR) recycling
The hydrophobic, acidic phospholipid, BMP, also called lysobisphosphatidic acid (LBPA), has been shown to accumulate within NPC1 cells (30,31). To test the possibility that restoration of acid SMase activity could have a broad corrective effect on NPC cells, we investigated whether BMP accumulation was also diminished in SMPD1-transfected CT60 cells. Consistent with the data cited above, CT60 cells accumulated much more BMP than 25RA cells (Figure 3A, a–b). Cells transfected with non-SMPD1-containing control vector also accumulated large amounts of BMP (Figure 3A, c). In contrast, cells transfected with WT or C629S SMPD1 had greatly diminished accumulation of BMP (Figure 3A, d–e). The quantified data are shown in Figure 3B. Thus, defective BMP accumulation, like defective cholesterol accumulation, can be partially corrected by restoration of acid SMase activity in NPC1-deficient cells.
Figure 3.
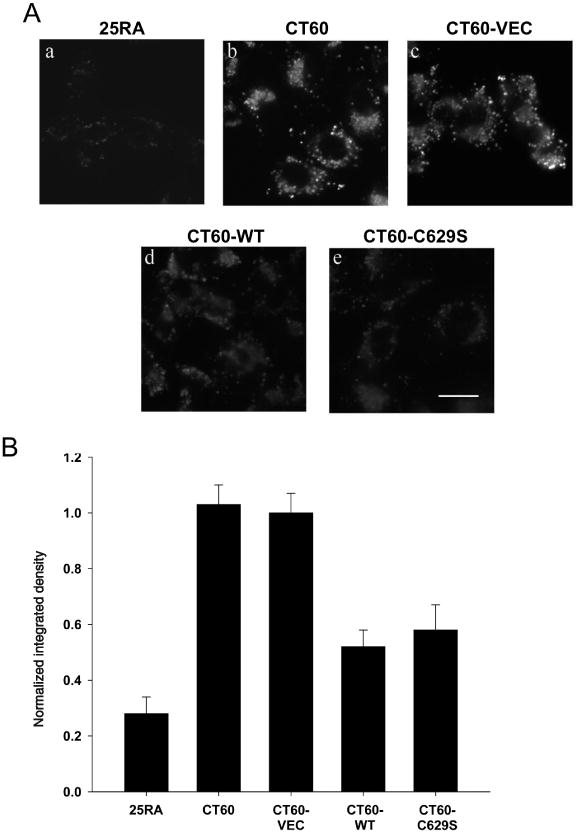
Decrease in BMP accumulation in CT60 cells by restoration of acid SMase activity. A) Anti-BMP immunofluorescence in 25RA, CT60, CT60-VEC, CT60-WT and CT60-C629S cells. Scale bar, 20 μm. B) Quantitation of anti-BMP immunofluorescence intensity. Each bar in the data quantification represents the average of 20 images from two independent experiments ±SEM. The values for CT60-WT and CT60-C629S cells were significantly different from that of CT60 and CT60-VEC cells (p < 0.01). NB: As in Figure 1, the cells used here express the hTfR, but they have the same level of BMP accumulation in LSOs and the same response to acid SMase restoration as the cells not expressing the TfR (data not shown).
We reported previously that the recycling rates of TfRs are decreased in NPC1 cells compared with that in normal fibroblasts (30,32). Using 25RA and CT60 cells that express the human transferrin receptor (hTfR) (30), we were able to assess TfR cycling in the control and SMPD1-transfected cells and to determine whether the decrease in recycling in NPC1-deficient cells could be corrected by restoration of acid SMase activity. To assay TfR recycling, the cells were incubated with [125I]Tf to achieve steady-state occupancy of the TfR with Tf. The Tf bound to the cell surface was then removed, and the release of internal Tf from the cells was monitored as a function of time. The [125I]Tf released into the medium reflects the return of TfR from endosomes to plasma membrane. The fraction of [125I]Tf remaining in the cells decreases as a function of time as a first-order process, and therefore the decrease in cell-associated [125I]Tf fits an exponential decay curve. We previously reported that the Tf efflux kinetics yielded a t1/2 of 11.3 min for 25RA cells and 21.7 min for CT60 cells (30), which is consistent with our current results (Figure 4). As expected, the TfR recycling defect persisted in CT60-VEC cells. In contrast, cells expressing either WT or C629S acid SMase displayed a marked improvement in Tf trafficking, showing a t1/2 value close to that of 25RA cells. Thus, the defect in TfR recycling in CT60 cells, similar to the defect in LSO cholesterol and BMP accumulation, can be substantially corrected by restoration of acid SMase activity.
Figure 4.
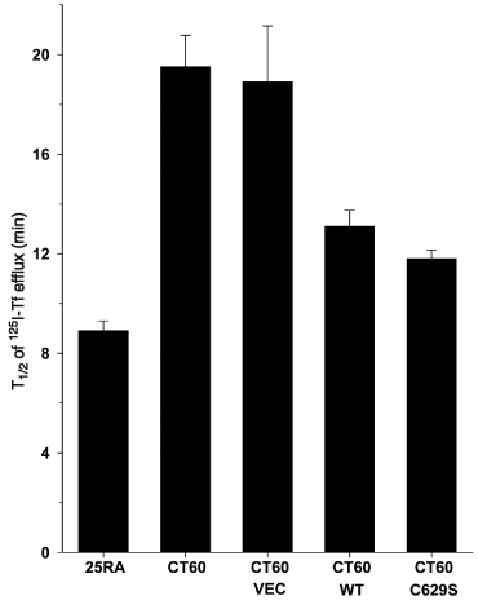
Recycling of the transferrin receptor in CT60 cells is improved by restoration of acid SMase activity. The efflux kinetics of [125I]-transferrin was measured in 25RA, CT60, CT60-VEC, CT60-WT and CT60-C629S cells expressing the hTfR, as described in Materials and Methods. The values for CT60-WT and CT60-C629S cells were significantly different from that of CT60 and CT60-VEC cells (p < 0.05).
Genetic restoration of acid SMase activity in human NPC1 cells leads to a dramatic decrease in cholesterol accumulation in LSOs
To the extent that the secondary defect in acid SMase activity observed in NPC1 cells and in tissues from NPC mice and humans contributes to one or more aspects of NPC disease pathology (see Discussion), the data in this report raise the possibility that correction of this enzyme activity defect may be clinically beneficial. Moreover, because lysosomal pH is the same in control and NPC1 fibroblasts (NPC Fbs) (4), the loss of acid SMase activity was not due to abnormal lysosomal pH. To explore this concept at the cellular level, we conducted a series of acid SMase restoration experiments using skin fibroblasts isolated from a compound heterozygous child with the late infantile form of NPC1 disease. NPC1 expression in these cells is undetectable by immunoblot analysis, and the cells display a severe defect in trafficking lipoprotein-derived cholesterol (33,34). As described previously (35), and verified by us (below), these fibroblasts also have a partial defect in acid SMase activity. Filipin staining of the cells revealed, as expected, intense LSO staining, indicative of LSO cholesterol accumulation (Figure 5A, a–f and first two bars of plot 5B). Cells transfected with the empty vector, which contained a GFP expression construct, showed marked filipin staining that was indistinguishable from untransfected cells (Figure 5A, g–i and third bar of plot 5B). When the cells were transfected with either the WT or the C629S SMPD1 cDNA construct, LSO filipin staining was dramatically reduced in cells that were successfully transfected (green) but not in cells that were not successfully transfected (non-green), which acted as an internal negative control (Figure 5A, j–o and last two bars of plot 5B).
Figure 5.
