

Abstract
BACKGROUND & AIMS
Severe polycystic liver disease can complicate adult dominant polycystic kidney disease, a genetic disease caused by defects in polycystin-1 (Pkd1) or polycystin-2 (Pkd2). Liver cyst epithelial cells (LCECs) express vascular endothelial growth factor (VEGF) and its receptor, VEGFR-2. We investigated the effects of VEGF on liver cyst growth and autocrine VEGF signaling in mice with Pkd1 and Pkd2 conditional knockouts.
METHODS
We studied mice in which Pkd1 or Pkd2 were conditionally inactivated following exposure to tamoxifen; these mice were called Pkd1flox/−:pCxCreER (Pkd1KO) and Pkd2flox/−:pCxCreER (Pkd2KO).
RESULTS
Pkd1KO and Pkd2KO mice developed liver defects; their LCECs expressed VEGF, VEGFR-2, hypoxia-inducible factor (HIF)-1α, phosphorylated extracellular signal–regulated kinase 1/2 (pERK1/2), and proliferating cell nuclear antigen (PCNA). In Pkd2KO but not Pkd1KO mice, exposure to the VEGFR-2 inhibitor SU5416 significantly reduced liver cyst development, liver/body weight ratio, and expression of pERK and PCNA. VEGF secretion and phosphorylation of ERK1/2 and VEGFR-2 were significantly increased in cultured LCECs from Pkd2KO compared with Pkd1KO mice. Inhibition of protein kinase A (PKA) reduced VEGF secretion and pERK1/2 expression. Addition of VEGF to LCECs from Pkd2KO mice increased phosphorylated VEGFR-2 and phosphorylated mitogen signal-regulated kinase (MEK) expression and induced phosphorylation of ERK1/2; this was inhibited by SU5416. Expression of HIF-1α increased in parallel with secretion of VEGF following LCEC stimulation. VEGF-induced cell proliferation was inhibited by the MEK inhibitor U1026 and by ERK1/2 small interfering RNA.
CONCLUSIONS
The PKA–ERK1/2–VEGF signaling pathway promotes growth of liver cysts in mice. In Pkd2-defective LCECs, PKA-dependent ERK1/2 signaling controls HIF-1α–dependent VEGF secretion and VEGFR-2 signaling. Autocrine and paracrine VEGF signaling promotes the growth of liver cysts in Pkd2KO mice. VEGF inhibitors might be used to treat patients with polycystic liver disease.
Autosomal dominant polycystic kidney disease (ADPKD) is a common hereditary disease characterized by the formation of innumerable cysts in the kidney that is accompanied in more than 90% of cases by bile duct–derived liver cysts.1 In the liver, multiple biliary micro-hamartomas progressively dilate to form macroscopic cysts scattered throughout the liver parenchyma.2 The progressive growth of the liver cysts may cause a mass effect and even lead to complications requiring partial hepatectomy or liver transplantation. There is strong interest in finding medical therapies for symptomatic liver disease.3,4 Aside from estrogen-related factors, the mechanisms promoting the growth of liver cysts in ADPKD are not well understood.
ADPKD is caused by mutations in PKD1, encoding polycystin-1 (PC1), or PKD2, encoding polycystin-2 (PC2).5 PC1 is a transmembrane protein localized in the primary cilium of epithelial cells, where it senses changes in apical flow and compositions and modulates signals involved in epithelial cell proliferation, morphogenesis, and differentiation.5 PC2 (or TRPP26) is a nonselective Ca2+ channel that interacts with PC1 in cilia.7 PC2 is also expressed in the endoplasmic reticulum, where it interacts with 2 important regulators of intracellular Ca2+ homeostasis: the ryanodine8 and the InsP3 receptors.9
Cholangiocytes lining the liver cysts in ADPKD present phenotypic and functional similarities with reactive ductules, that is, the immature cholangiocytes that proliferate in many forms of liver injury.10 Among them are an aberrant secretion of cytokines and chemokines11 as well as growth factors, including vascular endothelial growth factor A (VEGF).11,12 VEGF interacts with 2 receptor tyrosine kinases: VEGFR-1 and VEGFR-2. In endothelial cells, VEGFR-2 mediates the mitogenic effects of VEGF, signaling through the Raf/mitogen signal-regulated kinase (MEK)/extracellular signal–regulated kinase (ERK) cascade13; VEGF and VEGF receptors are expressed also by biliary cell progenitors during development and have an important function in regulating liver arterial neovasculogenesis.14
We12 and others15 have previously studied the expression of VEGF and its receptors in the liver cysts of patients with ADPKD and shown that VEGF, VEGFR-1, and VEGFR-2 are strongly up-regulated in the cystic epithelium.12 Cholangiocytes isolated from these cysts secreted VEGF and proliferated in response to administration of VEGF. We hypothesized that VEGF promotes the progression of polycystic liver disease via autocrine stimulation of cholangiocyte proliferation.
This study was designed to test this hypothesis and to address the mechanisms by which cystic cholangiocytes express more VEGF and are more responsive to VEGF. Using inducible knockout of either Pkd1 or Pkd2 in mice, we show that protein kinase A (PKA)-mediated activation of ERK1/2 is responsible for increased hypoxia-inducible factor (HIF)-1α–dependent VEGF production and increased VEGFR-2–mediated autocrine stimulation of cyst growth following inactivation of Pkd2. Administration of a competitive inhibitor of VEGFR-2 inhibits the phosphorylation of ERK1/2, reduces the proliferative activity of the cystic epithelium, and inhibits the growth of liver cysts following inactivation of Pkd2 in vivo.
Materials and Methods
Materials, Antibodies, and Immunohistochemistry
All materials, antibodies, reagents, and their providers are listed in the Supplementary Materials and Methods.
Animals and Treatment
We established an inducible model for Pkd1 and Pkd2 inactivation using conditional Pkd1flox and Pkd2flox alleles in combination with the tamoxifen inducible pCXCreER line. The Pkd1flox allele16 and the pCXCreER line have been reported previously.17 The pCXCreER transgene has a generalized promoter that achieves robust expression in bile ducts (Supplementary Figure 1A). The novel Pkd2flox allele (X. Tian, S. Somlo, manuscript in submission) introduced loxP sites flanking exons 3 and 4 of Pkd2. The Pkd2flox allele functions as a wild-type (WT) allele before Cre-mediated excision and as a null allele after excision of exons 3 and 4. Experimental mice with either Pkd1flox/−:pCxCreER (Pkd1KO) or Pkd2flox/−:pCxCreER (Pkd2KO) genotypes received tamoxifen (0.2 mg · g−1 · day−1) for 5 days beginning at postnatal day 28. These mice developed bile duct– derived liver cysts over the ensuing 8 weeks. Cre activity was present in the liver cyst linings of tamoxifen-treated Pkd2flox/−:pCxCreER;Rosa26R mice stained with β-galactosidase antibody (Supplementary Figure 1B), confirming the association of Cre-mediated gene inactivation with the formation of bile duct–derived cysts by a cellular recessive mechanism.
Animals were treated with the VEGFR-2 inhibitor SU5416 (12.5 mg/kg), or with vehicle (dimethyl sulfoxide) intraperitoneally twice a week for 8 weeks, starting 1 week after induction with tamoxifen. All experiments were performed according to protocols approved by the Yale University Institutional Animal Care and Use Committee.
