

Abstract
Mitochondrial reactive oxygen species (ROS) generation and the attendant mitochondrial dysfunction are implicated in a range of disease states. The objective of the present studies was to test the hypothesis that the mitochondrial β-oxidation pathway could be exploited to deliver and biotransform the prodrugs ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids to the corresponding phenolic antioxidants or methimazole. 3 -and 5-(Phenoxy)alkanoic acids and methyl-substituted analogs were biotransformed to phenols; rates of biotransformation decreased markedly with methyl-group substitution on the phenoxy moiety. 2,6-Dimethylphenol formation from the analogs 3-([2,6-dimethylphenoxy]methylthio)propanoic acid and 3-(2,6-dimethylphenoxy)acrylic acid was greater than that observed with ω-(2,6-dimethylphenoxy)alkanoic acids. 3- and 5-(1-Methyl-1H-imidazol-2-ylthio)alkanoic acids were rapidly biotransformed to the antioxidant methimazole and conferred significant cytoprotection against hypoxia-reoxygenation injury in isolated cardiomyocytes. Both 3-(2,6-dimethylphenoxy)propanoic acid and 3-(2,6-dimethylphenoxy)acrylic acid also afforded cytoprotection against hypoxia-reoxygenation injury in isolated cardiomyocytes. These results demonstrate that mitochondrial β-oxidation is a potentially useful delivery system for targeting antioxidants to mitochondria.
Keywords: Mitochondrial β-oxidation, Antioxidants, Mitochondrial prodrug targeting, Hypoxia-reoxygenation injury
1. Introduction
There is considerable contemporary interest in targeting antioxidants to mitochondria. Mitochondria account for >90% of cellular oxygen consumption and are a major source of reactive oxygen species (ROS); indeed, 1–2% of the oxygen consumed by mitochondria is converted to ROS.1 Moreover, a role for mitochondrial ROS in a range of disease states has been established.2
A range of strategies for targeting of antioxidants to mitochondria has been reported, including targeting based on the mitochondrial electrochemical potential, on binding to mitochondrial membrane components, on the unique mitochondrial expression of enzymes that catalyze the release of drugs from prodrugs, and on transporter-dependent delivery of prodrugs or drugs.3–8
The objective of the experiments reported herein was to test the hypothesis that the mitochondrial location of the enzymes of β-oxidation could be exploited to deliver phenolic chain-breaking and thiol-based antioxidants to mitochondria (Fig. 1). Accordingly, the mitochondrial β-oxidation of a series of ω-(phenoxy)alkanoic acids and 3-(phenoxy)acrylic acids to the corresponding phenols and of ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids to methimazole was undertaken (Fig. 2).
Figure 1.

Mitochondrial biotransformation of ω-(phenoxy)alkanoic acids. FACL, fatty-acid-CoA ligase; CPT-I, carnitine palmitoyltransferase-I; CACT, carnitine-acyl carnitine translocase; CPT-II, carnitine palmitoyltransferase-II; MCAD, medium-chain acyl-CoA dehydrogenase; SCAD, short-chain acyl-CoA dehydrogenase.
Figure 2.
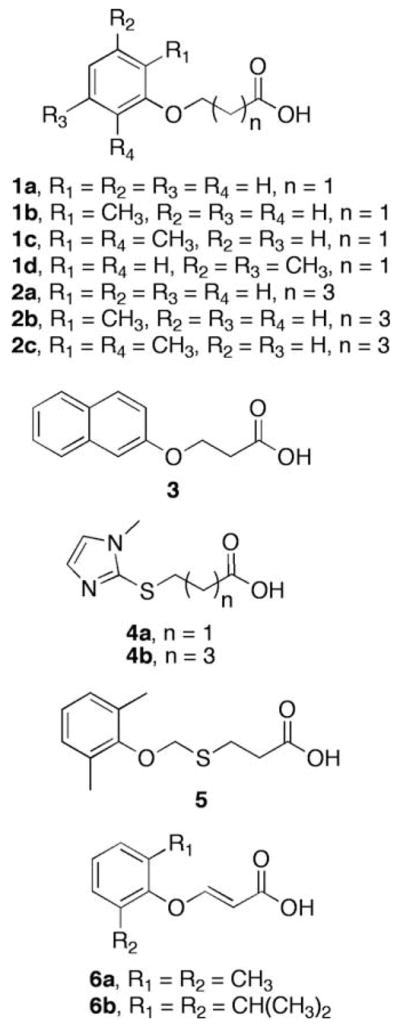
Structures of ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids.
2. Targeting strategy
The enzymes of mitochondrial β-oxidation catalyze the biotransformation of dietary and endobiotic fatty acids along with a range of xenobiotic alkanoic acids, including tianeptine, chlorophenoxybutyric acids, and 5-hydroxydecanoic acid.9–11 Moreover, the toxicity of several xenobiotic alkanoic acids, including haloalkene-derived 4-thiaalkanoates,12–14 4-bromo-2-butenoic acid,15 valproic acid,16 and hypoglycin,17 is associated with their bioactivation by the mitochondrial β-oxidation pathway.
3. Results and discussion
Studies on the metabolism of ω-phenylalkanoic acids by Knoop and others established the concept of fatty acid β-oxidation.18–22 These studies were extended by Levey and Lewis,23 who showed that 4-(phenoxy)butanoic acid and 6-(phenoxy)hexanoic acid are metabolized to phenoxyacetic acid. The present study exploits these seminal investigations to determine whether alkanoic acid-based prodrugs could be used to target antioxidants to mitochondria.
The phenolic substituents on ω-(phenoxy)alkanoic acids, that is, 2-methylphenol, 2,6-dimethylphenol, 3,5-dimethylphenol, and 2,6-diisopropylphenol, were chosen because of their antioxidant activity and because the compounds afforded insight into the substrate selectivity of the β-oxidation pathway.24–27 Finally, methimazole is an antioxidant and cytoprotective agent,28,29 and the methimazole prodrugs ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b were investigated as substrates and as cardioprotective agents.
The data presented herein show that ω-(phenoxy)alkanoic acids are substrates for mitochondrial β-oxidation, but that the rates of biotransformation differ markedly depending on the structure. All 3-(phenoxy)propanoic acids and 5-(phenoxy)pentanoic acids studied were biotransformed to phenolic products in isolated rat liver mitochondria (Table 1), but the rates of biotransformation differed markedly: with 3-(phenoxy)propanoic acids (1a–1d), the rates of biotransformation decreased with number and position of the methyl groups on the phenoxy moiety in the order: 1a ≫ 1b > 1d > 1c. A similar pattern was observed with 5-(phenoxy) pentanoic acids where the relative rates of biotransformation were 2a ≫ 2b > 2c > 2d. In general, the rates of biotransformation of 5-(phenoxy)pentanoic acids were lower than those of similarly substituted 3-(phenoxy)propanoic acids, except that the rates of biotransformation of ω-(2-methylphenoxy)alkanoic acids 1b and 2b were similar. The rate of biotransformation of all ω-(phenoxy) alkanoic acids studied was much lower than that of octanoic acid (Table 1).