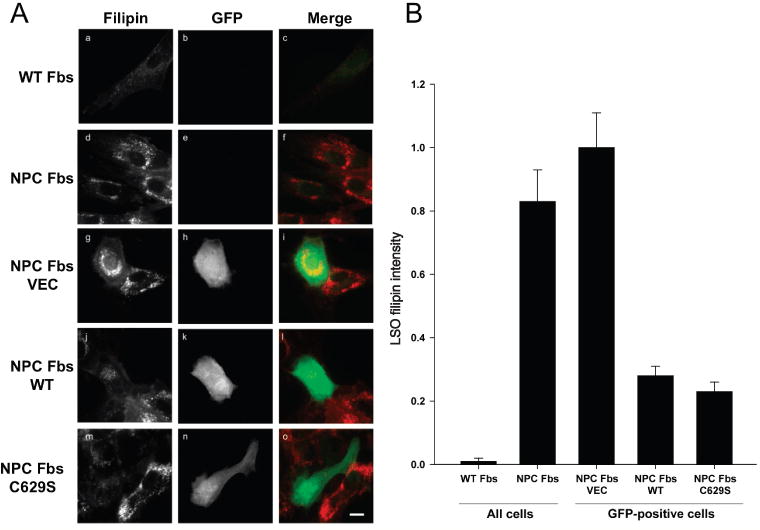
Decrease in cholesterol accumulation in LSOs by genetic restoration of acid SMase activity in human fibroblasts. A) Human WT (GM05659) and NPC (GM03123) Fbs were left untransfected or were transiently transfected with empty GFP-expressing vector (VEC) or with GFP-vector containing either WT or C629S SMPD1 cDNA constructs. Two days later, the cells were washed with PBS, fixed and stained with filipin. Standard UV and FITC filters were used for filipin imaging (all cells) or GFP imaging (transfected cells), respectively. The displayed filipin images and GFP images are on the same gray scale range, respectively. Scale bar, 30 μm. B) The bar graph shows LSO ratio values normalized to the NPC-VEC values (average of 20–30 images from three independent experiments ±SEM). The values for NPC and NPC-VEC fibroblasts were significantly different from those for NPC-WT and NPC-C629S fibroblasts (p < 0.0001).
Restoration of acid SMase activity in human NPC1 cells by exogenous acid SMase also leads to a decrease in cholesterol accumulation in LSOs
Lysosomal enzyme defects, unlike defects in lysosomal membrane proteins such as NPC1, can be corrected both in vitro and in vivo using enzyme replacement therapy (36,37). Indeed, this approach is currently being tested in humans with primary acid SMase deficiency (38) (http://www.genzyme.com/research/medical/medi_home.asp). This strategy takes advantage of the fact that cells can endocytose lysosomal enzymes and deliver them in a functionally active state to late endosomes and lysosomes (36). To apply this concept to human NPC Fbs, the cells were pre-treated in the absence or presence of recombinant human acid SMase (rhASM) and then assayed for acid SMase activity and LSO fluorescence after filipin staining. As shown in Figure 6A, the defect in acid SMase activity in the NPC Fbs was corrected by pre-treatment with rhASM. Most importantly, acid SMase replacement dramatically decreased LSO filipin fluorescence (Figure 6B). Similar results were obtained using NPC1 skin fibroblasts from a different donor (GM18453) (Figure 6C). Thus, restoration of acid SMase activity in NPC1 cells using two independent methods, genetic and enzyme replacement, markedly corrected the defect in LSO cholesterol accumulation.
Figure 6.
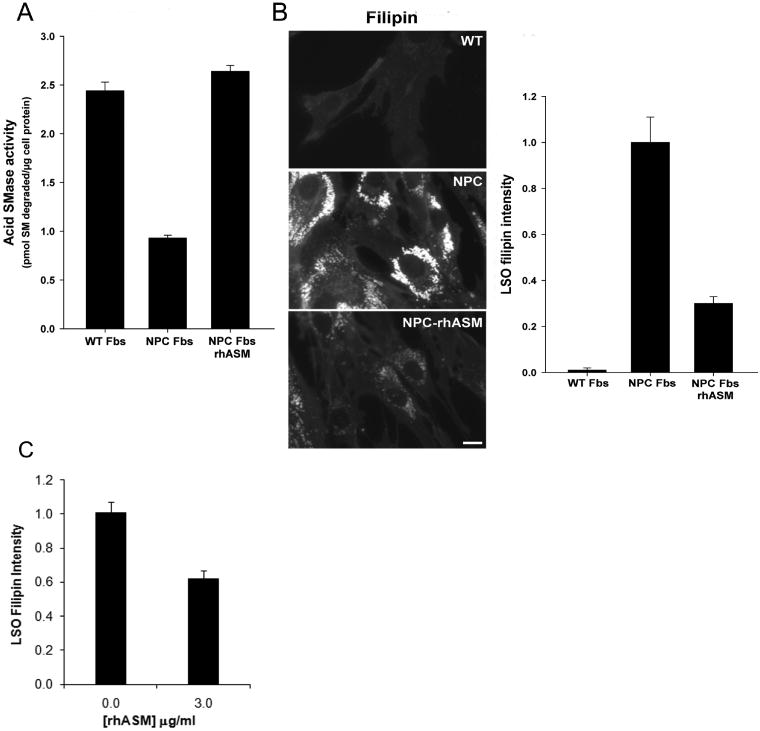
Exogenous acid SMase decreases cholesterol accumulation in LSOs in human NPC fibroblasts. Parallel sets of human WT (GM05659) and NPC (GM03123) Fbs were incubated in medium alone or, in the case of the NPC fibroblasts, medium containing 3 μg/mL recombinant human acid SMase (rhASM). Two days later, the cells were washed thoroughly with PBS and either lysed and assayed for acid SMase activity (A) or fixed and stained with filipin for imaging and quantification (B). The images are displayed with the same gray scale range. Scale bar, 15 μm. The quantified data in the bar graph represent LSO ratios normalized to the WT Fb values (average of 60–66 images from three independent experiments ±SEM). C) Another human NPC Fbs (GM18453) was incubated with 3 μg/mL rhASM for 24 h, unlike 48 h in NPC1 (GM03123). Quantified data shown in bar chart are the normalized LSO ratios (normalized to untreated NPC Fbs) in the presence and absence of recombinant enzyme. Data represents an average from two independent experiments and 30–36 images ±SEM, p < 0.0001. The values for NPC fibroblasts in panels A, B and C were significantly different from both the WT Fbs and the NPC fibroblasts treated with rhASM (p < 0.0001).
Sub-cellular localization of exogenously added rhASM
To ascertain whether exogenously added rhASM was localized and processed in late endosomal organelles, WT (GM05659) and NPC1 (GM03123) human fibroblasts were incubated for 24 h in the absence or presence of 3 μg/mL of Alexa555-labeled rhASM. To remove surface labeling, the cells were further incubated for 15 min in a medium without enzyme. The cells were then washed, fixed and stained with filipin for imaging and quantification. The uptake of rhASM–Alexa555 was completely blocked when the enzyme was added in the presence of excess mannose-6-phosphate (10 mm) (data not shown). Conjugation of Alexa555 to the enzyme did not affect its activity (data not shown). Shown in Figure 7 are filipin images (A, D and G) and Alexa555 images (B, E and H) for WT, NPC1 and NPC 1+rhASM–Alexa 555, respectively. Color overlays for filipin (green) and rhASM–Alexa555 (red) are shown in panels C, F and I of Figure 7. The images in the inset are the zoomed color overlays of the regions marked in Figure 7C,F,I. Exogenously added rhASM–Alexa555, presumably internalized via the mannose-6-phosphate receptor, specifically localized to cholesterol-laden storage organelles (as shown in Figure 7C,I) and was not visible in the plasma membrane or other cellular organelles. Quantification of LSO filipin and Alexa555–rhASM intensity after incubation with 0 or 3 μg/mL Alexa555 conjugated rhASM for 24 h in WT and NPC (GM03123) Fbs (±SEM) is shown in Figure 7J,K. Note that the higher level of uptake of the rhASM in the NPC Fbs can be explained by the previous finding that expression of IGF2/MPR is increased in human NPC fibroblast (39).