Quantization of Cystic Area and of Pancytokeratin-Positive Structures
The 2 main liver lobes were embedded in paraffin, and serial 5-μm sections were cut and immunostained with a pancytokeratin antibody to allow a correct discrimination of biliary cysts from vascular structures. We calculated the relative area covered by the biliary cysts as follows. (1) Five random nonoverlapping fields for each main liver lobe (10 fields/mouse) were recorded with a digital camera at 10× magnification, and the pancytokeratin-positive cystic areas (μm2) were manually measured using ImageJ software18 by 2 investigators blinded to the treatment code. (2) The same samples underwent computer-assisted morphometric analysis (MetaMorph software, Ontario, NY) using a motorized stage system able to scan, at 4× magnification, the whole liver lobes.18 In this case, data were expressed as the percentage of the whole liver lobe area occupied by pancytokeratin-positive cells. The setup consisted of a Nikon Eclipse TE2000U microscope (Nikon, Avon, MA), a motorized stage system (Roper Scientific, Trenton, NJ), and a Photometrics Cool Snap HQ digital camera (Photometrics, Tucson, AZ).
Measurement of Proliferating Cell Nuclear Antigen–Positive Cells
Liver sections from treated and untreated animals were immunostained with proliferating cell nuclear antigen (PCNA) antibody to assess the percentage of cystic cholangiocytes entering the cell cycle. Five random nonoverlapping fields per each slide were recorded by a digital camera at 40× magnification. An average of 500 nuclei was counted per each mouse, and the percentage of strongly PCNA-positive nuclei was then calculated.
Morphometric Quantization of Phosphorylated ERK and Cleaved Caspase-3
Immunohistochemistry for phosphorylated ERK (pERK) and for cleaved caspase-3 (CC3) was used to assess the activation of the ERK pathway and to detect cells undergoing apoptosis, respectively. The amount of pERK and CC3-positive structures was expressed as a percentage of the cytokeratin-19 –positive epithelial cell area.
Morphometric Quantization of Pericystic CD34-Positive Structures
Liver sections of Pkd2KO and Pkd1KO mice, in the presence and absence of SU5416, were stained with rat anti-CD3419 and counterstained with pancytokeratin. The biliary and vascular areas were calculated as reported in the Supplementary Materials and Methods.
Cell Isolation and Characterization
Mouse cholangiocytes and cystic epithelial cells were isolated from WT, Pkd1KO, and Pkd2KO mice and cultured essentially as previously described.12,20 Microdissected intrahepatic bile ducts or liver cysts were used to obtain WT cholangiocytes or cystic epithelium, respectively. Cells were then cultured on the top of rat tail collagen and subcultured when confluent. Cells were used up to the eighth passage. Method and characterization of the cell cultures are reported in the Supplementary Materials and Methods.
Western Blots
Western blots were performed essentially as previously described12 and as described in the Supplementary Materials and Methods.
Determination of ERK and pERK Expression in Cultured Cells
Twenty-four–hour serum-starved cells were exposed to 25 ng/mL of exogenous VEGF, a dose that we had previously shown to produce a 75% stimulation of cell proliferation in cystic cholangiocytes.12 After 5-, 10-, 15-, and 30-minute incubation times, cells were collected as previously described and nuclear and cytoplasmic extracts were then prepared using the NE-PER Nuclear and Cytoplasmic Extraction Protocol (Pierce Biotechnology, Rockford, IL) according to the manufacturer’s instructions. Each extract was subjected to Western blot analysis for the detection of pERK, total ERK1/2, and actin.
Determination of HIF-1α in Nuclear Fractions in Cultured Cells
Cells were incubated in the presence of 2-oxoglutarate analogues dimethyloxaloylglycine (DMOG) (3 mmol/L for 18 hours), with or without the MEK inhibitor U0126 (10 μmol/L, 1 hour pretreatment). Nuclear fractions of each sample were isolated using a nuclear extraction kit (NE-PER; Pierce Biotechnology). Protein concentration was determined using the Bradford method (Pierce Biotechnology). The amount of HIF-1α was measured by DuoSet enzyme-linked immunosorbent assay, following the protocol from the manufacturer (R&D Systems, Minneapolis, MN), and normalized to the amount of nuclear protein.
Measurement of VEGF Secretion in Cultured Cells
An enzyme-linked immunosorbent assay (Biosource International, Carlsbad, CA) was used to quantify VEGF in culture medium collected from cholangiocytes isolated from polycystic and control mice, as we previously described.12 Briefly, medium was incubated with a highly purified antibody coated onto 96-well plates. A VEGF standard curve was generated for each individual experiment. Readings were normalized for the total protein in the well.
Measurement of Cell Proliferation
WT and PKD2KO cholangiocytes were passaged and plated in a 96-multiwell plate (5000 cells/well) with quiescent medium (without fetal bovine serum).12 After 24 hours, cells were supplemented with VEGF (25 ng/mL) alone and with the MEK inhibitor U0126 (10 μmol/L). Proliferation was assessed by the CellTiter 96 AQueous One Solution (Promega, Madison, WI), which exploits the MTS tetrazolium compound colorimetric bioreduction by the cells.12
Silencing of ERK1/2 in Cultured Cells
Commercially available silencer small interfering RNA (siRNA) for ERK1/2 and a scramble negative control were purchased from Qiagen (Valencia, CA). Cultured cystic cholangiocytes were plated at 5000 cells per well and transfected using naked siRNA resuspended in OptiMEM medium (Invitrogen, Carlsbad, CA) as described21 (final siRNA concentration was 30 nmol/L). Twenty-four hours after transfection, cells were stimulated with VEGF (25 nmol/L) for an additional 24 hours. Proliferation was assessed by MTS assay.12 Efficiency of silencing was checked in cells cultured at 60% confluence and processed for the isolation of total protein 48 hours after tranfection. ERK1/2 expression was determined by Western blot. siRNA decreased ERK1/2 by 60% with respect to scramble siRNA.
Statistical Analysis
Results are shown as mean ± SD. Statistical comparisons were made using one-way analysis of variance or the Wilcoxon–Mann–Whitney 2-sample rank sum test, where appropriate. In the latter, the P value was obtained from the exact permutation null distribution. The statistical analysis was performed using SAS software (SAS Institute Inc, Cary, NC). P values <.05 were considered significant.
Results
Characterization of the Liver Phenotype in Conditional Polycystin Knockout Mice
Pkd1KO and Pkd2KO mice had normal-appearing bile ducts before tamoxifen-induced gene inactivation (data not shown) but developed a bile duct cystic liver phenotype similar to human ADPKD after activation of Cre-mediated recombination by tamoxifen (Figure 1). Liver cysts were evident 4 weeks after induction and progressively enlarged until the time the mice were killed (8 weeks after, 15 weeks of age). These findings show that Pkd1 and Pkd2 expression are required to maintain normal bile ducts in adult, postdevelopmental liver tissues. Consistent with our earlier observations in human ADPKD, VEGF, VEGFR-2, and HIF-1α were expressed in the cystic epithelium of both Pkd1KO and Pkd2KO mice (Figure 1). This establishes the Pkd1KO and Pkd2KO mice as appropriate models to study the role of VEGF on liver cyst growth in ADPKD. The liver phenotype was more severe in Pkd2KO than Pkd1KO mice (Figure 2). Cystic area in Pkd2KO mice was 2.78-fold higher than in Pkd1KO mice; liver/body weight ratio was 0.089 ± 0.017 (n = 6) in Pkd2KO mice versus 0.048 ± 0.014 (n = 4) in Pkd1KO mice (P < .003). In both mice, the cystic epithelium was strongly positive for PCNA, indicating ongoing epithelial proliferation. Consistent with the more severe phenotype, the percentage of PCNA-positive cystic cholangiocytes was significantly higher in Pkd2KO mice (57.74%) than in Pkd1KO mice (28.92%) (Figure 3). Furthermore, the percentage of phospho-ERK1/2–positive cells was higher in Pdk2KO mice (pERK-positive area in Pkd2KO mice was 3.17% ± 0.84% of the total lobe area vs 1.7% ± 1.2% in Pkd1KO mice; P < .05) (Figure 3). Pericystic CD34-positive structures were significantly higher in Pkd2KO mice as compared with Pkd1KO mice (Supplementary Figures 2 and 3).