Table 1.
Rates of mitochondrial biotransformation of ω-(phenoxy)alkanoic acids, 3-(phenoxy)acrylic acids, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acidsa
| Compound | Rate |
|---|---|
| 1a | 2.06 ± 0.50 |
| 1b | 0.39 ± 0.12 |
| 1c | 0.07 ± 0.02 |
| 1d | 0.10 ± 0.06 |
| 2a | 1.27 ± 0.37 |
| 2b | 0.39 ± 0.12 |
| 2c | 0.06 ± 0.01 |
| 2d | 0.01 ± 0.06 |
| 3 | 1.54 ± 0.17 |
| 4a | 2.50 ± 0.31 |
| 4b | 0.97 ± 0.16 |
| 5 | 0.54 ± 0.06 |
| 6a | 1.55 ± 0.14 |
| 6b | N.D. |
| Octanoic acid | 40.3 ± 6.7 |
The alkanoic acids (Fig. 2) were incubated with rat liver mitochondria, and product formation was quantified, as described in Section 5. Rates are expressed as nmol min−1 mg protein−1. Data are shown as mean ± SD, n = ≥ 3. Statistical analysis (unpaired t-test): 1a versus 2a, p <.05; 1b versus 2b, NS; 1c versus 2c, NS; 1d versus 2d, p <.05; 1a versus 3, p <.05; 2a versus 3, p >.05; 1c versus 5, p <.05; 1c versus 6a, p <0.05; 2c versus 5, p <.05; 1a versus 6a, p <.05. N.D., not detected.
Rates of biotransformation of ω-(phenoxy)alkanoic acids 1a and 2a decreased with increasing chain length, but this was not observed with ω-(2-methylphenoxy)- [1b vs 2b], ω-(2,6-dimethylphenoxy)- [1c vs 2c], or ω-(3,5-dimethylphenoxy)alkanoic acids [1d vs 2d] (Table 1). Indeed, the rates of biotransformation of the ω-(2,6-dimethylphenoxy)- [1c and 2c] and ω-(3,5-dimethylphenoxy) alkanoic acids [1d vs 2d] were low compared with the unsubstituted analogs.
3-(2-Naphthoxy)propanoic acid 3 was biotransformed at a rate slower than that of 3-(phenoxy)propanoic acid 1a but similar to that of the 5-(phenoxy)pentanoic acid 2a (Table 1).
Although the biotransformation of the alkanoic acids was studied in the presence of carnitine, the biotransformation of alkanoic acids 1a and 5 was studied in both the presence and absence of added carnitine, but no significant difference was observed (data not shown), perhaps because sufficient carnitine concentrations were present in isolated mitochondria.
The mitochondrial biotransformation of 5-(2,6-dimethylphenoxy) pentanoic acid 2c was accompanied by the formation of the carnitine ester of acid 2c (Fig. 3). In addition, acid 2c was biotransformed to the carnitine ester of acid 1c, but 3-(2,6-dimethylphenoxy) propanoic acid 1c itself was not observed. The formation of the product 2,6-dimethylphenol was, however, observed, indicating that acid 2c underwent two cycles of β-oxidation to release product (Fig. 1). This observation differs from the mitochondrial β-oxidation of dietary fatty acids, which apparently proceeds to product formation with no release of intermediates and is attributed to intermediate channeling.30,31 The formation of carnitine esters has, however, been observed with other xenobiotic alkanoic acids: the mitochondrial β-oxidation of elaidic acid (9-trans-octadecenoic acid) is accompanied by the formation of 5-trans-tetradecenoylcarnitine. 32 The formation of the 5-trans-tetradecenoylcarnitine is attributed to the low rate of dehydrogenation of 5-trans-tetradecanoyl-CoA by the acyl-CoA dehydrogenases, which results in the accumulation of the CoA thioester; the accumulated CoA thioester is converted to the carnitine ester by carnitine palmitoyltransferase-II. Indeed, carnitine ester formation is favored over hydrolysis to the acid.32 ω-(2,6-Dimethylphenoxy)alkanoic acids 1c and 2c were poor substrates for β-oxidation (Table 1). Hence, the observed accumulation of ω-(2,6-dimethylphenoxy)alkanoyl-CoA esters may have been accompanied by the carnitine palmitoyltransferase-II-catalyzed conversion to the corresponding carnitine esters.
Figure 3.

Biotransformation of 5-(2,6-dimethylphenoxy)pentanoic acid 2c to carnitine esters. Acid 2c was incubated with rat liver mitochondria, and a sample of the reaction mixture was analyzed by LC–MS, as described in Section 5. (A) 3-(2,6-Dimethylphenoxy)propanoylcarnitine; (B) 5-(2,6-dimethylphenoxy)pentanoylcarnitine; (C) 2,6-dimethylphenol; (D) 5-(2,6-dimethylphenoxy)pentanoic acid 2c.
5-(Phenoxy)pentanoic acids must undergo two cycles of β-oxidation to release a phenolic metabolite, whereas 3-(phenoxy)propanoic acids release a phenolic metabolite after one cycle of β-oxidation (Fig. 1). Moreover, 4-thiaalkanoic acids are better substrates for the medium-chain acyl-CoA dehydrogenase than 4-oxaalkanoic acids.33 Hence, 3-([2,6-dimethylphenoxy]methylthio)propanoic acid 5 was prepared to test the hypothesis that the insertion of a methylthio linking group between the 2,6-dimethylphenoxy group and the propanoic acid moiety would result in less steric interaction between the substrate and β-oxidation enzymes and, thereby, increase the rate of biotransformation. The intermediate (2,6-dimethylphenoxy)methanethiol formed from acid 5 after one cycle of β-oxidation would be expected to eliminate thioformaldehyde to afford 2,6-dimethylphenol. Indeed, 4-thiaalkanoic acid 5 proved to be amuchbetter substrate for β-oxidation than ω-(2,6-dimethylphenoxy)alkanoic acids 1c and 2c (Table 1), affording a several-fold increase in reaction rate.