Figure 7.
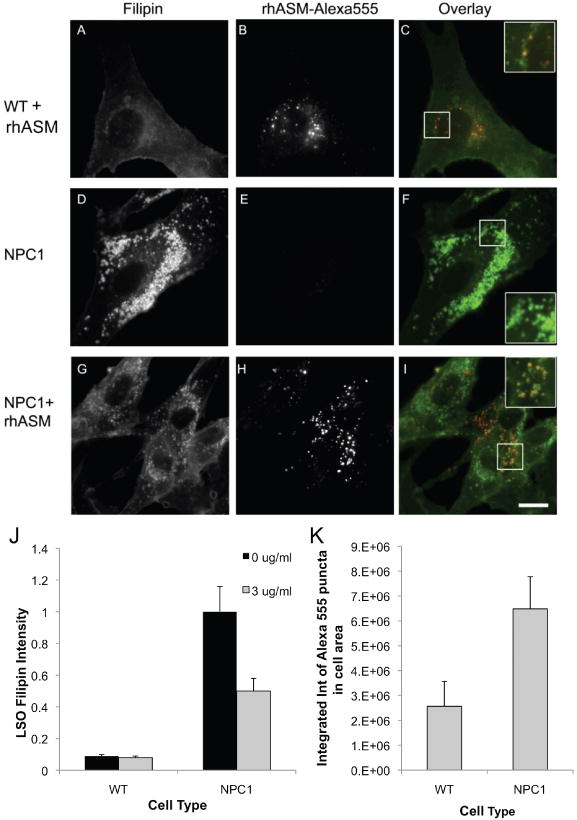
Addition of Alexa555-conjugated rhASM to demonstrate sub-cellular localization of exogenous acid SMase. WT (GM5659) and NPC (GM03123) Fbs were incubated with or without 3 μg/mL Alexa555-conjugated rhASM for 24 h. To remove surface-bound label, the cells were further incubated for 15 min with a growth medium without the enzyme. Finally, the cells were washed with PBS, fixed with 1.5% PFA and stained with filipin for imaging and quantification. The uptake of rhASM–Alexa555 was completely blocked when enzyme was added in the presence of excess mannose-6-phosphate (10 mm) (data not shown). Filipin images in panels (A, D and G) and the Alexa555 images in panel (B, E and H) for WT, NPC1 and NPC1 + rhASM –Alexa 555, respectively, are displayed on the same gray scale range. Color overlays for filipin (green) and rhASM–Alexa555 (red) are shown in panels (C, F and I). The images in the inset are the zoomed color overlays of the region marked in (C, F and I). Scale bar = 10 μm. Panel (J) shows the quantification of LSO filipin intensity after incubation with 0 or 3 μg/mL Alexa555-conjugated rhASM for 24 h in WT and NPC (GM03123) Fbs (values are ±SEM, p < 0.001). Conjugation of Alexa555 to the enzyme did not affect its activity. Panel (K) shows the quantification of rhASM uptake after incubation with 0 and 3 μg/mL Alexa555-conjugated rhASM for 24 h in WT and NPC (GM03123) Fbs (values are ±SEM, p < 0.05).
Overcoming secondary inactivation of acid SMase
To validate our hypothesis that loss of acid SMase activity is a secondary effect of cholesterol storage in NPC1 patients, we treated normal (GM05649) and NPC1 (GM03123) fibroblasts with 0, 0.2 and 1.8 μg/mL rhASM. Acid SMase activity was assayed in parallel with LSO filipin quantification as shown in Figure 8A,B. The acid SMase activity in wild-type fibroblasts (WT Fbs) (GM05659) increased slightly with 0.2 μg/mL rhASM (p < 0.05), and the activity did not increase further at 1.8 μg/mL (black bars in Figure 8A). The minimal amount of LSO filipin staining evident in normal fibroblasts (GM05659) remained low and constant for all doses of rhASM tested (black bars in Figure 8B). In contrast, the addition of 0.2 μg/mL rhASM to NPC Fbs (GM03123) increased acid SMase activity significantly with a corresponding modest decrease in LSO filipin intensity (~20%), which indicates a decrease in LSO cholesterol accumulation (hashed bars in Figure 8A,B). Most importantly, increasing the rhASM dose to 1.8 μg/mL resulted in a marked reduction of LSO filipin intensity (~60%). Our earlier experiments with genetic restoration of acid SMase in both CHO and human NPC1 mutant cells (Figures 1 and 5) showed an approximately 65–70% decrease in LSO filipin. Similarly, addition of rhASM back to human fibroblast at a higher (3 μg/mL) concentration (Figure 6) also resulted in ~65–70% decrease in LSO filipin intensity. Thus, the inhibited acid SMase activity in NPC1 cells can be rescued by high-dose exogenous rhASM treatment, which presumably overcomes the enzyme inactivation process.
Figure 8.
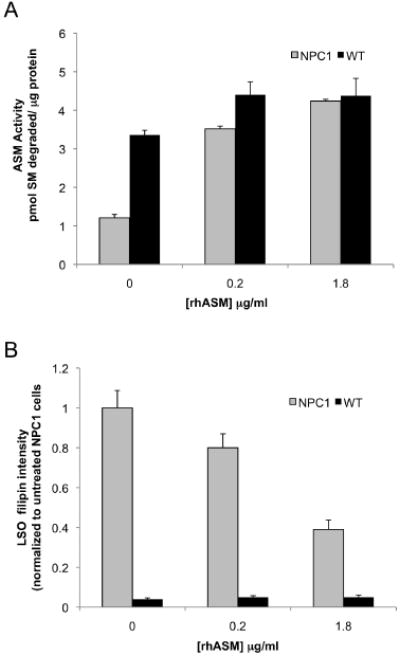
Decreased LSO cholesterol accumulation and increased acid SMase activity are achieved in NPC1 human fibroblasts by increasing amounts of rhASM. WT (GM5659) and NPC (GM03123) Fbs were incubated with 0, 0.2 and 1.8 μg/mL rhASM for 24 h. To remove surface-bound label, the cells were further incubated for 15 min with growth medium without the enzyme. The cells were washed thoroughly with PBS and either lysed and assayed for acid SMase activity (A) or fixed and stained with filipin for imaging and quantification (B). Each data point in plot (A) is representative of three wells in an experiment, p < 0.05. Plot (B) represents LSO ratios normalized to the untreated NPC Fb GM01323 values (average of 16–20 images from two independent experiments ±SEM), p < 0.005.
Discussion
The panoply of cellular perturbations triggered by dysfunctional mutations in individual lysosomal enzymes, co-factors or transport proteins reflects multiple adverse effects of excess substrate accumulation (1,2). For LSDs involving the accumulation of certain lipids in LSOs, physical or biochemical consequences of accumulation of these excess lipids may lead to secondary effects on lysosomal/late endosomal processes that, in turn, could amplify the original defect or otherwise contribute to cellular pathology (Figure 9). In any given LSD, one or more of these secondary defects may be particularly important, and those involving defects in lysosomal enzymes may be more amenable to correction than those involving the primary mutation (36,37). Thus, identification of such processes could reveal potentially promising therapeutic opportunities for certain LSDs. As an example, several sphingolipid LSDs acquire a secondary defect in late endosomal cholesterol trafficking which can exacerbate the lipid storage (3,4). In cell culture, correction of this secondary defect by cellular cholesterol depletion ameliorates a key defect in these cells, namely, abnormal trafficking of plasma membrane sphingolipids to lysosomes instead of the Golgi (3). However, it remains unclear how sufficient cellular cholesterol depletion could be achieved to treat patients.
Figure 9.