Figure 1.
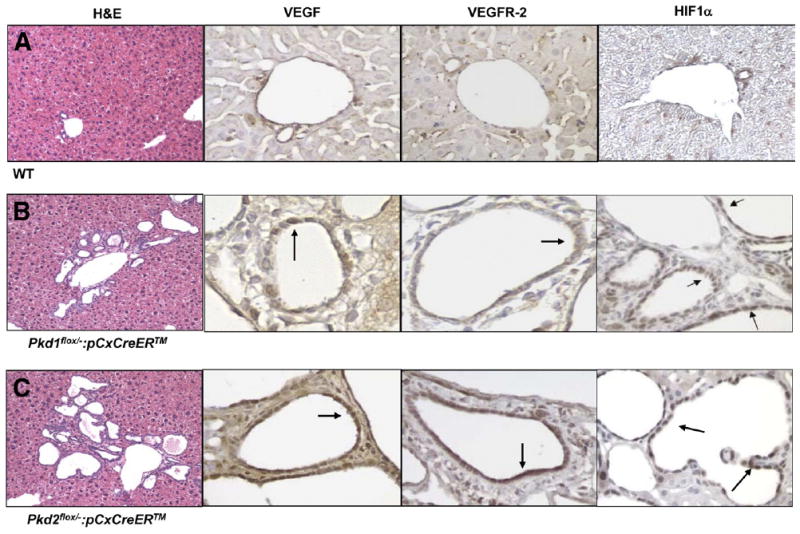
Expression of VEGF, VEGFR-2, and HIF-1α in ADPKD mice. (A) WT, (B) Pkd1KO, and (C) Pkd2KO mice were stained with H&E or with specific antibodies against VEGF, VEGFR-2, and HIF-1α. In cystic epithelium (B and C), immunoreactivity for VEGF, VEGFR-2, and HIF-1α (arrows) is clearly positive. H&E stain: original magnification 10×. VEGF, VEGFR-2, and HIF-1α: original magnification 40×. See Supplementary Materials and Methods for methodological details.
Figure 2.
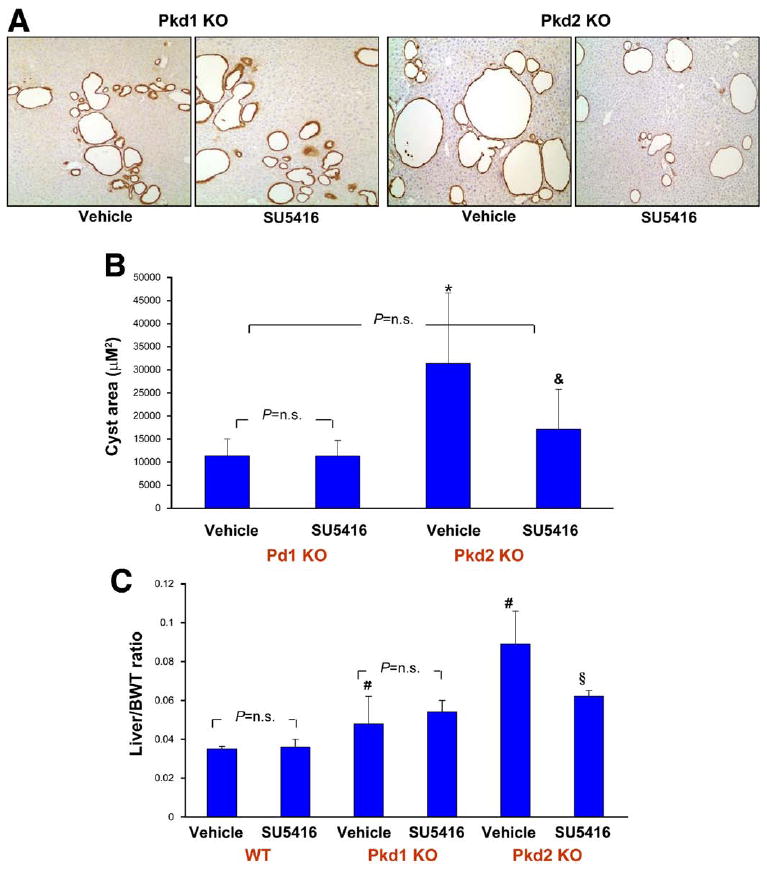
Inhibition of VEGFR-2 (SU5416) reduces cystic area in Pkd2KO mice. (A) Paraffin-embedded liver sections were labeled with a pancytokeratin antibody to discriminate between vessels and cystic structures. Micrographs are representative of vehicle- (left) and SU5416-treated mice (right). (B) Morphometric analysis was performed as described in Materials and Methods and Supplementary Materials and Methods. A significant reduction in cystic area was observed in Pkd2KO animals. (C) The decrease in cyst development is reflected also in the significant reduction in liver/body weight ratio. *P < .05 vs Pkd1KO vehicle; &P < .01 vs Pkd2KO vehicle; #P < .05 vs WT vehicle; §P < .05 vs Pkd2KO vehicle.
Figure 3.
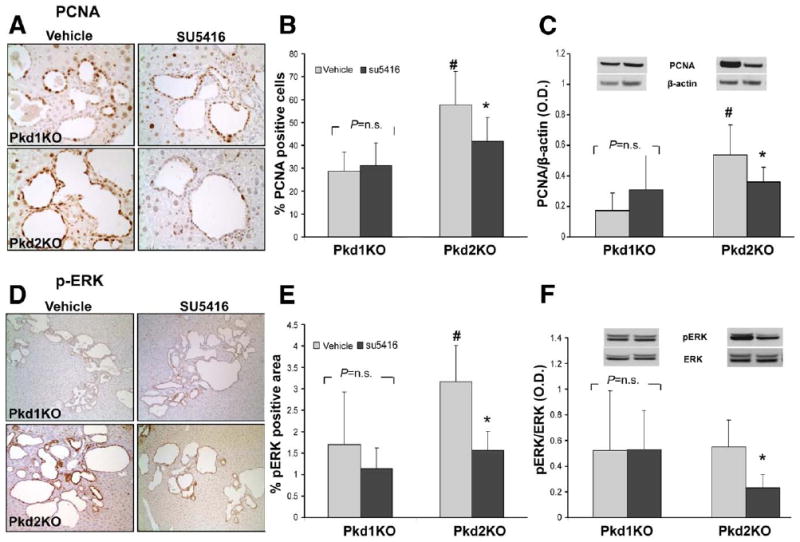
PCNA and pERK are reduced after SU5416 treatment in Pkd2KO mice. Cystic cholangiocytes showed strong proliferative activity (A, PCNA staining) and (D) pERK expression, particularly in Pkd2KO mice. As shown in the bar graphs, a significant reduction in (B) PCNA and (E) pERK expression, assessed by (B and E) morphometric analysis and by (C and F) Western blot, was observed in Pkd2KO-treated animals, while no significant changes were observed in Pkd1KO mice. #P < .05 vs Pkd1KO vehicle; *P < .05 vs Pkd2KO vehicle.
Effects of the VEGFR-2 Receptor Inhibitor SU5416 in Conditional Polycystin Knockout Mice
Effect on cyst growth
To understand the role of VEGF and VEGFR-2, we inhibited VEGFR-2 signaling in vivo and examined the growth of liver cysts. Mice were given SU5416 or vehicle (dimethyl sulfoxide) starting 1 week after tamoxifen induction. The dosing regimen was based on the median inhibitory concentration and the volume of distribution of SU5416 in mice.22,23 After 8 weeks, mice were killed and cystic area was measured. Preliminary studies showed no changes in the liver of WT mice that received the same doses of tamoxifen. Pkd1KO and Pkd2KO mice responded differently to inhibition of VEGFR-2 signaling. In Pkd2KO mice, liver cyst area was reduced by 56%; the liver/body weight ratio was also significantly reduced by 31% (Figure 2). SU5416 had no significant effect on cyst growth in Pkd1KO mice. Measurement of the amount of cytokeratin-positive structures confirmed these data (Pkd2KO control mice: 4.1% ± 1.1% vs 2.0% ± 0.9% in SU5416-treated mice, n = 6, P < .003; Pkd1KO control mice: 2.8% ± 0.2% [n = 4] vs 3.1% ± 0.4% [n = 5] in SU5416-treated mice, P = NS). SU5416 was well tolerated, without significant differences in liver and kidney function tests between treated and untreated mice and between Pkd1KO and Pkd2KO mice (Supplementary Table 1).