The mitochondrial biotransformation of endobiotic and xenobiotic alkanoic acids requires the medium-chain acyl-CoA dehydrogenase-catalyzed formation of enoyl-CoA intermediates. (E)-3-(2,6-Dimethylphenoxy)acrylic acid 6a was prepared to test the hypothesis that circumventing the potential barrier formed by the medium-chain acyl-CoA dehydrogenase might overcome the apparent steric impediment to biotransformation observed with ω-(2,6-dimethylphenoxy)alkanoic acids 1c and 2c (Table 1). The rate of biotransformation of (E)-3-(2,6-dimethylphenoxy)acrylic acid 6a was approximately 20-fold greater than that of the saturated analog 3-(2,6-dimethylphenoxy)propanoic acid 1c, indicating that steric effects play a larger role for the medium-chain acyl-CoA dehydrogenase than for enoyl-CoA hydratase (Table 1). Moreover, no biotransformation of (Z)-3-(2,6-dimethylphenoxy) acrylic acid was detected (data not shown); this is consistent with the known selectivity of enoyl-CoA hydratase, which catalyzes the conversion of (E)-2-alkenoyl-CoA thioesters, but not (Z)-2-alkenoyl-CoA thioesters, to 3-hydroxyalkanoyl-CoA thioesters. 34 No quantifiable biotransformation was observed with the bulky analog (E)-3-(2,6-diisopropylphenoxy)acrylic acid 6b (Table 1), indicating that steric factors also affect enoyl-CoA hydratase-catalyzed reactions. This is the first demonstration of the utility of using ω-(phenoxy)acrylic acids as prodrugs for antioxidant delivery to mitochondria.
Depolarization of the mitochondrial membrane potential with carbonylcyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) significantly decreased the biotransformation of ω-(phenoxy)alkanoic acids 1a and 2a (1a: 1.69 ± 0.19, 1a + FCCP: 0.41 ± 0.12, p<.05; 2a: 1.17 ± 0.27, 2a + FCCP: 0.64 ± 0.10, p <.05; rates are expressed as nmol product min−1 mg protein−1 and are shown as mean ± SD, n = 3). The mechanism by which FCCP decreased the biotransformation of ω-(phenoxy)alkanoic acids was not investigated, but may be similar to that of Arochlor 1254, which, like FCCP,35 decreases both the mitochondrial membrane potential and the β-oxidation of palmitate.36 The effect of Arochlor 1254 on β-oxidation was attributed to Arochlor 1254-induced impairment of the electron transport chain.37 Several compounds, including perhexiline, amiodarone, benzarone, and benzbromarone, also inhibit mitochondrial β-oxidation,38,39 but the mechanism by which these drugs inhibit β-oxidation has not been elucidated. Although FCCP may perturb the function of the carnitine shuttle and, hence, the uptake of xenobiotic alkanoic acids by mitochondria, the effect of FCCP on the carnitine shuttle has apparently not been reported. The decreased mitochondrial membrane potential that may accompany pathological mitochondrial dysfunction may, however, reduce the rate of biotransformation of alkanoic-acid-based antioxidant prodrugs.
The cytoprotective potential of the thiol-based antioxidant prodrugs 4a and 4b and of 2,6-dimethylphenol prodrugs 1c and 6a was tested in a hypoxia-reoxygenation protocol with isolated cardiomyocytes, which is a model system relevant to ischemia/reperfusion injury. Incubation of cardiomyocytes with either ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a or 4b conferred significant cytoprotection in cardiomyocytes incubated in a hypoxia-reoxygenation protocol (Fig. 4). The cytoprotective effect of ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a or 4b was blocked by etomoxir, which inhibits carnitine palmitoyl transferase-I and, thereby, prevents their delivery to mitochondria.40 Incubation of cardiomyocytes under the hypoxia-reoxygenation protocol with etomoxir alone exerted no cytoprotective effect. Significantly, incubation of cardiomyocytes under the hypoxia-reoxygenation protocol with methimazole alone failed to provide cytoprotection. Hence, the methimazole prodrugs ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a or 4b allow the delivery and release of methimazole to mitochondria and provide cytoprotection that is superior to that afforded by the methimazole itself.
Figure 4.
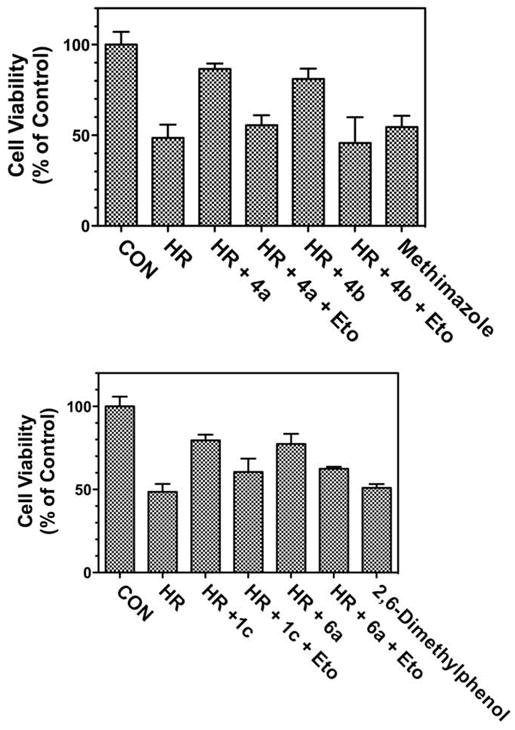
Cytoprotective effects of ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a or 4b and 3-(2,6-dimethylphenoxy)propanoic acid 1c and 3-(2,6-dimethylphenoxy) acrylic acid 6a in isolated rat cardiomyocytes. Upper panel: Alkanoic acids 4a and 4b (1 μM), methimazole (1 μM), and etomoxir (20 μM) were incubated with cardiomyocytes in a hypoxia-reoxygenation protocol, as described in Section 5. CON, control; HR, hypoxia-reoxygenation; Eto, etomoxir. Statistical analysis (one-way ANOVA, n = 3): CON versus HR, p <.05; HR versus HR + 4a, p <.05; CON versus HR + 4a, NS; HR + 4a versus HR + 4a + Eto, p <.05; HR versus HR + 4b, p <.05; HR + 4b versus HR + 4b + Eto, p <.05; HR versus Methimazole, NS. Lower panel: Alkanoic acids 1c and 6a (1 μM), 2,6-dimethylphenol (1 μM), and etomoxir (20 μM) were incubated with cardiomyocytes in a hypoxia-reoxygenation protocol, as described in Section 5. CON, control; HR, hypoxia-reoxygenation; Eto, etoximir. Statistical analysis (one-way ANOVA, n = 3): CON versus HR, p <.05; HR versus HR + 1c, p <.05; CON versus HR + 1c, p <.05; HR + 1c versus HR + 1c + Eto, p <.05; HR versus HR + 6a, p <.05; HR + 6a versus HR + 6a + Eto, p <.05; HR versus 2,6-dimethylphenol, NS.
Similarly, both 3-(2,6-dimethylphenoxy)propanoic acid 1c and 3-(2,6-dimethylphenoxy)acrylic acid 6a were cytoprotective in a hypoxia-reoxygenation protocol, and the cytoprotective effects were blocked by etomoxir (Fig. 4).
