

Abstract
Two distinct families of small molecules were discovered as novel α7 nicotinic acetylcholine receptor (nAChR) antagonists by pharmacophore-based virtual screening. These novel antagonists exhibited selectivity for the neuronal α7 subtype over other nAChRs and good brain penetration. Neuroprotection was demonstrated by representative compounds 7i and 8 in a mouse seizure-like behavior model induced by the nerve agent diisopropylfluorophosphate (DFP). These novel nAChR antagonists have potential use as antidote for organophosphorus nerve agent intoxication.
Keywords: Nicotinic acetylcholine receptors, α7 antagonists, Pharmacophore, Neuroprotection
Nicotinic acetylcholine receptors (nAChRs) belong to the Cys-loop subfamily of pentameric ligand-gated ion channels and can be classified into muscle-type and neuronal subtypes. The neuronal nAChRs comprise twelve subunits (α2–α10 and β2–β4) with different arrangements, while the muscle-type is consisted of four subunits in a single arrangement of α1γα1β1δ (γ is replaced by ε in the adult).1 Two major neuronal receptors α4β2 and α7 have been identified in the central nervous system.2,3 The neuronal α7 nAChR has been proposed as a potential therapeutic target for a broad range of neurodegenerative and psychiatric diseases, including Alzheimer's disease, schizophrenia, anxiety, and epilepsy.4–8 A variety of selective partial and full agonists have been developed for the α7 nAChR as potential therapeutics.4 Several α7 nAChR selective agonists (e.g., TC-5619 and MEM-3454) have advanced to clinical trials for Alzheimer's disease and schizophrenia.9–11
Although extensive efforts have been taken to identify selective α7 nAChR agonists, the development of α7 selective antagonists is relatively limited. Naturally derived compounds have been reported as α7 selective antagonists. For example, the krait Bungarus multicinctus derived peptide toxin α-bungarotoxin (α-BTX) and the seeds of Delphinum isolated nonpetide toxin methyllycaconitine (MLA) are two frequently used α7 selective antagonists.12,13 Unfortunately, α-BTX is a potent antagonist for muscle-type nAChRs as well, and both compounds also inhibit nAChR subtypes α9 and α9α10.4,14 Nevertheless, subtype-selective antagonists are valuable tools to define the roles played by α7 nAChRs in the physiological and pathophysiological processes. Moreover, α7 nAChR selective antagonists recently have been explored as potential treatments for non-small cell lung cancer,15 and for organophosphorus nerve agent intoxication.16 Exposure to nerve agents, as part of military or terrorist activities, or due to chronic low-level exposure to pesticides, has devastating health consequences. Clearly, there is a need for better α7 antagonists to probe the receptor's pharmacological functions in the brain and potential clinical uses.
In our effort to search for novel α7 nAChR selective ligands, we developed three dimensional (3D) pharmacophore models and conducted ligand-based virtual screening. Six potent and selective α7 ligands were selected as the training set for the pharmacophore model (Fig. 1).17–20 The structures of all compounds were built with Molecular Operating Environment (MOE).21 Energy minimization was conducted for the protonated forms using AM1 since all these compounds could be protonated at physiological conditions (pH 7.4). Flexible structural alignments were performed to identify the common chemical features responsible for the α7 receptor binding using the Flexible Alignment module within MOE. This alignment method uses a stochastic search algorithm to simultaneously explore the conformation space of all compounds in the training set. This operation generated several scores to quantify the quality of each alignment with lower scores indicating better alignments. In our study, the alignment with the lowest S score was selected for the 3D pharmacophore development. A six-point pharmacophore model was obtained based on the unified scheme within MOE (Fig. 2). Feature F1 is a hydrogen bond acceptor with radius 1.5 Å. Feature F2 is a cation atom (radius: 1.0 Å)—the basic nitrogen, which exists in most known nAChR ligands. Features F3 and F4 are characterized as aromatic rings with radius 1.5 Å. Features F5 and F6 cover hydrophobic regions (radius: 1.0 Å).
Figure 1.