Effect on PCNA expression
In SU5416-treated Pkd2KO mice, the percentage of PCNA-positive cystic cholangiocytes was significantly reduced (57.74% ± 14.72% [n = 6] in untreated Pkd2KO mice vs 41.91% ± 10.33% [n = 6] in treated Pkd2KO mice; P < .05). The reduction of PCNA expression was confirmed by Western blot of liver homogenates from treated and control mice (Figure 3) (ratio of PCNA/β-actin: controls, 0.53 ± 0.19 [n = 6]; treated mice, 0.36 ± 0.09 [n = 6]; P < .05). No significant differences were found between treated and untreated Pkd1KO mice both by immunohistochemistry (Pkd1KO untreated mice, 28.92 ± 8.14 [n = 4]; treated mice, 31.21 ± 10.03 [n = 5]; P = .15) and by Western blot (ratio of PCNA/β-actin: controls, 0.17 ± 0.12 [n = 4]; treated mice, 0.31 ± 0.23 [n = 5]; P = .316). These data suggest that inhibition of VEGFR-2 receptor in vivo reduces the proliferative activity of the cystic epithelium in mice with defective PC2 but not PC1.
Effect on CC3 expression
Immunohistochemical expression of CC3 was used to investigate if VEGFR-2 inhibition stimulated apoptosis. CC3 positivity was higher in Pkd2KO compared with Pkd1KO mice (Pkd2KO mice, 0.49% ± 0.18% of the total lobe area [n = 6]; Pkd1KO mice, 0.15% ± 0.01% of the total lobe area [n = 4]; P < .003), consistent with higher cell proliferation and turnover. After SU5416 treatment, changes in CC3 positivity were not significant.
Effect on pERK1/2 expression
In SU5416-treated Pkd2KO mice, pERK1/2 expression in the cystic epithelium was significantly reduced (pERK-positive area was 3.17% ± 0.84% of the total lobe area vs 1.14% ± 0.45% in vehicle- vs SU5416-treated mice, respectively; n = 6; P < .05) (Figure 3). Western blots of protein extracts confirmed that the pERK/ERK ratio was significantly reduced in SU5416-treated Pdk2KO mice compared with vehicle-treated control mice (0.55 ± 0.2 vs 0.23 ± 0.1 in SU5416- vs vehicle-treated mice, respectively; n = 6; P < .05) (Figure 3).
Effect on pericystic CD34-positive vascular structures
Treatment with SU5416 significantly decreased the amount of pericystic CD34-positive structures in both mice (Pkd1KO: from 0.57% ± 0.27% to 0.19% ± 0.01%; Pkd2KO: from 1.17% ± 0.3% to 0.41% ± 0.2%) (Supplementary Figures 2 and 3).
Cultured Cystic Cholangiocytes From Pkd2KO Mice Secrete More VEGF and Overexpress VEGFR-2 and Phosphorylated VEGFR-2
We next studied the mechanisms leading to increased VEGF secretion and increased responsiveness to VEGF in vitro. Primary cultures were generated from the liver cysts of Pkd1KO (n = 6) and Pkd2KO mice (n = 8) and from bile ductules of WT mice (n = 5).12 Extensive characterization is presented in the Supplementary Materials and Methods and Supplementary Figure 4. Using this preparation, we studied the differences in VEGF production between Pkd1KO, Pkd2KO, and WT cholangiocytes. As shown in Figure 4A, the amount of VEGF secreted by Pkd2KO cells was significantly higher than that secreted by WT and Pkd1KO cells. Increased production of VEGF by cultured Pkd2KO cells shows that VEGF overexpression in vivo is not a consequence of tissue hypoxia. The expression of VEGFR-2 was studied by Western blot. We found (Figure 4B) that Pkd2KO cholangiocytes express significantly higher levels of VEGFR-2 and of its active phosphorylated form (pVEGFR-2 as compared with WT and Pkd1KO mice Figure 4C and D). In many cells, VEGFR-2 expression is positively regulated by the levels of VEGF. We found a significant increase in phosphorylated VEGFR-2 after administration of VEGF (25 ng/mL, 10 minutes) in Pkd2KO cholangiocytes (Figure 4C and D), confirming that the receptor is functionally active.
Figure 4.
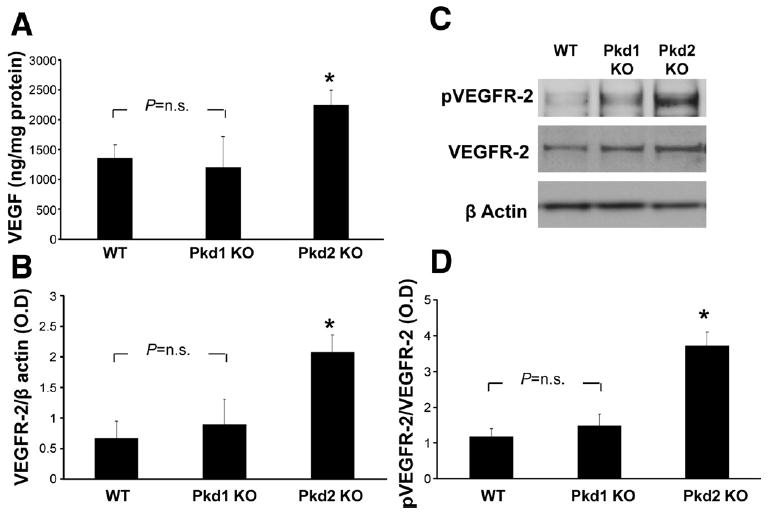
Pkd2KO cystic cholangiocytes significantly secrete more VEGF and express more VEGFR-2 in respect to Pkd1KO. (A) Cystic cholangiocytes isolated from Pkd2KO mice secreted a significantly higher amount of VEGF in respect to cells isolated from Pkd1KO and WT mice. (C) VEGFR-2 expression and (D) phosphorylation after treatment with VEGF (25 ng/mL, 10 minutes) are significantly higher in Pkd2KO compared with Pkd1KO mice (n = 4). (B) Representative example of Western blot. *P < .05 vs WT and Pkd1KO mice.
HIF-1α Is Overexpressed in Pkd2KO Cystic Cholangiocytes and Induces VEGF Secretion
HIF-1α, the main transcription factor regulating VEGF gene expression, is overexpressed in cyst cells (Figure 1C). We blocked HIF-1α degradation with DMOG (3 mmol/L), a selective prolyl 4-hydroxylases inhibitor, and then measured the nuclear accumulation of HIF-1α and VEGF secretion in the different cell lines (Supplementary Figure 5). Nuclear HIF-1α and VEGF secretion were significantly higher in Pkd2KO than in WT and Pkd1KO cells, both at baseline and in the presence of DMOG. DMOG-induced HIF-1α accumulation and VEGF release were inhibited by MEK inhibitor (U0126; 10 μmol/L), consistent with the key role of the MEK/ERK1/2 pathway. These results indicate that in Pkd2KO cells VEGF secretion is controlled by HIF-1α, the overexpression of which is mediated by ERK1/2.