The observed cytoprotective effects of prodrugs 1c, 4a, 4b, and 6a may be attributed to their antioxidant actions. Cardiac ischemia/reperfusion injury is characterized by the formation of reactive oxygen species, including superoxide, hydrogen peroxide, and oxygen- and carbon-centered free radicals.41–43 Accordingly, there is interest in the development of mitochondrial-targeted antioxidants for the management of cardiac ischemia/reperfusion injury.44 A range of phenols, particularly ortho-substituted phenols, are chain-breaking antioxidants that react with a range of free-radical species.45,24 2,6-Dimethylphenol, which is released from prodrugs 1c and 6a, is a chain-breaking antioxidant.26 Methimazole, which is released from prodrugs 4a and 4b, scavenges superoxide, hydrogen peroxide, hydroxyl radical, and free radicals.46–48,29 Hence, the cytoprotective actions of prodrugs 1c, 4a, 4b, and 6a are consistent with the pathophysiology of ischemia/reperfusion injury and with the known antioxidant chemistry of 2,6-dimethylphenol and methimazole.
4. Summary
The data presented herein demonstrate that the mitochondrial biotransformation of xenobiotic alkanoic acids can be exploited for the delivery of phenol- and thiol-based antioxidant prodrugs to mitochondria. The rates of biotransformation of ω-(phenoxy) alkanoic acids were highly dependent on the number and position of methyl groups on the phenoxy moiety. The finding that ω-(phenoxy)alkanoic acid 1c, 3-(phenoxy)acrylic acids 6a, and ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b undergo mitochondrial β-oxidation and afford protection against hypoxia-reoxygenation injury in cardiomyocytes demonstrate the potential utility of the use of antioxidant-based xenobiotic alkanoic acids as a mitochondrial delivery system. This is the first investigation of the structural requirements for the mitochondrial β-oxidation of ω-(phenoxy)alkanoic acids and the first demonstration that alkanoic acid-based antioxidant prodrugs are cytoprotective.
5. Experimental
5.1. General
HEPES, EGTA (Sigma E4378), sucrose, ATP disodium salt (Sigma A7699), CoA (Sigma C3144), KCl, MgCl2·6H2O, and L-carnitine were purchased from Sigma–Aldrich (St. Louis, MO). KH2PO4 was obtained from JT Baker; and D,L-dithiothreitol was purchased from TCI America (Boston, MA). Chemicals for organic synthesis were obtained from Aldrich (Milwaukee, WI).
5.2. Chemicals and syntheses (the structures are shown in Fig. 2)
3-(Phenoxy)propanoic acid 1a was purchased from Aldrich.
3-(2-Methylphenoxy)propanoic acid 1b was obtained from UkrOrgSynthesis (Kiev, Ukraine).
3-(2,6-Dimethylphenoxy)propanoic acid 1c was prepared as described by Lichtenberger and Geyer.49
3-(3,5-Dimethylphenoxy)propanoic acid 1d was purchased from UkrOrgSynthesis.
5-(Phenoxy)pentanoic acid 2a was obtained from TCI America (Boston, MA).
The new compound 5-(2-methylphenoxy)pentanoic acid 2b was prepared from 2-methylphenol and methyl 5-bromopentanoate by the method described by Sanders et al.50 1H NMR: (400 MHz, CDCl3) 1.83–1.90 (m, 4H), 2.22 (s, 3H), 2.44–2.48 (t, 2H, J = 7.0 Hz), 3.96–3.99 (t, 2H, J = 5.8 Hz), 6.78–7.15 (m, 4H), 11 (br s, 1H). MS (ESI): 231 [M+Na]+.
The new compound 5-(2,6-dimethylphenoxy)pentanoic acid 2c was prepared in the same manner as 2b,50 except that 2,6-dimethylphenol was used as the starting material. 1H NMR: (400 MHz, CDCl3): 7.0–7.1 (m, 3H), 3.83 (t, 2H, J = 6 Hz), 2.37 (t, 2H, J = 6 Hz), 2.73 (s. 6H), 1.92 (m, 4H). MS (ESI): 223 [M+H]+; 245 [M+Na]+.
The new compound 5-(3,5-dimethylphenoxy)pentanoic acid 2d was prepared in the same manner as 2b,50 except that 3,5-dimethylphenol was used as the starting material. 1H NMR (CDCl3, 400 MHz): 6.58 (s, 1H), 6.51 (s, 2H), 3.94 (t, 2H, J = 5.6 Hz), 2.44 (t, 2H, J = 7.2 Hz), 2.27 (s, 6H), 1.83 (m, 4H). MS (ESI): 245 [M+Na]+.
3-(2-Naphthoxy)propanoic acid 3 was prepared as described by Bel and Duewel.51
The new compound 3-(1-methyl-1H-imidazol-2-ylthio)propanoic acid 4a was prepared as described for 5-(1-methyl-1H-imidazol-2-ylthio)pentanoic acid,52 except that 3-bromopropanoic acid was used as the starting material. 1H NMR (400 MHz, CDCl3): 2.62–2.65 (t, 2H, J = 6.4 Hz), 3.13–3.16 (t, 2H, J = 6.4 Hz), 3.73 (s, 3H), 7.34–7.35 (d, 1H, J = 2.1 Hz), 7.38–7.39 (d, 1H, J = 2.1 Hz), 11 (br s, 1H). MS (ESI): 187 [M+H]+.
5-(1-Methyl-1H-imidazol-2-ylthio)pentanoic acid 4b was synthesized as described by Tweit et al.52
The new compound 3-([2,6-dimethylphenoxy]methylthio)propanoic acid 5 was prepared by reaction of 2-(chloromethoxy)-1,3-dimethylbenzene with methyl 3-mercaptopropanoate followed by hydrolysis of the ester. 2-(Chloromethoxy)-1,3-dimethylbenzene: A three-necked, 500-mL flask equipped with thermometer, reflux condenser, argon inlet, and a magnetic stirring bar was charged with 6.12 g (0.05 mol) of 2,6-dimethylphenol, 100 mL dry tetrahydrofuran, and 4.0 g (0.1 mol) sodium hydroxide powder. The mixture was heated at 66 °C for 1 h. The mixture was cooled to room temperature, and 98 mL (1.5 mol) of bromochloromethane was added. The suspension that formed was cooled to room temperature, filtered through Celite®, and the solvent was removed under reduced pressure. The oily residue was purified by vacuum distillation (1–1.5 torr, bp, 66 °C) to afford an overall yield of 6.50 g (76%) of 2-(chloromethoxy)-1,3-dimethylbenzene. 1H NMR (400 MHz, CDCl3): 2.33 (s, 6H), 5.80 (s, 2H), 6.99–7.06 (m, 3H).