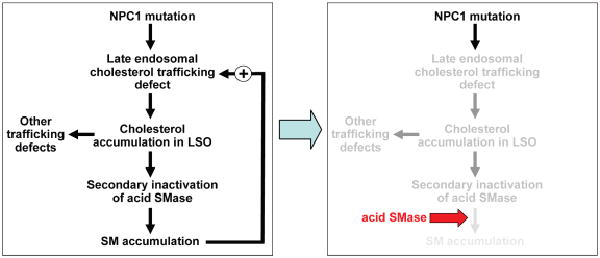
Working model of how acid SMase ameliorates the trafficking defects in NPC1-deficient cells. The primary defect in cholesterol trafficking caused by mutant NPC1 leads to a secondary decrease in acid SMase activity. The decrease in acid SMase activity causes an increase in intracellular SM, presumably in late endosomes and possibly other sites, which amplifies the original cholesterol trafficking defect. Moreover, cholesterol accumulation in the LSO and perhaps other effects of NPC1 deficiency are associated with trafficking defects in other lipids, such as BMP, and perturbation of vesicular trafficking of membrane proteins, including TfR recycling. Thus, these defects would also be amplified by the secondary decrease in acid SMase activity. Restoring the defect in acid SMase activity breaks the amplification cycle and thus helps correct the aforementioned lipid and protein trafficking defects. See text for details.
We considered the converse possibility that restoring SM hydrolysis in LSOs might reduce cholesterol storage and restore normal cellular membrane traffic. As a test case for this concept, we studied NPC1 deficient cells. These cells lack a membrane-bound late endosomal protein NPC1, but have a secondary defect in a soluble lysosomal enzyme acid SMase that is amenable to reconstitution. Our data show that correction of the secondary acid SMase defect in NPC1-deficient cells markedly reverses the accumulation of two lipids, cholesterol and BMP, and helps restore membrane TfR recycling – all in the face of complete absence of the NPC1 protein (Figures 3 and 4).
The hypothesis that correction of the acid SMase activity defect in NPC1 cells would have a beneficial effect on NPC cellular pathology is based on extensive biophysical data focused on SM–cholesterol interactions (25,40,41). It is well known, for example, that SMase treatment of the plasma membrane can release cholesterol for delivery to internal organelles (42). Cholesterol and SM can mutually stabilize each other in membrane bilayers, so removal of excess SM should enable movement of cholesterol. In the case of late endosomal membranes, Lobel, Storch and colleagues (5) suggested that lipoprotein-derived cholesterol is delivered to the late endosomal membrane via a collisional mechanism involving cholesterol in complex with the endosomal cholesterol-binding protein NPC2. The authors showed that acceptor membranes enriched in SM were relatively poor acceptors of the NPC2–cholesterol complex, a finding that would predict the benefit of acid SMase correction (5). Thus, just as cholesterol depletion reduces SM accumulation (3), SM depletion might reduce cholesterol accumulation in LSOs. Furthermore, accumulation of ceramide, a product of the acid SMase reaction, can displace cholesterol from ordered membrane domains. The mechanism may involve competition for sites in the membrane bilayer by small polar head groups, which are constituents of both cholesterol and ceramide (26). A very recent report provides additional insight into how NPC1 and NPC2 might co-operate in a cholesterol exchange reaction to transfer endocytosed cholesterol from the lumen to the membrane of late endosomes (6). Other studies have also implicated a possible role for a BMP-interacting protein called Alix in this transfer process (13). Whether restoring acid SMase activity in NPC1 cells enhances one or more of these processes remain to be determined.
The demonstration in this study that defective acid SMase activity in NPC1 cells is functionally important provides a strong rationale for a future study addressing the mechanism of acid SMase inactivation. Thomas et al. (15)showed that acid SMase activity in NPC1 cells grown in a medium containing 13% FBS was three times lower than that in WT cells. However, when NPC Fbs are grown in a medium containing delipidated serum, acid SMase activity was restored to the normal WT level. These data indicate that the increment in cellular lipid loading caused by incubation in 13% serum is enough to inactivate acid SMase. In terms of mechanism, it is possible that a relatively low level of lipid accumulation caused initially by mutant NPC1 may lead to defective processing of the C-terminal Cys of acid SMase, which, according to the data of Qiu et al. (24), would decrease enzyme activity. This defect would then amplify the lipid trafficking defect, as explained in this study (Figure 9).
If the suppression of acid SMase activity in NPC1 cells is a secondary effect, increasing levels of the enzyme may help overcome the inactivation. In support of this concept, we observed that adding increasing amounts of exogenous rhASM has no effect on cholesterol levels in WT cells. On the contrary, there is a dose-dependent decrease in LSO cholesterol with the addition of increasing dose of rhASM to NPC1 cells. The treatment is effective only after the secondary inactivation of acid SMase in these cells is overcome. Once this secondary defect is relieved by excess acid SMase, there is only 25–30% residual cholesterol accumulation, presumably because of the primary NPC1 mutation.
In terms of the possible pathophysiological importance of our findings, hepatosplenomegaly with liver disease is a major cause of NPC morbidity in the early years (43), and liver and spleen SM accumulation is substantial in NPC disease (9,11,12). With regard to NPC neuronal disease, investigators are uncertain as to the role of cholesterol versus other lipids, notably gangliosides, and other processes, such as defective neurosteroidogenesis (44-46). Nonetheless, a number of laboratories have found that NPC neurons accumulate cholesterol in cell bodies (47-49), and Walkey and colleagues demonstrated that cholesterol and gangliosides accumulate in the cerebral cortex, cerebellum and hippocampus of NPC brains (44). Moreover, a specific pattern of cerebellar Purkinje cell loss observed in NPC disease is also seen in Niemann-Pick A/B disease, where acid SMase deficiency is the primary defect (50,51).
To the extent that SM accumulates in at least some of the tissues of NPC patients, what is the evidence that suppressed acid SMase activity is involved? There is some direct evidence of defective acid SMase activity in NPC tissues, including the first description of the disease in NPC mice and subsequent follow-up studies (14-16). However, some in vitro studies have not found a decrease in acid SMase activity in certain cells and tissues from NPC subjects (52). These observations might be explained by the finding of Reagan et al. (16), and confirmed by us, that the enzymatic defect requires cellular cholesterol accumulation. Thus, a loss of acid SMase activity in vivo may be apparent only after the disease has progressed to the point where LSOs accumulate a threshold amount of excess cholesterol. If so, acid SMase inactivation might be an important amplification factor in advanced NPC disease progression (Figure 9).
In summary, we have shown that key pathological features of an LSD cell can be markedly improved by correcting a secondary abnormality despite complete absence of the protein responsible for the primary defect. In the case of NPC1 disease, the secondary defect of acid SMase inactivity is a more feasible therapeutic target than the primary mutation (36,37). The data in Figure 6 demonstrate this concept at the cell level by showing that the LSO cholesterol trafficking defect in two different human NPC Fbs can be markedly corrected by acid SMase enzyme replacement. A reduction of up to 70% of the cholesterol overload in LSOs might translate into clinical improvement. Indeed, replacement therapy with acid SMase is being tested in humans who have NPA and B disease with primary mutations in this protein (38) (http://www.genzyme.com/research/medical/medi_home.asp). The challenge with NPC disease is to achieve expression in the brain, but recent advances using SMPD1-containing adeno-associated virus vectors in mice have shown promise in this regard (53,54). We propose that careful examination of other LSDs may reveal similar findings, and some of these findings may suggest new approaches to therapy.
Materials and Methods
The tissue culture plasticware used in these experiments was purchased from Fisher Scientific Co. Tissue culture media and other tissue culture reagents were purchased from Invitrogen Corp. FBS was obtained from Gemini Bio-Products. LDL (d, 1.020–1.063 g/mL) from fresh human plasma was isolated by preparative ultracentrifugation as described elsewhere (55). Radiochemicals were purchased from either Perkin-Elmer Life and Analytical Sciences, Inc., or American Radiolabeled Chemicals, Inc. All restriction enzymes, Antarctic phosphatase and T4 DNA ligase were purchased from New England BioLabs. Recombinant human acid SMase was prepared from transfected CHO cells and purified as previously described (56). All other chemicals and reagents were from Sigma-Aldrich, and all organic solvents were from Fisher Scientific Co.