ERK1/2 Phosphorylation and VEGF Secretion in Pkd2 Cholangiocytes Is Dependent on PKA
To better understand the mechanisms leading to ERK1/2 overactivation in Pkd2KO cells, we treated normal and cystic cholangiocytes with the PKA inhibitor PKI (1 μmol/L) and measured pERK expression and VEGF secretion. We found that PKI significantly inhibits pERK and VEGF secretion in PKI-treated cells (Figure 5). Basal phosphorylation of kinases upstream of ERK, such as pRaf and pMEK, were not significantly reduced by PKI (data not shown), suggesting that PKA directly stimulates ERK.
Figure 5.
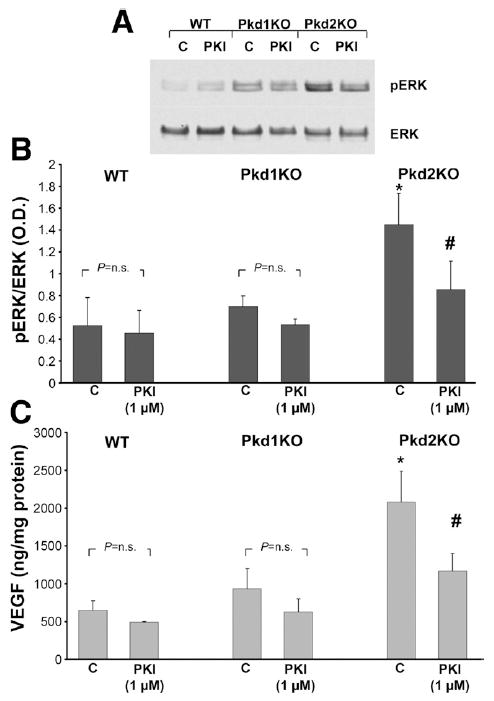
Overactivation of ERK and VEGF secretion in Pkd2 cholangiocytes is PKA dependent. Normal and cystic cholangiocytes were treated with the PKA inhibitor PKI (1 μmol/L), and pERK expression was assessed by Western blot while VEGF secretion was assessed by enzyme-linked immunosorbent assay. (A) Representative blot showing the reduction of pERK/ERK ratio in cells treated with PKI (1 μmol/L). (B) Data from 3 different experiments are shown in the bar graph. (C) In a similar manner, PKI inhibited VEGF secretion. *P < .05 vs WT and Pkd1KO controls; #P < .05 vs Pkd2KO controls.
VEGF Signaling and Cell Proliferation in Cystic Cholangiocytes Is Mediated by the MEK/ERK1/2 Pathway
Administration of VEGF (25 ng/mL) to cells isolated from Pkd1KO and Pkd2KO mice caused a time-dependent increase in ERK phosphorylation that was inhibited by treatment with the VEGFR-2 inhibitor SU5416 (5 μmol/L) (Figure 6). Both at basal and after stimulation with VEGF, the amount of pERK was significantly higher in Pkd2KO than in Pkd1KO and WT mice (Figure 6 and Supplementary Figure 6). Administration of VEGF (25 ng/mL) to cells isolated from WT and Pkd2KO mice induced cell proliferation. Proliferation was inhibited by the MEK inhibitor U0126 (10 μmol/L) and siRNA (Figure 7). These data indicate that VEGFR-2 signaling in cholangiocytes is mediated by MEK/ERK1/2.
Figure 6.
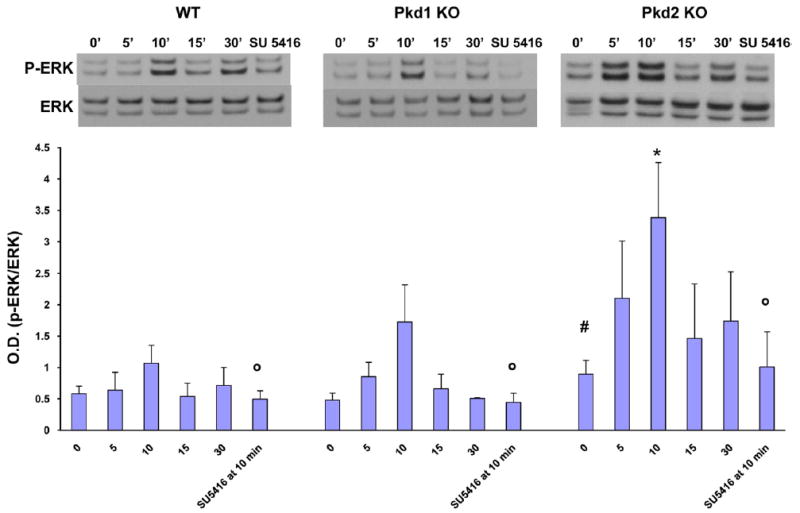
VEGF-induced ERK phosphorylation is significantly higher in Pkd2KO cystic cholangiocytes. Cells were treated with VEGF 25 ng/mL for 5-10-15-30 minutes or 10 minutes in presence of SU5416, a VEGFR-2 inhibitor, and then lysated for Western blot analysis. A clear time course in ERK phosphorylation with a significantly more pronounced effect in Pkd2 cystic cholangiocytes is shown. SU5416 (5 μmol/L) completely inhibited ERK phosphorylation in cells treated with VEGF for 10 minutes (n = 3). #P < .05 vs WT and Pkd1KO mice; *P < .05 vs WT and Pkd1KO 10 minutes; ◦P < .05 vs VEGF 10 minutes.
Figure 7.
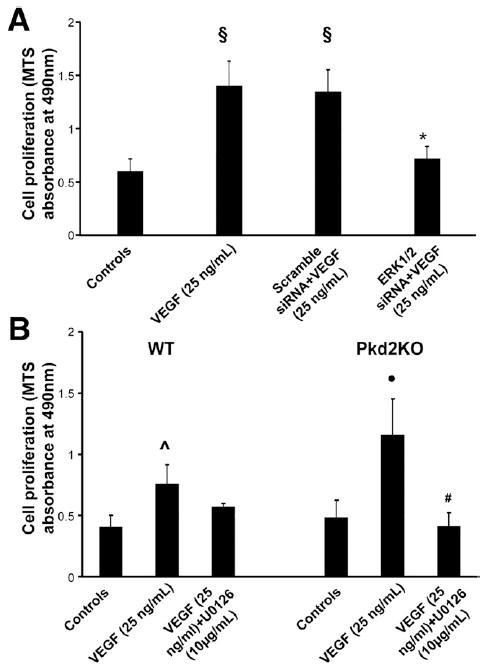
VEGF-induced cell proliferation is MEK/ERK mediated. VEGF significantly enhances cell proliferation in both WT cholangiocytes and Pkd2KO cystic cholangiocytes, but this effect is significantly greater in Pkd2KO cells. VEGF-induced cell proliferation was inhibited by (A) ERK1/2 siRNA or by (B) treatment with an MEK inhibitor (U1026, 10 μmol/L). §P < .01 vs controls; *P < .01 vs Scramble+VEGF; ●P < .01 vs controls; #P < .01 vs VEGF-treated cells; ˆP < .05 vs controls.