Methyl 3-[(2,6-dimethylphenoxy)methylthio]propanoate
A mixture of 2.40 g (20 mmol) of methyl 3-mercaptopropionate, 2.58 g (20 mmol) of diisopropylethylamine, and 1.70 g (100 mmol) of 2-(chloromethoxy)-1,3 dimethylbenzene in 40 mL of dry tetrahydrofuran under argon was heated at reflux for 52 h. The reaction mixture was cooled, diluted with 60 mL methylene chloride, and extracted with 5% HCl. The organic phase was extracted twice with 30 mL of distilled water, then with 40 mL of saturated sodium chloride solution, and dried with anhydrous magnesium sulfate. The solvent was removed under reduced pressure, and 3.20 g of yellowish oil was obtained. The product was purified by normal-phase chromatography (CombiFlash, Teledyne Isco, Inc., Lincoln, NE) with a hexane/ethyl acetate gradient (100:0, 5 min, 95:5, 15 min) to afford 2.20 g (87%) of methyl 3-[(2,6-dimethylphenoxy) methylthio]propanoate. 1H NMR (400 MHz, CDCl3): 2.32 (s, 6H), 2.78–2.82 (t, 2H, J = 7.2 Hz), 3.03–3.07 (t, 2H, J = 7.2 Hz), 3.70 (s, 3H), 4.07 (s, 2H), 6.92–7.02 (m, 3H).
3-[(2,6-Dimethylphenoxy)methylthio]propanoic acid 5
A 50-mL flask equipped with reflux condenser and a magnetic stirring bar was charged with 0.254 g (1 mmol) of methyl 3-[(2,6-dimethylphenoxy) methylthio]propanoate and 0.362 g (2 mmol) of trimethyltin hydroxide dissolved in 25 mL of 1,2-dichloroethane and was refluxed overnight under an argon atmosphere. The solvent was removed under reduced pressure, the organic residue was dissolved in 40 mL of ethyl acetate, extracted three times with 10 mL 5% HCl, then with 10 mL brine, and dried with anhydrous magnesium sulfate. The solvent was removed under reduced pressure, and the reddish residue was purified by normal-phase (CombiFlash) chromatography with a hexane/ethyl acetate (95:5, 7 min; 95:40, 15 min) to afford 0.2 g (83%) of 3-[(2,6-dimethylphenoxy)methylthio] propanoic acid 5. 1H NMR (400 MHz, CDCl3): 2.33 (s, 6H), 2.78–2.82 (t, 2H, J = 7.2 Hz), 3.03–3.07 (t, 2H, J = 7.2 H), 4.99 (s, 2H), 6.94–7.04 (m, 3H), 10.98 (br, OH). MS (ESI): 241 [M+H]+; 263 [M+Na]+.
The new compound (E)-3-(2,6-dimethylphenoxy)acrylic acid 6a was prepared by the general method of Fan et al.:53 ethyl (E/Z)-3-(2,6-dimethylphenoxy)acrylate: 2,6-Dimethylphenol (1.5 g, 12.3 mmol) and 1,4-diazabicyclo[2.2.2]octane (100 mg, 0.9 mmol) and 2 mL of methylene chloride were combined with magnetic stirring. Ethyl propiolate (1.2 mL, 12.6 mmol) in 3 mL of methylene chloride was added drop wise over 5–10 min. Thin-layer chromatography of the reaction mixture (silica gel, hexane) showed the formation of two products. The reaction mixture was diluted with ~70 mL of ethyl acetate and extracted with 10% potassium hydroxide, and then with saturated sodium chloride solution. The mixture was then extracted with 10% hydrochloric acid solution and then again with saturated sodium chloride solution. The ethyl acetate extract was dried with anhydrous magnesium sulfate, filtered, and evaporated to dryness to afford 2.9 g (100%) of product. A sample of this material (1.0 g) was purified by normal-phase chromatography (CombiFlash) to afford 650 mg of ethyl (E)-3-(2,6-dimethylphenoxy)acrylate [1H NMR (400 MHz, CDCl3): 1.24 (t, 3H, J = 7.2 Hz), 2.17 (s, 6H), 4.13 (q, 2H, J = 7.2 Hz), 5.00 (d, 1H, J = 12.4 Hz), 7.04 (m, 3H), 7.74 (d, 1H, J = 12.4 Hz], and 300 mg of the ethyl (Z)-3-(2,6-dimethylphenoxy)acrylate [1H NMR (400 MHz, CDCl3): 1.31 (t, 3H, J = 7.2 Hz), 2.24 (s, 6H), 4.23 (q, 2H, J = 7.2 Hz), 5.02 (d, 1H, J = 6.8 Hz), 6.46 (d, 1H, J = 6.8 Hz), 7.02 (m, 3H)]. (E)-3-(2,6-Dimethylphenoxy)acrylic acid 6a. Three hundred milligrams of ethyl (E)-3-(2,6-dimethylphenoxy)acrylate was combined with 0.5 g of KOH in 10 mL of a 20:80 mixture of water/ethanol and heated at reflux for 2.5 h. The progress of the reaction was monitored by thin-layer chromatography (silica gel, hexane/ethyl acetate, 95:5). When the reaction had gone to completion, the reaction mixture was removed from heat and the ethanol was removed under reduced pressure. The reaction mixture was acidified with 5% hydrochloric acid solution and extracted with three 30-mL portions of ethyl acetate. The ethyl acetate solution was extracted with saturated sodium chloride solution, dried with anhydrous sodium sulfate, and the solvent was removed under reduced pressure. The solid that formed was titurated with hexane and collected by vacuum filtration to afford 100 mg of (E)-3-(2,6-dimethylphenoxy)acrylic acid 6a. NMR (400 MHz, CDCl3): 2.17 (s, 6H), 5.00 (d, 1H, J = 12.4 Hz), 7.05 (s, 3H), 7.80 (d, 1H, J = 12.4 Hz), 11.0 (br s, 1H). MS (ESI): 215 [M+Na]+.
The new compound (E)-3-(2,6-diisopropylphenoxy)acrylic acid 6b was also prepared by the general method of Fan et al.:53 ethyl (E/Z)-3-(2,6-diisopropylphenoxy)acrylate. 2,6-Diisopropylphenol (2.0 g, 11.3 mmol), 1,4-diazabicyclo[2.2.2]octane (122 mg, 1 mmol), and 10 mL of methylene chloride were combined with magnetic stirring. Ethyl propiolate (1.20 mL, 116 mmol) in 4 mL of methylene chloride was added drop wise over 10 min. The reaction mixture was refluxed overnight and was then was diluted with 85 mL of ethyl acetate and extracted with 10% potassium hydroxide solution, with 10% hydrochloric acid solution, and then with saturated sodium chloride solution. The ethyl acetate layer was dried with anhydrous magnesium sulfate and filtered. The solvent was removed under reduced pressure to afford 2.72 g (87% yield) of a mixture of E- and Z-isomers of the product. The isomeric mixture (500 mg) was separated and purified by normal-phase chromatography (CombiFlash) with a hexane/ethyl acetate gradient (95:5, 6 min; 80:20, 10 min) to afford 325 mg (65%) of ethyl (E)-3-(2,6-diisopropylphenoxy)acrylate. 1H NMR (400 MHz, CDCl3): 1.17–1.19 (d, 12H), 1.22–1.26 (t, 3H, J = 7.2), 3.00–3.07 (m, 2H), 4.11–4.17 (q, 2H), 5.06–5.09 (d, 1H, J = 12.4 Hz), 7.14–7.22 (m, 3H), 7.74–7.77 (d, 1H, J = 12.4 Hz).