Cells
CHO, 25RA (57)and CT60 cells (22,23)were grown in monolayer cultures in Ham’s F12 medium containing 10% FBS. Human WT (GM05659) and NPC (GM03123) Fbs, from Coriell Institute of Medical Research, were cultured in Modified Eagle’s Medium (MEM) containing 10% FBS. The NPC Fbs were derived originally from a 9-year-old compound heterozygote female, with missense mutations in exon 6 of one allele (P237S) and exon 21 of the other allele (I1061T). The fibroblasts from this subject expressed no detectable NPC1 protein by immunoblot analysis and, as expected, had a severe defect in the trafficking lipoprotein-derived cholesterol (33,34). The GM18453 NPC Fbs were derived from a male donor with a homozygous mutation at I1061T. All cells were plated at a minimum of 24 h prior to commencement of the experiment.
Acid SMase activity assay
Cell extracts were prepared by scraping cells into ice-cold 250 mm sucrose, followed by sonication on ice twice for 10 seconds each using a Branson Sonifier 450. To prepare the substrate, solvent from 0.1 μCi [choline-methyl-14C] SM (52 mCi/mmol; Perkin-Elmer Life and Analytical Sciences) was evaporated, and the labeled SM was resuspended in 20 μL of assay buffer (100 mm sodium acetate pH 5.0, 100 μm ZnCl2) containing 2.7% Triton X-100 and vortexed for 2 min. The assay solution, which was added to 1.5-mL microcentrifuge tubes on ice, consisted of 50 μL assay buffer, 20 μL substrate and 20 μL of cell extract. After incubation for 60 min at 37°C, the reaction was terminated by adding 125 μL of chloroform:methanol (2:1, v/v). The assay tubes were vortexed for 1 min and then centrifuged 5000 xg for 5 min at 4°C. A 50-μL aliquot of the upper aqueous phase was removed for scintillation counting to determine the amount of [14C]phosphorylcholine released from [14C]SM. The protein content of the cell extracts was assayed using the method of Lowry (58).
Cellular cholesterol mass determination by gas chromatography
In Ham’s F12 growth medium supplemented with 10% FBS, 25RA, CT60, CT60-VEC, CT60-WT and CT60-C629 cells were grown. Lipids were extracted from the cells with hexane:2-propanol (3:2) and separated on a Varian Factor Four capillary column (VF-1 ms 30 m × 0.25 mm ID DF 0.25) using Varian 4000 GC/MS/MS system. The injector temperature was 270°C. The following temperature gradient was used: initial temperature was 115°C, which was raised to 260°C at 9°C/min and held for 2.89 min, then raised to 269°C at 3°C/min and again to 290°C at 9°C/min and held for 4.67 min. Flow rate was 5 mL/min He(g). Electron ionization was used with the current set at 10 μA. Total ionic current was used for detection (50 1000 m/z). β-Sitosterol was used as an internal standard for quantification of μg free cholesterol per mg cell protein. Protein concentration was determined with modified Lowry reagent (Bio-Rad).
Sphingomyelin mass assay
Total cellular SM mass was quantified in lipid extracts of cells using the TLC-Bartlett procedure as previously described (59,60), except that the scraped thin-layer chromatography (TLC) spot was extracted with methanol:chloroform (2:1)
Preparation of WT or C629S SMPD1 constructs
A plasmid containing the human cDNA for acid SMase (SMPD1) (56)was digested with EcoRI to release the cDNA. The cDNA was then ligated to phosphatase-treated, EcoRI-digested pBSIISK (Stratagene) to generate pBS.SMPD1. To obtain the 3′ ends of the cDNA designed to encode WT or C629S acid SMase, polymerase chain reaction (PCR) was conducted using the high-fidelity Platinum Pfx DNA polymerase (Invitrogen) and the pBS.SMPD1 plasmid as the template. For both constructs, a NotI site was created after the stop codon to assist in the cloning process. The primers for the WT construct consisted of the sense primer SMPD11469 (5′-ACTGTCTGAAGAGCTGGAGCT-3′) and the antisense primer 5′-TTTTATTGCGGCCGCCTAGCAAAACAGTGGCCTTGG-3′. The sense primer for the C629S construct was SMPD11469 and the antisense primer was 5′-TTTTATTGCGGCCGCCTAGGAAAACAGTGGCCTTGG-3′ (the codon for serine in position 629 is underlined). Each PCR product was digested with SphI (which cuts at position 1860 of SMPD1 cDNA) and NotI, and then separately ligated to the 4.8-kb SphI-NotI fragment from pBS.SMPD1 to generate pBS.WT or pBS.C629S, in which the polyadenylation sequence was removed from the SMPD1 cDNA. The final vectors were created in the expression vector pIRES-hrGFP II (Stratagene) by ligating the 6-kb EcoRI–NotI fragment from the vector with the 2-kb EcoRI–NotI fragment from either pBS.WT or pBS.C629S to generate pIRES.WT or pIRES.C629S. The DNA sequence was confirmed for each construct. The vector pIRES-hrGFP II consists of the SV40 promoter driving the expression of the neomycin-resistance gene and a bicistronic expression cassette under the control of the human cytomegalovirus promoter which contains a multiple cloning site followed by an internal ribosome entry site (IRES) that is linked to the GFP coding sequence.
Transfection of cells with the WT or C629S SMPD1 constructs
For transient transfection, CT60 cells in 16-mm wells were transfected with 340 ng empty vector (pIRES-hrGFP II; VEC), pIRES.WT or pIRES.C629S using Lipofectamine 2000. For stable transfection, CT60 cells were transfected with the empty vector, pIRES.WT or pIRES.C629S, and then selected for growth in G418 (0.5 mg/mL) and named, respectively, CT60-VEC, CT60-WT and CT60-C629S. Human fibroblasts were transiently transfected using a reverse format with Effectene (Qiagen, Valencia, CA) as the transfection reagent. Briefly, VEC, WT and C629S vectors (above) were mixed with Effectene transfection reagent, enhancer and manufacturer-supplied buffer (EC) in the ratio recommended by the manufacturer and dispensed in different wells of a six-well plate. The fibroblasts were then added to the above mixture in suspension and incubated overnight at 37°C in a tissue culture incubator. Fresh growth medium was added to the cells and, after an additional 6 h of incubation, the cells were fixed and analyzed for filipin and/or GFP staining (as described below).
Acid SMase enzyme replacement
Normal (GM05659) and NPC1 human fibroblasts (GM03123) were incubated in a medium containing 3 μg/mL recombinant human acid SMase (rhASM). Two days later, the cells were washed thoroughly with PBS, and either lysed and assayed for acid SMase activity or fixed and stained with filipin for imaging and quantification. NPC1 human fibroblasts GM18453 were treated with 3 μg/mL rhASM for 24 h before fixing, staining with filipin, imaging and quantifying.
Alexa555-conjugated rhASM enzyme replacement
For identifying the localization of rhASM in the cells, recombinant enzyme was conjugated with Alexa555 (Invitrogen) according to the manufacturer’s protocol. Normal (GM05659) and NPC1 human fibroblasts (GM03123) were incubated in a medium containing 3 μg/mL Alexa555–rhASM for 24 h prior to fixing and staining with filipin. Images were acquired using standard UV and TRITC filters for filipin and Alexa555-labeled rhASM, and quantified.
Filipin staining
Cells were plated onto poly-d-lysine-coated 35-mm coverslip dishes in a medium containing 10% FBS. After 1–2 days of growth, the cell monolayers were washed three times with PBS and then fixed with 3% paraformaldehyde in PBS for 20 min at room temperature, followed by three more washes with PBS. To detect free cholesterol, filipin was added to the fixed cells (50 μg/mL in PBS) for 45 min at room temperature. Finally, the cells were washed three times with PBS, and images were acquired immediately after labeling.