Discussion
Progressive enlargement of liver cysts is responsible for severe complications in polycystic liver disease.2 The mechanism of cyst enlargement in ADPKD is not well understood, but the consensus is that a major determinant is the excessive proliferation of the cystic epithelium.5
Our previous studies have indicated that estrogens, insulin-like growth factor-1, and VEGF are among the factors able to stimulate cholangiocyte proliferation that are overexpressed in the cystic epithelium of the liver in ADPKD.12,24 Also, VEGFR-2 is overexpressed in liver cysts,12,15 indicating that VEGF may stimulate the proliferation of the cystic epithelium through an autocrine mechanism. The main focus of our study was to understand the role and mechanisms of VEGF signaling in liver cyst growth. Our strategy was to study the effects of VEGF signaling inhibition in vivo and to analyze the mechanisms of VEGF/VEGFR-2 overexpression and signaling in vitro using cholangiocytes lacking in either PC1 or PC2. This approach also allowed us to differentially study the effects of PC1 and PC2 deficiency on cholangiocyte physiology. In fact, PC1 and PC2 appear to work in concert for certain functions25 but not for others.26
The 2 novel mouse models used in this study produced adult-onset liver cystic disease after conditional inactivation of either Pkd1 or Pkd2. These models have a very consistent phenotype and present a number of advantages: (1) the effect of homozygous polycystin gene inactivation can be studied, avoiding embryonic lethality; (2) genes can be inactivated at the desired time points, allowing a synchronization of experimental interventions; and (3) direct comparison of Pkd1 with Pkd2 can be performed. We found that both Pkd1KO and Pkd2KO mice expressed HIF-1α, VEGF, and VEGFR-2 in the liver, similarly to what we described earlier in human ADPKD.12 However, we have found interesting differences between Pkd1KO and Pkd2KO mice. The liver phenotype is more severe in Pkd2KO mice, which also shows higher pERK1/2 and PCNA expression in the cystic epithelium and increased expression of VEGF by isolated cystic cells. Consistent with increased VEGF expression, pericystic vascular structures, as detected by CD34 immunohistochemistry, were higher in Pkd2KO livers. Interestingly, the 2 models responded differently to inhibition of VEGFR-2 signaling in vivo.
A variety of agents able to inhibit VEGF or VEGF receptors have been developed.27-29 The reported inhibitory effect of rapamycin in ADPKD may also be due to inhibition of VEGF secretion.3 To study the role of VEGF signaling on cyst growth in vivo, we treated mice with SU5416 for 8 weeks and compared them with vehicle-treated mice. SU5416 is an inhibitor of the tyrosine kinase activity of VEGFR-2 and VEGFR-1.22,23,30 SU5416 was previously used in mice and shown to inhibit angiogenesis in vivo.2,15,22,23,31 Biochemical and clinical studies have shown that SU5416 has long-lasting inhibitory activity when administered twice a week.31
SU5416 was well tolerated, without histologic and biochemical signs of liver toxicity, and without clinical evidence of pulmonary and cardiovascular toxicity. The safety of long-term administration of SU5616 to rodents with ADPKD was first shown by Amura et al, who also reported preliminary evidence of cyst reduction in Pkd2(ws25/−)mice.15 In Pkd2KO mice, SU5416 significantly decreased the area of the liver occupied by cysts, the amount of pancytokeratin-positive structures, the liver/body weight ratio, and the expression of pERK and PCNA. This 56% reduction in cyst area is particularly significant considering that VEGF is likely not the only factor involved in cyst growth (estrogens, insulin-like growth factor 1, and epidermal growth factor, among others, also play a role15). However, we did not find a significant effect of SU5416 in Pkd1KO mice, even though the reduction in pericystic vascular structures confirmed that SU5416 was active in both Pkd1KO and Pkd2KO mice.
To better understand these mechanisms, we cultured and characterized cystic cholangiocytes from Pkd1KO and Pkd2KO mice (Supplementary Figure 4). Using this preparation, we confirmed our12 and others’32 earlier observation that cystic cholangiocytes produce more VEGF and express more VEGFR-2 than normal cholangiocytes. Increased secretion of VEGF by cultured cells indicates that VEGF expression in ADPKD livers is not an effect of tissue ischemia. VEGF production, VEGFR-2 expression, ERK1/2 phosphorylation, and cell proliferation before and after administration of VEGF, as well as HIF-1α and VEGF production after DMOG, were all higher in cultured cholangiocytes from Pkd2KO mice rather than from Pkd1KO mice. These findings suggest that distinct mechanisms regulate cyst growth in Pkd1KO and Pkd2KO mice and that Pkd2 deficiency up-regulates pathways involved in the modulation of VEGF signaling that are normally inhibited in the presence of Pkd2.
In most tissues, VEGF production is controlled by the transcription factor HIF-1. In hypoxic conditions, HIF-1α degradation is blocked and HIF-1α translocates into the nucleus, where it binds on the HRE (hypoxia responsive element) present on the VEGF promoter.33 In normoxic conditions, HIF-1α transcription can be stimulated by a number of growth factors, cytokines, and extracellular mediators (interleukin-1, interleukin-6, epidermal growth factor, hepatocyte growth factor, 17β-estradiol, insulin-like growth factor 1). These mediators can stabilize or phosphorylate HIF-1α via (1) phosphatidylinositol 3-kinase/AKT/tuberin/mTOR or Raf/MEK/ERK or (2) STAT3 of mitogen-activated protein kinases. 32-34 We found that Pkd2 cystic cholangiocytes express more HIF-1α and that, in the presence of DMOG, HIF-1α expression and VEGF secretion were inhibited by treatment with the MEK inhibitor. In these conditions, the production of VEGF consistently followed the changes in HIF-1α expression. These results indicate that, in Pkd2KO mice, VEGF expression is controlled by the MEK–ERK1/2–HIF-1α pathway.
The mechanisms linking polycystin dysfunction to overexpression of VEGF are not known. Baseline expression of ERK1/2 was significantly higher in Pkd2KO mice than in WT and Pkd1KO mice. Previous studies in renal epithelial cells have shown that overexpression of WT PC2 inhibits pERK and cell proliferation.9 We hypothesize that up-regulation of ERK1/2 in cholangiocytes lacking PC2 function may result from Pkd2-dependent changes in cellular Ca2+ homeostasis. In cystic epithelial cells of the kidney, a reduction in intracellular Ca2+ decreases the Ca2+-mediated inhibition of adenyl cyclase 6 expressed in cilia, thereby increasing cellular adenosine 3′,5′-cyclic monophosphate (cAMP) levels.35 cAMP can activate ERK, either by direct PKA-mediated phosphorylation or by PKA-independent, Epac1/2-mediated phosphorylation of Raf, as shown in PCK rats, a genetically distinct polycystic liver model.36 As shown in Figure 5, we found that administration of PKI (a PKA inhibitor) to Pkd2KO cells reduced both ERK and phosphorylation and VEGF secretion, suggesting a functional connection between Pkd2 defect, increased PKA, ERK phosphorylation, and VEGF secretion (Figure 8). In contrast to ERK1/2, baseline phosphorylation of B-Raf and MEK was not up-regulated and did not decrease significantly after PKI administration, suggesting that PKA directly phosphorylates ERK.
Figure 8.
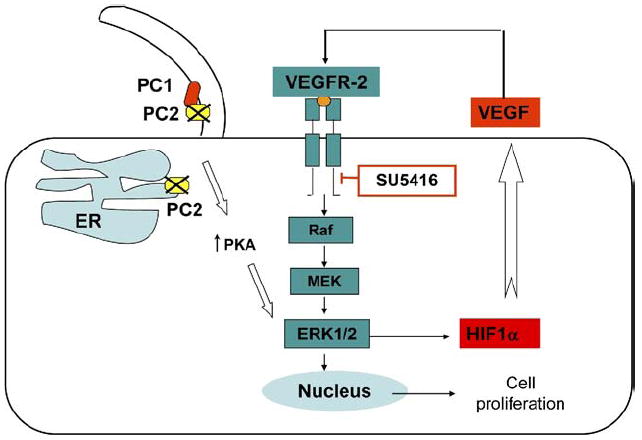
ERK-mediated VEGF secretion and cell proliferation in Pkd2KO cystic cholangiocytes: working model. Defects in PC2 function may influence VEGF signaling via PKA-mediated stimulation of ERK1/2. As a consequence, VEGF production is enhanced by HIF-1α induction and, through an autocrine effect on cystic cholangiocytes, stimulates cell proliferation.