(E)-3-(2,6-Diisopropylphenoxy)acrylic acid 6b. Ethyl (E)-3-(2,6-diisopropylphenoxy)acrylate (250 mg, 0.91 mmol) was combined with 0.5 g of solid KOH in 10 mL of a mixture of water/ethanol (20:80), and the mixture was refluxed overnight. The solvent was removed under reduced pressure. The residue was acidified with 5% HCl solution and then extracted with three 25 mL portions of ethyl acetate. The ethyl acetate solution was extracted with saturated sodium chloride solution and dried with anhydrous sodium sulfate. The solvent was removed under reduced pressure to yield 150 mg (60%) of (E)-3-(2,6-diisopropylphenoxy)acrylic acid 6b. 1H NMR (400 MHz, CDCl3) 1.17–1.19 (d, 12H), 2.98–3.05 (m, 2H), 5.05–5.08 (d, 1H, J = 12.4 Hz), 7.14–7.22 (m, 3H), 7.81–7.85 (d, 1H, J = 12.4 Hz). MS (ESI): 271 [M+Na]+.
5.3. Mitochondrial biotransformation of alkanoic-acid-based prodrugs
Rat liver mitochondria were isolated from livers freshly collected from adult male Sprague-Dawley rats (190–225 g) and were used as soon as possible after isolation.54,55 The animal-use protocol was reviewed and approved by the University of Rochester University Committee on Animal Resources. Livers were removed immediately after CO2 euthanasia of the animal, minced, and homogenized with a glass Dounce homogenizer with loose-fitting pestle in liver mitochondria isolation medium that contained 0.25 M sucrose, 2.0 mM HEPES, 1.0 mM EGTA (pH 7.4) at 4 °C. The homogenate was centrifuged once at 1000g for 3 min at 4 °C to remove cellular debris. After removal of visible surface fat, the resulting supernatant was centrifuged at 10,000g for 10 min at 4 °C. The supernatant was discarded after removing visible surface fat, and the pellet was suspended in ice-cold liver mitochondria isolation medium and centrifuged again at 10,000g for 10 min at 4 °C. The mitochondrial pellet thus obtained was suspended in Ca2+-depletion buffer (1 mM EGTA, 10 mM NaCl, 5 mM succinate, 195 mM mannitol, 25 mM sucrose, 40 mM HEPES, pH 7.2) at room temperature) and incubated with gentle stirring first at room temperature for 10 min and then in an ice bath for 5 min. Ca2+-Depletion buffer serves to chelate calcium, activate mitochondrial respiration, and activate the Na+/Ca2+ exchanger. The mitochondrial suspension was then centrifuged at 10,000g for 10 min at 4 °C, and the pellet was suspended in liver swelling buffer that contained 195 mM mannitol, 25 mM sucrose, 40 mM HEPES (pH 7.2) at 4 °C; liver swelling buffer allows for ionic/osmotic equilibrium through the remainder of the preparation. The suspension was centrifuged at 10,000g for 10 min at 4 °C, followed by the removal of light mitochondria and a final centrifugation at 10,000g for 10 min at 4 °C. The mitochondrial pellet was suspended in liver swelling buffer at 4 °C. Protein concentrations were determined with the Lowry reagent.56
The quality of mitochondrial preparations was determined by estimating the respiration control ratio (RCR, i.e., the ratio of state 3 vs state 4 respiration rates) with a Clark-type oxygen electrode (Oxygraph, Hansatech Instruments, Norfolk, England). The incubation mixtures contained 120 mMKCl, 25 mMsucrose, 5 mMMgCl2, 5mM KH2PO4, 1 mM EGTA, 10 mM HEPES, 10 mM succinate, 2.5 mg/mL fat-free bovine serum albumin, 0.5–0.8 mg/mL mitochondrial protein, 5 μM rotenone, and 100 μM ADP (pH 7.4) and were incubated at 37 °C. RCR ranged from 4.5 to 6.5.
Reaction mixtures were prepared in disposable glass culture tubes and contained 20 mM HEPES, 1 mM EGTA, 100 mM KCl, 5mM KH2PO4, 10 mM MgCl2, 25 mM sucrose, 2 mM L-carnitine (if added), 5 mM ATP, 10 mg/mL bovine serum albumin, 1.4 mM dithiothreitol, 0.13 mM CoA, and 1 mM substrate (pH 7.4). Reaction mixtures were incubated at 37 °C with gentle stirring. Mitochondrial protein (approximately 5 mg/mL) was added, and the samples were incubated for 5 min with gentle mixing, which was maintained during the incubation period, to keep the homogenate suspended. After 5 min of incubation, the substrates were added. The substrates were dissolved in methanol and added to the reaction mixtures to a final concentration of 1 mM, unless otherwise stated; the final methanol concentration was ≤0.5%. ω-(1-Methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b were dissolved in distilled water and added to the reaction mixtures to a final concentration of 1 mM. Samples were incubated at 37 °C for 30 min with constant gentle mixing, and samples were collected for analysis at t = 0, 10, 20, and 30 min.
5.4. Analyses
Phenolic metabolite formation, that is, phenol, 2-methylphenol, 2,6-dimethylphenol, 3,5-dimethylphenol, and 2,6-diisopropylphenol, from ω-(phenoxy)alkanoic acids, was quantified by HPLC analysis. Two hundred microliters of the reaction mixture was added to 200 μL of acetonitrile; the mixtures were mixed and then sonicated in a Branson ultrasonic water bath (Danbury, CT) for 5–10 min. The denatured sample mixtures were centrifuged at 15,000–16,000g for 5 min. Supernatants were analyzed with an Agilent 1100 series HPLC (Santa Clara, CA) with UV detection at 265 and 272 nm. A 5 μL sample of the supernatant was injected onto a Phenomenex Synergi Hydro-RP analytical column (150 mm × 2.0 mm, 4 μm particle size, 80 Å pore, 30 °C). The column was eluted at a flow rate of 0.4 mL/min with 0.1% (v/v) formic acid (solvent A) and acetonitrile (solvent B) with this gradient: 15% B for 5 min, increased to 95% B over 20 min, held at 95% B for 10 min, and returned to 15% B in 2 min. The column was equilibrated with 15% B for 10 min before the next sample was analyzed.