Fluorescence microscopy
Fluorescence microscopy and digital image acquisition were carried out using a Leica DMIRB microscope (Leica Mikroscopie und Systeme GmbH) equipped with a cooled Charge Coupled Device (CCD) camera (Princeton Instruments) and driven by MetaMorph Imaging System acquisition software (MDS Analytical Technologies). All images were acquired using an oil immersion objective (63×, 1.25 NA). Filipin was imaged using an A4 filter cube obtained from Chroma Technology Corp.: 360-nm (40-nm bandpass) excitation filter; 365 DCLP (DiChroic Long Pass) filter and 480-nm (40-nm bandpass) emission filter. To minimize photo-bleaching of the filipin signal, a neutral density filter transmitting 1.5% light was used to acquire images. GFP was imaged using a standard FITC filter cube. Fluorescence crossover from one channel to another was measured using single-labeled samples of each probe and was found to be insignificant.
Image analysis
Images were analyzed using Metamorph (version 7.0 r4) image analysis software from Molecular Devices (MDS Analytical Technologies). All images were corrected for background before analysis. The average filipin and LSO ratio was calculated as described previously (61). The LSO ratio was calculated based on low-thresholded region and high-thresholded intensities in the filipin image as described previously (61). All data were normalized to vector control values or, in the case of the human fibroblast – rhASM reconstitution experiment (Figure 5C), to the WT Fb value.
Cholesterol efflux assay
[3H]cholesteryl ester (CE)-labeled LDL was prepared as described by Krieger (62). Briefly, the lipid core of LDL was replaced with [1,2,6,7-3H(N)] cholesteryl oleate (American Radiolabeled Chemicals, Inc.). The specific activity of the labeled LDL was 13.7 cpm/ng of protein. Cells were plated in 24-well plates and incubated for 2 days in F12 medium containing 10% lipoprotein-deficient serum. The cells were labeled by incubation for 4 h in F12 medium containing 0.2% BSA and 10 μg/mL [3H]CE-labeled LDL. At the end of this pulse period, the cells were washed and the medium was replaced with F12/BSA medium containing 50 μg/mL HDL3. At the indicated time points, 100 μL of media was removed and centrifuged for 5 min at 14000 xg to remove cellular debris, and the radioactivity in this portion of the media was determined by liquid scintillation counting. After the last time point, the cells were washed and the monolayer was dissolved in 250 μL of 0.1n NaOH at room temperature for a minimum of 4 h. A 100-μL aliquot of the cell lysate was measured, and the percent efflux was calculated as [(media cpm)/(cell + media cpm)] × 100. To obtain the value for HDL3-stimulated efflux, the percent efflux in the absence of HDL3 was subtracted from the percent efflux in the presence of acceptor.
Transferrin efflux kinetics as a measure of TfR recycling
CT60 cells expressing the human TfR (CT60hTfR cells) (30)were stably transfected with the empty vector pIRES-hrGFPII, pIRES.WT or pIRES.C629S and sorted for high levels of green fluorescence. The cells were maintained in a medium containing G418 (0.5 mg/mL). The resulting stable cell lines were named, respectively, CT60hTfR-VEC, CT60hTfR-WT and CT60hTfR-C629S. The recycling of TfR from endosomes to cell surface was measured as previously described elsewhere (63). Briefly, the cells were plated in 12-well culture dishes in bicarbonate-buffered McCoy’s medium. For each experiment and for each cell type, six plates were prepared for six time points (3, 5, 10, 15, 30 and 60 min). Four wells in each plate were pulsed with 5 μg/mL [125I]-labeled Tf, and the remaining two wells were pulsed with 5 μg/mL [125I]-labeled Tf, with a 200-fold excess of unlabeled Tf to ascertain non-specific binding. All plates were incubated for 30 min and washed once with serum-free McCoy’s medium, once with acid wash buffer (pH 5.0) and twice with efflux medium. Finally, the efflux medium was added to each plate and incubated for varying time periods. At the end of each chase time, the efflux medium was transferred to collection tubes, and the cells were quickly washed once with the same medium, which was then added to the collection tube. Solubilization solution was added to each well, triturated and transferred to another collection tube. The cells were washed with water, and the washings were added to the solubilization medium. The amount of [125I]-labeled Tf was measured for all the efflux and cellular fractions, and the recycling rate of TfR was determined.
BMP labeling
Cells seeded in poly-d-lysine-coated coverslip dishes and grown for 2 days were fixed with 3% paraformaldehyde in PBS for 20 min at room temperature. Fixed cells were subsequently permeabilized with 0.5% saponin (to ensure complete immunolabeling of BMP within multivesicular lysosomal compartments) and incubated with primary murine anti-BMP antibody (Echelon Biosciences Inc.) for 45 min. The cell monolayers were washed three times with PBS and then incubated with Alexa546-labeled goat anti-mouse IgG for 1 h in the presence of 0.1% saponin. Finally, the cells were washed three times with PBS. Images were acquired using wide-field epi-fluorescence microscopy at 63x magnification and standard TRITC filters.
Assay for uptake of LDL-derived cholesterol
Cells were incubated for 2 h with 5 μg/mL [14C]CE-labeled LDL, which was produced using the same method described above for [3H]CE-labeled LDL. Lipids were extracted in hexane:isopropanol (3:2) and subjected to thin-layer chromatography to separate free cholesterol and CE.
Statistics
Data are presented as mean ± SEM of triplicate experiments. Statistical significance was determined using the Student’s t-test with unequal variance or one-way anova and the Tukey’s multiple comparison test using GraphPad Prism version 4.03 for Windows (GraphPad Software).
Acknowledgments
We thank Dr T. Y. Chang from Dartmouth University for providing the CT60 and 25RA cells. This work was supported by NIH grant HL-57560 to F. R. M. and I. T. and by NIH grant DK-27083 to F. R. M.