PC2, in contrast to PC1, is expressed in the endoplasmic reticulum, where it participates in endoplasmic reticulum calcium regulation.37,38 A recent study suggests that a decrease in endoplasmic reticulum calcium concentration may directly activate cAMP production independently from cytoplasmic Ca2+, a mechanism called store-operated cAMP signaling.39 The role of store-operated cAMP signaling in ERK/HIF-1α/VEGF signaling is currently being addressed.
To understand if ERK is also involved in the downstream signaling of VEGFR-2 in cystic cholangiocytes, we studied the effects of VEGF administration on cystic cholangiocytes. Administration of VEGF increased phospho-VEGFR-2 levels, indicating that the receptor is functionally active in cholangiocytes. VEGF increased ERK1/2 phosphorylation in a SU5416-inhibitable manner. VEGF administration also stimulated proliferation of Pkd2KO cholangiocytes, an effect that was inhibited by the MEK inhibitor U1026 and by silencing ERK1/2 expression. All together, these data establish that in Pkd2KO mice the MEK/ERK1/2 pathway also mediates VEGF autocrine effects on the cystic epithelium.
It is interesting to note that mice deficient in PC2 and PC1 differ in terms of VEGF production/response, yet both mice have cystic disease. This suggests that lack of PC1 and PC2 causes liver cystic disease through a common mechanism that is VEGF independent. The MEK/ERK activation and VEGF signaling caused by PC2 deficiency represents an additive PC1-independent proliferative signal that results in incremental cyst growth in PC2 deficiency. VEGF inhibition eliminates this latter effect, reducing cyst growth rates to the same levels of PC1 deficiency. As shown in Figure 2, VEGFR-2 inhibition suppresses cyst growth to the same level as that in the Pkd1KO mice. This hypothesis explains both the higher rate of bile duct cyst growth in PC2 and the unique responsiveness of PC2 cysts to the SU5416 compound. It also raises the novel concept that both tissue-specific and gene-specific factors can impact disease progression in ADPKD/polycystic liver disease.
The implications of these studies extend beyond polycystic liver disease. Cholangiocytes react to many forms of liver damage40 and acquire a phenotype characterized by the expression of multiple cytokines and growth factors. 40 VEGF is expressed by reactive cholangiocytes in a number of acute and chronic liver diseases and cholangiopathies, 32,41 suggesting that epithelial expression of angiogenic factors is important in liver repair mechanisms. One may speculate that, under physiologic conditions, PC2 expression by negatively regulating the MEK/ERK1/2 pathway inhibits VEGF secretion and signaling as well as cholangiocyte proliferation.10,15,32,42,43
In summary, we have shown that up-regulation of VEGF signaling promotes liver cyst growth in a mice model of ADPKD. Furthermore, our findings shed light on substantial differences in the pathophysiologic consequences of Pkd1 and Pkd2 deficiencies. The cystic epithelium of PC-2 mice is characterized by an increased activity of the ERK pathway,44 which actually mediates both the increased secretion of VEGF and the increased response to VEGF. These studies improve our understanding of the mechanism leading to the progressive growth of cysts in ADPKD, provide proof of concept for the potential use of antiangiogenic therapy in ADPKD, and improve our understanding of the mechanisms of regulation of epithelial VEGF/VEGFR-2 during liver repair.
Supplementary Material
Acknowledgments
The authors thank Corinne Lobe (University of Toronto, Toronto, Ontario, Canada) for the pCX-CreER mice.
Funding Supported by National Institutes of Health (NIH) grant DK079005, PKD Foundation (to M.S.), Yale University Liver Center (NIH grant K34989 to M.S. and C.S.), and NIH grants DK51041 and DK54053 (to S.S.). C.S. is a recipient of an American Liver Foundation/American Association for the Study of Liver Diseases Liver Scholar Award. The support of Fondazione S. Martino (Bergamo, Italy) is gratefully acknowledged.
Abbreviations used in this paper
- ADPKD
autosomal dominant polycystic kidney disease
- CC3
cleaved caspase-3
- DMOG
2-oxoglutarate analogues dimethyloxaloylglycine
- ERK
extracellular signal–regulated kinase
- HIF
hypoxia-inducible factor
- MEK
mitogen signal-regulated kinase
- PC1
polycystin-1
- PC2
polycystin-2
- PCNA
proliferating cell nuclear antigen
- pERK
phosphorylated extracellular signal–regulated kinase
- PKA
protein kinase A
- Pkd1KO
Pkd1flox/−:pCxCreER
- Pkd2KO
Pkd2flox/−:pCxCreER
- PKI
protein kinase A inhibitor 14–22 amide
- siRNA
small interfering RNA
- VEGF
vascular endothelial growth factor A
- VEGFR
vascular endothelial growth factor A receptor
- WT
wild-type
Footnotes
Supplementary Data Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2009.09.005.
Conflicts of interest The authors disclose no conflicts.
References
- 1.Bae KT, Zhu F, Chapman AB, et al. Magnetic resonance imaging evaluation of hepatic cysts in early autosomal-dominant polycystic kidney disease: the Consortium for Radiologic Imaging Studies of Polycystic Kidney Disease cohort. Clin J Am Soc Nephrol. 2006;1:64–69. doi: 10.2215/CJN.00080605. [DOI] [PubMed] [Google Scholar]
- 2.Everson GT, Taylor MR, Doctor RB. Polycystic disease of the liver. Hepatology. 2004;40:774–782. doi: 10.1002/hep.20431. [DOI] [PubMed] [Google Scholar]
- 3.Qian Q, Du H, King BF, et al. Sirolimus reduces polycystic liver volume in ADPKD patients. J Am Soc Nephrol. 2008;19:631–638. doi: 10.1681/ASN.2007050626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Masyuk TV, Masyuk AI, Torres VE, et al. Octreotide inhibits hepatic cystogenesis in a rodent model of polycystic liver disease by reducing cholangiocyte adenosine 3′,5′-cyclic monophosphate. Gastroenterology. 2007;132:1104–1116. doi: 10.1053/j.gastro.2006.12.039. [DOI] [PubMed] [Google Scholar]
- 5.Torres VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2:40–55. doi: 10.1038/ncpneph0070. [DOI] [PubMed] [Google Scholar]
- 6.Qian F, Noben-Trauth K. Cellular and molecular function of mucolipins (TRPML) and polycystin 2 (TRPP2) Pflugers Arch. 2005;451:277–285. doi: 10.1007/s00424-005-1469-4. [DOI] [PubMed] [Google Scholar]
- 7.Aguiari G, Campanella M, Manzati E, et al. Expression of polycystin-1 C-terminal fragment enhances the ATP-induced Ca2+ release in human kidney cells. Biochem Biophys Res Commun. 2003;301:657–664. doi: 10.1016/s0006-291x(02)03011-5. [DOI] [PubMed] [Google Scholar]
- 8.Anyatonwu GI, Estrada M, Tian X, et al. Regulation of ryanodine receptor-dependent calcium signaling by polycystin-2. Proc Natl Acad Sci U S A. 2007;104:6454–6459. doi: 10.1073/pnas.0610324104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Wright JM, Qian F, et al. Polycystin 2 interacts with type I inositol 1,4,5-trisphosphate receptor to modulate intracellular Ca2+ signaling. J Biol Chem. 2005;280:41298–41306. doi: 10.1074/jbc.M510082200. [DOI] [PubMed] [Google Scholar]
- 10.Strazzabosco M, Fabris L, Spirli C. Pathophysiology of cholangiopathies. J Clin Gastroenterol. 2005;39:S90–S102. doi: 10.1097/01.mcg.0000155549.29643.ad. [DOI] [PubMed] [Google Scholar]