In some experiments, phenolic metabolites of ω-(phenoxy)alkanoic acids were analyzed by LC–MS on an Agilent 1100 LC/MSD ion trap with electrospray ionization (ESI) interface in either positive- or negative-ion, smart-tune mode (nebulizer pressure, 30 psi; dry gas flow (nitrogen), 8 Lpm; dry temperature, 350 °C) with the same HPLC parameters as described above.
Methimazole formation from ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b was quantified by adding 200 μL of the reaction mixture to 10 μL of 70% perchloric acid followed by mixing. The mixtures were centrifuged at 15,000–16,000g for 5 min. Samples of the supernatants were analyzed by HPLC (Agilent 1100 series) with UV detection at 250 nm. A 5 μL sample of the supernatant was injected onto a Phenomenex Synergi Hydro-RP analytical column (150 mm × 2.0 mm, 4 μm particle size, 80 Å pore) held at 30 °C. The column was eluted with 5 mM sodium 1-heptanesulfonate monohydrate (Fluka) (solvent A) and acetonitrile (solvent B) at a flow rate of 0.4 mL/min with this gradient: 100% A for 5 min, increased to 30% B over 15 min, held at 30% B for 5 min, increased to 90% B over 5 min, held at 90% B for 5 min, and returned to 100% A over 3 min. The column was equilibrated with 100% A for 10 min before the next sample was analyzed.
The mitochondrial biotransformation of octanoic acid was measured as described by Bjorge and Baillie.16
5.5. Biological evaluation
The cytoprotective effects of ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b, 3-(2,6-dimethylphenoxy)propanoic acid 1c, and 3-(2,6-dimethylphenoxy)acrylic acid 6a were investigated in cardiomyocytes. Primary ventricular cardiomyocytes were isolated from adult rats and viability was assessed as described previously. 54 All cells were used within 1 h of isolation and were 80–85% viable and rod-shaped prior to the experiments. Cell viability was quantified with the Trypan Blue exclusion test. The cells were subjected to a hypoxia-reoxygenation protocol designed to simulate cardiac ischemia-reperfusion injury. The cells were divided into four treatment groups: (i) control: cells were incubated under an atmosphere of 95% O2/5% CO2 in Krebs–Henseleit buffer, pH 7.4; (ii) hypoxia-reoxygenation (HR): cells were incubated for 1 h under an atmosphere of 95% N2/5% CO2 in glucose-free 50 mM HEPES buffer, pH6.5, followed by incubation for 30 min in the reoxygenation medium (95% O2/5% CO2, glucose-containing Krebs–Henseleit buffer, pH 7.4); (iii) HR conditions except that ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b, 3-(2,6-dimethylphenoxy)propanoic acid 1c, and 3-(2,6-dimethylphenoxy)-acrylic acid 6a were added 20 min before incubation under HR conditions; (iv) HR conditions (see iii) except that etomoxir was added 10 min before ω-(1-methyl-1H-imidazol-2-ylthio)alkanoic acids 4a and 4b, 3-(2,6-dimethylphenoxy) propanoic acid 1c, and 3-(2,6-dimethylphenoxy)-acrylic acid 6a and before incubation under HR conditions. Note that with this protocol, it was necessary to isolate the cells by centrifugation (2 min. at 31g) to change buffers; hence, the added compounds were not present during incubation under hypoxic conditions.
5.6. Statistical analyses
Data were analyzed with GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA). Data are shown as means ± SD. A value of p <.05 was used for the rejection of the null hypothesis.
Acknowledgments
The authors thank James L. Robotham for support of this research and Horst Schulz for helpful discussions.
References and notes
- 1.Turrens JF. J Physiol. 2003;552:335. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brookes PS, Yoon Y, Robotham JL, Anders MW, Sheu SS. Am J Physiol. 2004;287:C817. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 3.Sheu SS, Nauduri D, Anders MW. Biochim Biophys Acta. 2006;1762:256. doi: 10.1016/j.bbadis.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 4.Anders MW, Robotham JL, Sheu SS. Expert Opin Drug Metab Toxicol. 2006;2:71. doi: 10.1517/17425255.2.1.71. [DOI] [PubMed] [Google Scholar]
- 5.Murphy MP, Smith RA. Annu Rev Pharmacol Toxicol. 2007;47:629. doi: 10.1146/annurev.pharmtox.47.120505.105110. [DOI] [PubMed] [Google Scholar]
- 6.Cocheme HM, Kelso GF, James AM, Ross MF, Trnka J, Mahendiran T, Asin-Cayuela J, Blaikie FH, Manas AR, Porteous CM, Adlam VJ, Smith RA, Murphy MP. Mitochondrion. 2007;7:S94. doi: 10.1016/j.mito.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Victor VM, Rocha M. Curr Pharm Des. 2007;13:845. doi: 10.2174/138161207780363077. [DOI] [PubMed] [Google Scholar]
- 8.Mattarei A, Biasutto L, Marotta E, De Marchi U, Sassi N, Garbisa S, Zoratti M, Paradisi C. ChemBioChem. 2008;9:2633. doi: 10.1002/cbic.200800162. [DOI] [PubMed] [Google Scholar]
- 9.Van Peteghem CH, Heyndrickx AM. Bull Environ Contam Toxicol. 1975;14:632. doi: 10.1007/BF01683383. [DOI] [PubMed] [Google Scholar]
- 10.Fromenty B, Freneaux E, Labbe G, Deschamps D, Larrey D, Letteron P, Pessayre D. Biochem Pharmacol. 1989;38:3743. doi: 10.1016/0006-2952(89)90580-7. [DOI] [PubMed] [Google Scholar]
- 11.Hanley PJ, Drose S, Brandt U, Lareau RA, Banerjee AL, Srivastava DK, Banaszak LJ, Barycki JJ, Van Veldhoven PP, Daut J. J Physiol. 2005;562:307. doi: 10.1113/jphysiol.2004.073932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitzsimmons ME, Baggs RB, Anders MW. J Pharmacol Exp Ther. 1994;271:515. [PubMed] [Google Scholar]