References
- 1.Brady RO. Lysosomal storage diseases. Pharmacol Ther. 1982;19:327–336. doi: 10.1016/0163-7258(82)90072-9. [DOI] [PubMed] [Google Scholar]
- 2.Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- 3.Puri V, Watanabe R, Dominguez M, Sun X, Wheatley CL, Marks DL, Pagano RE. Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases. Nat Cell Biol. 1999;1:386–388. doi: 10.1038/14084. [DOI] [PubMed] [Google Scholar]
- 4.Lloyd-Evans E, Morgan AJ, He X, Smith DA, Elliot-Smith E, Sillence DJ, Churchill GC, Schuchman EH, Galione A, Platt FM. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat Med. 2008;14:1247–1255. doi: 10.1038/nm.1876. [DOI] [PubMed] [Google Scholar]
- 5.Cheruku SR, Xu Z, Dutia R, Lobel P, Storch J. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J Biol Chem. 2006;281:31594–31604. doi: 10.1074/jbc.M602765200. [DOI] [PubMed] [Google Scholar]
- 6.Infante RE, Wang ML, Radhakrishnan A, Kwon HJ, Brown MS, Goldstein JL. NPC2 facilitates bidirectional transfer of cholesterol between NPC1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc Natl Acad Sci U S A. 2008;105:15287–15292. doi: 10.1073/pnas.0807328105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urano Y, Watanabe H, Murphy SR, Shibuya Y, Geng Y, Peden AA, Chang CC, Chang TY. Transport of LDL-derived cholesterol from the NPC1 compartment to the ER involves the trans-Golgi network and the SNARE protein complex. Proc Natl Acad Sci U S A. 2008;105:16513–16518. doi: 10.1073/pnas.0807450105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liscum L, Klansek JJ. Niemann-Pick disease type C. Curr Opin Lipidol. 1998;9:131–135. doi: 10.1097/00041433-199804000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Ory DS. The Niemann-Pick disease genes; regulators of cellular cholesterol homeostasis. Trends Cardiovasc Med. 2004;14:66–72. doi: 10.1016/j.tcm.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee S, Maxfield FR. Lipid and cholesterol trafficking in NPC. Biochim Biophys Acta. 2004;1685:28–37. doi: 10.1016/j.bbalip.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 11.Pentchev PG. Niemann-Pick C research from mouse to gene. Biochim Biophys Acta. 2004;1685:3–7. doi: 10.1016/j.bbalip.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 12.Brady RO, Filling-Katz MR, Barton NW, Pentchev PG. Niemann-Pick disease types C and D. Neurol Clin. 1989;7:75–88. [PubMed] [Google Scholar]
- 13.Chevallier J, Chamoun Z, Jiang G, Prestwich G, Sakai N, Matile S, Parton RG, Gruenberg J. Lysobisphosphatidic acid controls endosomal cholesterol levels. J Biol Chem. 2008;283:27871–27880. doi: 10.1074/jbc.M801463200. [DOI] [PubMed] [Google Scholar]
- 14.Pentchev PG, Gal AE, Booth AD, Omodeo-Sale F, Fouks J, Neumeyer BA, Quirk JM, Dawson G, Brady RO. A lysosomal storage disorder in mice characterized by a dual deficiency of sphingomyelinase and glucocerebrosidase. Biochim Biophys Acta. 1980;619:669–679. doi: 10.1016/0005-2760(80)90116-2. [DOI] [PubMed] [Google Scholar]
- 15.Thomas GH, Tuck-Muller CM, Miller CS, Reynolds LW. Correction of sphingomyelinase deficiency in Niemann-Pick type C fibroblasts by removal of lipoprotein fraction from culture media. J Inherit Metab Dis. 1989;12:139–151. doi: 10.1007/BF01800716. [DOI] [PubMed] [Google Scholar]
- 16.Reagan JW, Jr, Hubbert ML, Shelness GS. Posttranslational regulation of acid sphingomyelinase in Niemann-Pick type C1 fibroblasts and free cholesterol-enriched Chinese hamster ovary cells. J Biol Chem. 2000;275:38104–38110. doi: 10.1074/jbc.M005296200. [DOI] [PubMed] [Google Scholar]
- 17.Vanier MT, Rodriguez-Lafrasse C, Rousson R, Gazzah N, Juge MC, Pentchev PG, Revol A, Louisot P. Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim Biophys Acta. 1991;1096:328–337. doi: 10.1016/0925-4439(91)90069-l. [DOI] [PubMed] [Google Scholar]
- 18.Tamura H, Takahashi T, Ban N, Torisu H, Ninomiya H, Takada G, Inagaki N. Niemann-Pick type C disease: novel NPC1 mutations and characterization of the concomitant acid sphingomyelinase deficiency. Mol Genet Metab. 2006;87:113–121. doi: 10.1016/j.ymgme.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 19.Harzer K, Massenkeil G, Frohlich E. Concurrent increase of cholesterol, sphingomyelin and glucosylceramide in the spleen from non-neurologic Niemann-Pick type C patients but also patients possibly affected with other lipid trafficking disorders. FEBS Lett. 2003;537:177–181. doi: 10.1016/s0014-5793(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 20.Dvorakova L, Sikora J, Hrebicek M, Hulkova H, Bouckova M, Stolnaja L, Elleder M. Subclinical course of adult visceral Niemann-Pick type C1 disease. A rare or underdiagnosed disorder? J Inherit Metab Dis. 2006;29:591. doi: 10.1007/s10545-006-0330-z. [DOI] [PubMed] [Google Scholar]
- 21.Leventhal AR, Chen W, Tall AR, Tabas I. Acid sphingomyelinase-deficient macrophages have defective cholesterol trafficking and efflux. J Biol Chem. 2001;276:44976–44983. doi: 10.1074/jbc.M106455200. [DOI] [PubMed] [Google Scholar]
- 22.Cadigan KM, Spillane DM, Chang T-Y. Isolation and characterization of Chinese hamster ovary cell mutants defective in intracellular low density lipoprotein-cholesterol trafficking. J Cell Biol. 1990;110:295–308. doi: 10.1083/jcb.110.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Watari H, Blanchette-Mackie EJ, Dwyer NK, Glick JM, Patel S, Neufeld EB, Brady RO, Pentchev PG, Strauss JF., III Niemann-Pick C1 protein: obligatory roles for N-terminal domains and lysosomal targeting in cholesterol mobilization. Proc Natl Acad Sci U S A. 1999;96:805–810. doi: 10.1073/pnas.96.3.805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qiu H, Edmunds T, Baker-Malcolm J, Karey KP, Estes S, Schwarz C, Hughes H, Van Patten SM. Activation of human acid sphingomyelinase through modification or deletion of C-terminal cysteine. J Biol Chem. 2003;278:32744–32752. doi: 10.1074/jbc.M303022200. [DOI] [PubMed] [Google Scholar]
- 25.Ridgway ND. Interactions between metabolism and intracellular distribution of cholesterol and sphingomyelin. Biochim Biophys Acta. 2000;1484:129–141. doi: 10.1016/s1388-1981(00)00006-8. [DOI] [PubMed] [Google Scholar]
- 26.Megha, London E. Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): implications for lipid raft structure and function. J Biol Chem. 2004;279:9997–10004. doi: 10.1074/jbc.M309992200. [DOI] [PubMed] [Google Scholar]
- 27.Qin C, Nagao T, Grosheva I, Maxfield FR, Pierini LM. Elevated plasma membrane cholesterol content alters macrophage signaling and function. Arterioscler Thromb Vasc Biol. 2006;26:372–378. doi: 10.1161/01.ATV.0000197848.67999.e1. [DOI] [PubMed] [Google Scholar]
- 28.Bartz F, Kern L, Erz D, Zhu M, Gilbert D, Meinhof T, Wirkner U, Erfle H, Muckenthaler M, Pepperkok R, Runz H. Identification of cholesterol-regulating genes by targeted RNAi screening. Cell Metab. 2009;10:63–75. doi: 10.1016/j.cmet.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 29.Maguire JA, Reagan JW., Jr Silencing of the mutant SCAP allele accounts for restoration of a normal phenotype in CT60 cells selected for NPC1 expression. J Lipid Res. 2005;46:1840–1848. doi: 10.1194/jlr.M500198-JLR200. [DOI] [PubMed] [Google Scholar]
- 30.Pipalia NH, Hao M, Mukherjee S, Maxfield FR. Sterol, protein and lipid trafficking in Chinese hamster ovary cells with Niemann-Pick type C1 defect. Traffic. 2007;8:130–141. doi: 10.1111/j.1600-0854.2006.00513.x. [DOI] [PubMed] [Google Scholar]
- 31.Salvioli R, Scarpa S, Ciaffoni F, Tatti M, Ramoni C, Vanier MT, Vaccaro AM. Glucosylceramidase mass and subcellular localization are modulated by cholesterol in Niemann-Pick disease type C. J Biol Chem. 2004;279:17674–17680. doi: 10.1074/jbc.M313517200. [DOI] [PubMed] [Google Scholar]