- 11.Nichols MT, Gidey E, Matzakos T, et al. Secretion of cytokines and growth factors into autosomal dominant polycystic kidney disease liver cyst fluid. Hepatology. 2004;40:836–846. doi: 10.1002/hep.20401. [DOI] [PubMed] [Google Scholar]
- 12.Fabris L, Cadamuro M, Fiorotto R, et al. Effects of angiogenic factor overexpression by human and rodent cholangiocytes in polycystic liver diseases. Hepatology. 2006;43:1001–1012. doi: 10.1002/hep.21143. [DOI] [PubMed] [Google Scholar]
- 13.Shibuya M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J Biochem Mol Biol. 2006;39:469–478. doi: 10.5483/bmbrep.2006.39.5.469. [DOI] [PubMed] [Google Scholar]
- 14.Fabris L, Cadamuro M, Libbrecht L, et al. Epithelial expression of angiogenic growth factors modulate arterial vasculogenesis in human liver development. Hepatology. 2008;47:719–728. doi: 10.1002/hep.22015. [DOI] [PubMed] [Google Scholar]
- 15.Amura C, Brodsky KS, Groff R, et al. VEGF receptor inhibition blocks liver cyst growth in pkd2(WS25/−) mice. Am J Physiol Cell Physiol. 2007;293:C419–C428. doi: 10.1152/ajpcell.00038.2007. [DOI] [PubMed] [Google Scholar]
- 16.Shibazaki S, Yu Z, Nishio S, et al. Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum Mol Genet. 2008;17:1505–1516. doi: 10.1093/hmg/ddn039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo C, Yang W, Lobe CG. A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis. 2002;32:8–18. doi: 10.1002/gene.10021. [DOI] [PubMed] [Google Scholar]
- 18.Torres VE, Wang X, Qian Q, et al. Effective treatment of an orthologous model of autosomal dominant polycystic kidney disease. Nat Med. 2004;10:363–364. doi: 10.1038/nm1004. [DOI] [PubMed] [Google Scholar]
- 19.Jebreel A, England J, Bedford K, et al. Vascular endothelial growth factor (VEGF), VEGF receptors expression and microvascular density in benign and malignant thyroid diseases. Int J Exp Pathol. 2007;88:271–277. doi: 10.1111/j.1365-2613.2007.00533.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spirli C, Fiorotto R, Song L, et al. Glibenclamide stimulates fluid secretion in rodent cholangiocytes through a cystic fibrosis transmembrane conductance regulator-independent mechanism. Gastroenterology. 2005;129:220–233. doi: 10.1053/j.gastro.2005.03.048. [DOI] [PubMed] [Google Scholar]
- 21.Masyuk AI, Masyuk TV, Splinter PL, et al. Cholangiocyte cilia detect changes in luminal fluid flow and transmit them into intracellular Ca2+ and cAMP signaling. Gastroenterology. 2006;131:911–920. doi: 10.1053/j.gastro.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ye C, Sweeny D, Sukbuntherng J, et al. Distribution, metabolism, and excretion of the anti-angiogenic compound SU5416. Toxicol In Vitro. 2006;20:154–162. doi: 10.1016/j.tiv.2005.06.047. [DOI] [PubMed] [Google Scholar]
- 23.Sukbuntherng J, Cropp G, Hannah A, et al. Pharmacokinetics and interspecies scaling of a novel VEGF receptor inhibitor, SU5416. J Pharm Pharmacol. 2001;53:1629–1636. doi: 10.1211/0022357011778232. [DOI] [PubMed] [Google Scholar]
- 24.Alvaro D, Onori P, Alpini G, et al. Morphological and functional features of hepatic cyst epithelium in autosomal dominant polycystic kidney disease. Am J Pathol. 2008;172:321–332. doi: 10.2353/ajpath.2008.070293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barr MM. Caenorhabditis elegans as a model to study renal development and disease: sexy cilia. J Am Soc Nephrol. 2005;16:305–312. doi: 10.1681/ASN.2004080645. [DOI] [PubMed] [Google Scholar]
- 26.Pennekamp P, Karcher C, Fischer A, et al. The ion channel polycystin-2 is required for left-right axis determination in mice. Curr Biol. 2002;12:938–943. doi: 10.1016/s0960-9822(02)00869-2. [DOI] [PubMed] [Google Scholar]
- 27.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of antitumour activity. Nat Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 28.Lee CB, Socinski MA. Vascular endothelial growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer: a review of recent clinical trials. Rev Recent Clin Trials. 2007;2:117–120. doi: 10.2174/157488707780599401. [DOI] [PubMed] [Google Scholar]
- 29.Veeravagu A, Hsu AR, Cai W, et al. Vascular endothelial growth factor and vascular endothelial growth factor receptor inhibitors as anti-angiogenic agents in cancer therapy. Recent Patents Anticancer Drug Discov. 2007;2:59–71. doi: 10.2174/157489207779561426. [DOI] [PubMed] [Google Scholar]
- 30.Itokawa T, Nokihara H, Nishioka Y, et al. Antiangiogenic effect by SU5416 is partly attributable to inhibition of Flt-1 receptor signaling. Mol Cancer Ther. 2002;1:295–302. [PubMed] [Google Scholar]
- 31.Mendel DB, Schreck RE, West DC, et al. The angiogenesis inhibitor SU5416 has long-lasting effects on vascular endothelial growth factor receptor phosphorylation and function. Clin Cancer Res. 2000;6:4848–4858. [PubMed] [Google Scholar]
- 32.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–1282. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 33.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 34.Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1) Mol Pharmacol. 2006;70:1469–1480. doi: 10.1124/mol.106.027029. [DOI] [PubMed] [Google Scholar]
- 35.Belibi FA, Reif G, Wallace DP, et al. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004;66:964–973. doi: 10.1111/j.1523-1755.2004.00843.x. [DOI] [PubMed] [Google Scholar]
- 36.Banales JM, Masyuk TV, Gradilone SA, et al. The cAMP effectors Epac and protein kinase A (PKA) are involved in the hepatic cystogenesis of an animal model of autosomal recessive polycystic kidney disease (ARPKD) Hepatology. 2009;49:160–174. doi: 10.1002/hep.22636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anyatonwu GI, Ehrlich BE. Calcium signaling and polycystin-2. Biochem Biophys Res Commun. 2004;322:1364–1373. doi: 10.1016/j.bbrc.2004.08.043. [DOI] [PubMed] [Google Scholar]
- 38.Geng L, Boehmerle W, Maeda Y, et al. Syntaxin 5 regulates the endoplasmic reticulum channel-release properties of polycystin-2. Proc Natl Acad Sci U S A. 2008;105:15920–15925. doi: 10.1073/pnas.0805062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lefkimmiatis K, Srikanthan M, Maiellaro I, et al. Store-operated cyclic AMP signalling mediated by STIM1. Nat Cell Biol. 2009;11:433–442. doi: 10.1038/ncb1850. [DOI] [PubMed] [Google Scholar]
- 40.Lazaridis KN, Strazzabosco M, Larusso NF. The cholangiopathies: disorders of biliary epithelia. Gastroenterology. 2004;127:1565–1577. doi: 10.1053/j.gastro.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 41.Strazzabosco M, Fabris L. Functional anatomy of normal bile ducts. Anat Rec (Hoboken) 2008;291:653–660. doi: 10.1002/ar.20664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.LeSage G, Glaser S, Alpini G. Regulation of cholangiocyte proliferation. Liver. 2001;21:73–80. doi: 10.1034/j.1600-0676.2001.021002073.x. [DOI] [PubMed] [Google Scholar]
- 43.Alvaro D, Mancino MG, Glaser S, et al. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology. 2007;132:415–431. doi: 10.1053/j.gastro.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 44.Grimm DH, Karihaloo A, Cai Y, et al. Polycystin-2 regulates proliferation and branching morphogenesis in kidney epithelial cells. J Biol Chem. 2006;281:137–144. doi: 10.1074/jbc.M507845200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.