- 13.Fitzsimmons ME, Anders MW. Chem Res Toxicol. 1993;6:662. doi: 10.1021/tx00035a011. [DOI] [PubMed] [Google Scholar]
- 14.Fitzsimmons ME, Thorpe C, Anders MW. Biochemistry. 1995;34:4276. doi: 10.1021/bi00013a017. [DOI] [PubMed] [Google Scholar]
- 15.Olowe Y, Schulz H. J Biol Chem. 1982;257:5408. [PubMed] [Google Scholar]
- 16.Bjorge SM, Baillie TA. Drug Metab Dispos. 1991;19:823. [PubMed] [Google Scholar]
- 17.Lai MT, Li D, Oh E, Liu HW. J Am Chem Soc. 1993;115:1619. [Google Scholar]
- 18.Knoop F. Beitr Chem Physiol Pathol. 1905;6:150. [Google Scholar]
- 19.Dakin HD. J Biol Chem. 1909;6:221. [Google Scholar]
- 20.Raper HS, Wayne EJ. Biochem J. 1928;22:188. doi: 10.1042/bj0220188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quick AJ. J Biol Chem. 1928;80:515. [Google Scholar]
- 22.Quick AJ. J Biol Chem. 1928;77:581. [Google Scholar]
- 23.Levey S, Lewis HB. J Biol Chem. 1947;168:213. [PubMed] [Google Scholar]
- 24.Wright JS, Johnson ER, DiLabio GA. J Am Chem Soc. 2001;123:1173. doi: 10.1021/ja002455u. [DOI] [PubMed] [Google Scholar]
- 25.Rigobello MP, Stevanato R, Momo F, Fabris S, Scutari G, Boscolo R, Folda A, Bindoli A. Free Radical Res. 2004;38:315. doi: 10.1080/03079450310001652031. [DOI] [PubMed] [Google Scholar]
- 26.Ogata M, Shin-Ya K, Urano S, Endo T. Chem Pharm Bull. 2005;53:344. doi: 10.1248/cpb.53.344. [DOI] [PubMed] [Google Scholar]
- 27.Nantasenamat C, Isarankura-Na-Ayudhya C, Naenna T, Prachayasittikul VJ. Mol Graphics Modell. 2008;27:188. doi: 10.1016/j.jmgm.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 28.Sausen PJ, Elfarra AA, Cooley AJ. J Pharmacol Exp Ther. 1992;260:393. [PubMed] [Google Scholar]
- 29.Kim H, Lee TH, Hwang YS, Bang MA, Kim KH, Suh JM, Chung HK, Yu DY, Lee KK, Kwon OY, Ro HK, Shong M. Mol Pharmacol. 2001;60:972. doi: 10.1124/mol.60.5.972. [DOI] [PubMed] [Google Scholar]
- 30.Nada MA, Rhead WJ, Sprecher H, Schulz H, Roe CR. J Biol Chem. 1995;270:530. doi: 10.1074/jbc.270.2.530. [DOI] [PubMed] [Google Scholar]
- 31.Liang X, Le W, Zhang D, Schulz H. Biochem Soc Trans. 2001;29:279. doi: 10.1042/0300-5127:0290279. [DOI] [PubMed] [Google Scholar]
- 32.Yu W, Liang X, Ensenauer RE, Vockley J, Sweetman L, Schulz H. J Biol Chem. 2004;279:52160. doi: 10.1074/jbc.M409640200. [DOI] [PubMed] [Google Scholar]
- 33.Lau SM, Brantley RK, Thorpe C. Biochemistry. 1988;27:5089. doi: 10.1021/bi00414a021. [DOI] [PubMed] [Google Scholar]
- 34.Wu WJ, Feng Y, He X, Hofstein HA, Raleigh DP, Tonge PJ. J Am Chem Soc. 2000;122:3987. [Google Scholar]
- 35.Benz R, McLaughlin S. Biophys J. 1983;41:381. doi: 10.1016/S0006-3495(83)84449-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aly HAA, Domenech O. Toxicology. 2009;262:175. doi: 10.1016/j.tox.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Latipää PM, Kärki TT, Hiltunen JK, Hassinen IE. Biochim Biophys Acta. 1986;875:293. doi: 10.1016/0005-2760(86)90179-7. [DOI] [PubMed] [Google Scholar]
- 38.Deschamps D, DeBeco V, Fisch C, Fromenty B, Guillouzo A, Pessayre D. Hepatology. 1994;19:948. [PubMed] [Google Scholar]
- 39.Kaufmann P, Török M, Hänni A, Roberts P, Gasser R, Krähenbühl S. Hepatology. 2005;41:925. doi: 10.1002/hep.20634. [DOI] [PubMed] [Google Scholar]
- 40.Declercq PE, Falck JR, Kuwajima M, Tyminski H, Foster DW, McGarry JD. J Biol Chem. 1987;262:9812. [PubMed] [Google Scholar]
- 41.Zweier JL, Flaherty JT, Weisfeldt ML. Proc Natl Acad Sci USA. 1987;84:1404. doi: 10.1073/pnas.84.5.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tompkins AJ, Burwell LS, Digerness SB, Zaragoza C, Holman WL, Brookes PS. Biochim Biophys Acta. 2006;1762:223. doi: 10.1016/j.bbadis.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 43.Lambert AJ, Brand MD. In: Methods in Molecular Biology. Walker JM, editor. Humana Press; New York: 2009. p. 165. [Google Scholar]
- 44.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. FASEB J. 2005;19:1088. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 45.Howard JA, Ingold KU. Can J Chem. 1963;41:2800. [Google Scholar]
- 46.Taylor JJ, Willson RL, Kendall-Taylor P. FEBS Lett. 1984;176:337. [Google Scholar]
- 47.Petry TW, Eling TE. J Biol Chem. 1987;262:14112. [PubMed] [Google Scholar]
- 48.Lagorce JF, Moulard T, Rousseau A, Comby F, Buxeraud J, Raby C. Pharmacology. 1997;55:173. doi: 10.1159/000139525. [DOI] [PubMed] [Google Scholar]
- 49.Lichtenberger J, Core J, Geyer R. Bull Soc Chim Fr. 1962:997. [Google Scholar]
- 50.Sanders BG, Kline K, Hurley L, Gardner R, Menchaca M, Yu W, Ramanan P, Liu S, Israel K. 6, 770, 672. US Patent. 2004
- 51.Bel KH, Duewel H. Aust J Chem. 1963;16:101. [Google Scholar]
- 52.Tweit RC, Kreider EM, Muir RD. J Med Chem. 1973;16:1161. doi: 10.1021/jm00268a021. [DOI] [PubMed] [Google Scholar]
- 53.Fan M-J, Li G-Q, Li L-H, Yang S-D, Liang Y-M. Synthesis. 2006:2286. [Google Scholar]
- 54.Nadtochiy SM, Burwell LS, Brookes PS. J Mol Cell Cardiol. 2007;42:812. doi: 10.1016/j.yjmcc.2007.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffman DL, Salter JD, Brookes PS. Am J Physiol. 2007;292:H101. doi: 10.1152/ajpheart.00699.2006. [DOI] [PubMed] [Google Scholar]
- 56.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. J Biol Chem. 1951;193:265. [PubMed] [Google Scholar]