- 32.Choudhury A, Sharma DK, Marks DL, Pagano RE. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol Biol Cell. 2004;15:4500–4511. doi: 10.1091/mbc.E04-05-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamamoto T, Ninomiya H, Matsumoto M, Ohta Y, Nanba E, Tsutsumi Y, Yamakawa K, Millat G, Vanier MT, Pentchev PG, Ohno K. Genotype-phenotype relationship of Niemann-Pick disease type C: a possible correlation between clinical onset and levels of NPC1 protein in isolated skin fibroblasts. J Med Genet. 2000;37:707–712. doi: 10.1136/jmg.37.9.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pentchev PG, Comly ME, Kruth HS, Vanier MT, Wenger DA, Patel S, Brady RO. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc Natl Acad Sci U S A. 1985;82:8247–8251. doi: 10.1073/pnas.82.23.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maziere JC, Maziere C, Mora L, Routier JD, Polonovski J. In situ degradation of sphingomyelin by cultured normal fibroblasts and fibroblasts from patients with Niemann-Pick disease type A and C. Biochem Biophys Res Commun. 1982;108:1101–1106. doi: 10.1016/0006-291x(82)92113-1. [DOI] [PubMed] [Google Scholar]
- 36.Neufeld EF. The uptake of enzymes into lysosomes: an overview. Birth Defects Orig Artic Ser. 1980;16:77–84. [PubMed] [Google Scholar]
- 37.Brady RO. Enzyme replacement for lysosomal diseases. Annu Rev Med. 2006;57:283–296. doi: 10.1146/annurev.med.57.110104.115650. [DOI] [PubMed] [Google Scholar]
- 38.Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis. 2007;30:654–663. doi: 10.1007/s10545-007-0632-9. [DOI] [PubMed] [Google Scholar]
- 39.Kobayashi T, Beuchat MH, Lindsay M, Frias S, Palmiter RD, Sakuraba H, Parton RG, Gruenberg J. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat Cell Biol. 1999;1:113–118. doi: 10.1038/10084. [DOI] [PubMed] [Google Scholar]
- 40.Radhakrishnan A, Anderson TG, McConnell HM. Condensed complexes, rafts, and the chemical activity of cholesterol in membranes. Proc Natl Acad Sci U S A. 2000;97:12422–12427. doi: 10.1073/pnas.220418097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang J, Feigenson GW. A microscopic interaction model of maximum solubility of cholesterol in lipid bilayers. Biophys J. 1999;76:2142–2157. doi: 10.1016/S0006-3495(99)77369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Slotte JP, Bierman EL. Depletion of plasma-membrane sphingomyelin rapidly alters the distribution of cholesterol between plasma membranes and intracellular cholesterol pools in cultured fibroblasts. Biochem J. 1988;250:653–658. doi: 10.1042/bj2500653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yerushalmi B, Sokol RJ, Narkewicz MR, Smith D, Ashmead JW, Wenger DA. Niemann-Pick disease type C in neonatal cholestasis at a North American Center. J Pediatr Gastroenterol Nutr. 2002;35:44–50. doi: 10.1097/00005176-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 44.Zervas M, Dobrenis K, Walkley SU. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J Neuropathol Exp Neurol. 2001;60:49–64. doi: 10.1093/jnen/60.1.49. [DOI] [PubMed] [Google Scholar]
- 45.Griffin LD, Gong W, Verot L, Mellon SH. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat Med. 2004;10:704–711. doi: 10.1038/nm1073. [DOI] [PubMed] [Google Scholar]
- 46.Langmade SJ, Gale SE, Frolov A, Mohri I, Suzuki K, Mellon SH, Walkley SU, Covey DF, Schaffer JE, Ory DS. Pregnane X receptor (PXR) activation: a mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci U S A. 2006;103:13807–13812. doi: 10.1073/pnas.0606218103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Walkley SU, Suzuki K. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim Biophys Acta. 2004;1685:48–62. doi: 10.1016/j.bbalip.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 48.Reid PC, Sakashita N, Sugii S, Ohno-Iwashita Y, Shimada Y, Hickey WF, Chang TY. A novel cholesterol stain reveals early neuronal cholesterol accumulation in the Niemann-Pick type C1 mouse brain. J Lipid Res. 2004;45:582–591. doi: 10.1194/jlr.D300032-JLR200. [DOI] [PubMed] [Google Scholar]
- 49.Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem. 2002;83:1154–1163. doi: 10.1046/j.1471-4159.2002.01220.x. [DOI] [PubMed] [Google Scholar]
- 50.Sarna JR, Larouche M, Marzban H, Sillitoe RV, Rancourt DE, Hawkes R. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J Comp Neurol. 2003;456:279–291. doi: 10.1002/cne.10522. [DOI] [PubMed] [Google Scholar]
- 51.Sarna J, Miranda SR, Schuchman EH, Hawkes R. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann-Pick type A/B disease. Eur J Neurosci. 2001;13:1873–1880. doi: 10.1046/j.0953-816x.2001.01564.x. [DOI] [PubMed] [Google Scholar]
- 52.Vanier MT, Revol A, Fichet M. Sphingomyelinase activities of various human tissues in control subjects and in Niemann-Pick disease – development and evaluation of a microprocedure. Clin Chim Acta. 1980;106:257–267. doi: 10.1016/0009-8981(80)90309-5. [DOI] [PubMed] [Google Scholar]
- 53.Passini MA, Macauley SL, Huff MR, Taksir TV, Bu J, Wu IH, Piepenhagen PA, Dodge JC, Shihabuddin LS, O’Riordan CR, Schuchman EH, Stewart GR. AAV vector-mediated correction of brain pathology in a mouse model of Niemann-Pick A disease. Mol Ther. 2005;11:754–762. doi: 10.1016/j.ymthe.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 54.Dodge JC, Clarke J, Song A, Bu J, Yang W, Taksir TV, Griffiths D, Zhao MA, Schuchman EH, Cheng SH, O’Riordan CR, Shihabuddin LS, Passini MA, Stewart GR. Gene transfer of human acid sphingomyelinase corrects neuropathology and motor deficits in a mouse model of Niemann-Pick type A disease. Proc Natl Acad Sci U S A. 2005;102:17822–17827. doi: 10.1073/pnas.0509062102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Havel RJ, Eder H, Bragdon J. The distribution and chemical composition of ultracentrifugally reported lipoproteins in human serum. J Clin Invest. 1955;34:1345–1353. doi: 10.1172/JCI103182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He X, Miranda SRP, Dagan A, Gatt S, Schuchman EH. Overexpression of human acid sphingomyelinase in Chinese hamster ovary cells. Purification and characterization of the recombinant enzyme. Biochim Biophys Acta. 1999;1432:251–264. doi: 10.1016/s0167-4838(99)00069-2. [DOI] [PubMed] [Google Scholar]
- 57.Chang TY, Limanek JS. Regulation of cytosolic acetoacetyl coenzyme A thiolase, 3-hydroxy-3-methylglutaryl coenzyme A synthase, 3-hydroxy-3-methylglutaryl coenzyme A reductase, and mevalonate kinase by low density lipoprotein and by 25-hydroxycholesterol in Chinese hamster ovary cells. J Biol Chem. 1980;255:7787–7795. [PubMed] [Google Scholar]
- 58.Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 59.Okwu AK, Xu X, Shiratori Y, Tabas I. Regulation of the threshold for lipoprotein-induced acyl-CoA:cholesterol O-acyltransferase stimulation in macrophages by cellular sphingomyelin content. J Lipid Res. 1994;35:644–655. [PubMed] [Google Scholar]
- 60.Bartlett GR. Phosphorus assay in column chromatography. J Biol Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 61.Pipalia NH, Huang A, Ralph H, Rujoi M, Maxfield FR. Automated microscopy screening for compounds that partially revert cholesterol accumulation in Niemann-Pick C cells. J Lipid Res. 2006;47:284–301. doi: 10.1194/jlr.M500388-JLR200. [DOI] [PubMed] [Google Scholar]
- 62.Krieger M. Reconstitution of the hydrophobic core of low-density lipoprotein. Methods Enzymol. 1986;128:608–613. doi: 10.1016/0076-6879(86)28094-5. [DOI] [PubMed] [Google Scholar]
- 63.Johnson AO, Lampson MA, McGraw TE. A di-leucine sequence and a cluster of acidic amino acids are required for dynamic retention in the endosomal recycling compartment of fibroblasts. Mol Biol Cell. 2001;12:367–381. doi: 10.1091/mbc.12.2.367. [DOI] [PMC free article] [PubMed] [Google Scholar]