α7 selective ligands used for the pharmacophore development.
Figure 2.
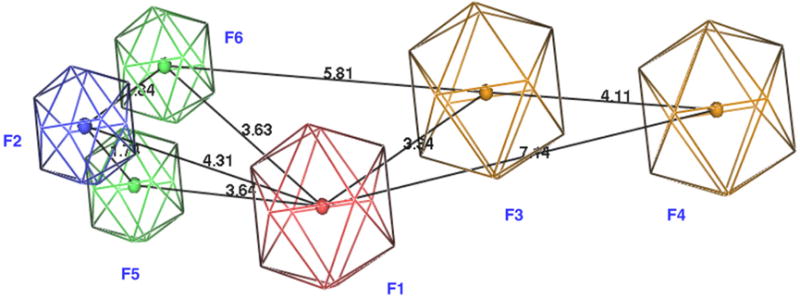
Pharmacophore model for α7 nAChR selective ligands. Blue: cation; red: hydrogen bond acceptors; green: hydrophobic groups; brown: aromatic groups. Six figures are labeled with F1–F6. Distances between features are indicated in Å.
The pharmacophore model was used to screen compounds assembled from different sources. In order to remove unwanted structures and accelerate the process of virtual screening, extended Lipinski's rules22 and three functional group filters were applied before the pharmacophore-based database searching. The work-flow is shown in Fig. 3. Extended Lipinski's rules include seven filters: 100 < molecular weight ≤ 500, −2 ≤ Clog P ≤ 5, number of hydrogen bond donors ≤5, number of hydrogen bond acceptors ≤10, topological polar surface area ≤120 Å2, number of rings ≤5, and number of rotatable bonds ≤10. These property filters were chosen to eliminate compounds that lacked sufficient drug-like properties to become drugs. Compounds that passed the above criteria were subjected to three functional group filters: absence of reactive groups, number of non-fluorine halogen atoms ≤4, and number of basic nitrogen atoms ≤1. Reactive groups are defined according to the Oprea set, including heteroatom–heteroatom single bonds, acyl halides, sulfonyl halides, perhalo ketone, and Michael acceptors, as identified previously.23 These groups can interfere with high-throughput biochemical screening assays and therefore often appear as false positives. The halogen filter was chosen to avoid pesticides that often contain many (>4) non-fluorine halogen atoms.24 The majority of known nAChR ligands contain a nitrogen atom protonated at physiological conditions (pH 7.4), and this nitrogen atom has been shown to be involved in extensive cation–π interactions between ligand and receptor.25,26 A basic nitrogen filter was selected to remove compounds that lack this chemical feature. This filter greatly reduced the size of the compound database and therefore improved the speed of conformer generation and pharmacophore matching. Altogether, compounds violating ≥2 Lipinski's rules or any functional group filters were eliminated from our selection. The resulting compounds were subjected to conformation sampling using the Conformational Import Module, a high-throughput method to generate 3D low-energy conformers in MOE. Recent studies revealed that this method performed as well as the established Catalyst FAST module.27 The ensemble of conformers were then screened by the six-point pharmacophore model by enabling exact match of features F1–F4 and partial match of features F5 and F6.
Figure 3.

Workflow of ligand-based virtual screening.
The consequent hits were subjected to database diversity and clustering analyses with the aim to remove close analogs and maximize the chemotypes of the selected compounds for biological tests. The MDL MACCS fingerprints implemented in MOE were calculated for all compounds and fingerprint-based clustering was carried out by using the Tanimoto coefficient (0.85) as a measure of fingerprint similarity. No more than three representative compounds in the same cluster were selected for the final collection. About 300 compounds were acquired from different commercial sources for in vitro biological screening, including compounds from Maybridge, Chembridge, Enamine. The binding affinity of these compounds for the native α7 nAChR in rat brain membranes was measured using previously reported methods with [125I]α-BTX as the radioligand.28
From the preliminary screening, various chemotypes were found to exhibit ≥50% inhibition on the α7 nAChR in this assay. Two of them (compound 7 and 8, Tables 1 and 2) exhibited low micromolar inhibition on brain α7 nAChR with an IC50 of 1.6 μM and 2.9 μM, respectively (Fig. 4). The structures of 7 and 8 were confirmed by UPLC–HRMS and found to have purities of 98% and 97%. Several analogs of 7 and 8 with different substitutions at positions of R1, R2, R3, and R4 were identified by substructure searches or chemical synthesis (Tables 1 and 2). Structure–activity relationships (SARs) were developed for both compounds using the binding assay. For example, substitution (e.g., methyl and chlorine) at R3 is tolerated for analogs of compound 7. Both benzyl (7i) and isobutyl (7k) at R1 were more favorable than the more flexible phenethyl (7d). For analogs of compound 8, a bulky benzyl group at R1 yielded more potent receptor binding than a less bulky methyl or free base (8 vs 8h and 8i). More hydrophobic substitutions at R3 improved receptor binding (8f vs 8h). The R4 position tolerated different substitutions (e.g., hydrogen and halogen).
Table 1. Neuronal α7 nAChR inhibition of compound 7 and its analogs.
 | ||||
|---|---|---|---|---|
| ID | R1 | R2 | R3 | Percent inhibition at 10 μMa (mean ± SD) |
| 7 | Benzyl | H | 3-Cl | 83.0 ± 1.7 |
| 7a | Benzyl | H | 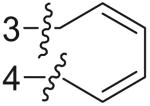 |
70.5 ± 3.1 |
| 7b | Phenethyl | H | 4-OMe | 9.5 ± 3.7 |
| 7c | 2-Methoxyethyl | H | 4-Cl | 27.3 ± 0.7 |
| 7d | Phenethyl | H | 2-Cl | 23.4 ± 2.4 |
| 7e | Isobutyl | Methyl | 2,5-F,F | 59.2 ± 0.2 |
| 7f | Phenethyl | H | 4-Cl | 18.3 ± 1.3 |
| 7g | Cyclopentyl | H | 2-Cl | 21.6 ± 2.6 |
| 7h | 2-Methoxyethyl | Methyl | 2-Cl | 43.0 ± 0.0 |
| 7i | Benzyl | Methyl | 2-Cl | 79.7 ± 1.9 |
| 7j | Phenethyl | H | 2-Me | 25.2 ± 1.8 |
| 7k | Isobutyl | Methyl | 2-Cl | 72.3 ± 1.0 |
The binding affinity of these compounds for the native α7 nAChR in rat brain membranes was measured at 10 μM using [125I]α-BTX as the radioligand.28 Nonspecific binding was determined with 1 μM MLA.
Table 2. Neuronal α7 nAChR inhibition of compound 8 and its analogs.
 | |||||
|---|---|---|---|---|---|
| ID | R1 | R2 | R3 | R4 | Percent inhibition at 10 μMa (Mean ± SD) |
| 8 | Benzyl | H | Propyl | H | 83.8 ± 2.3 |
| 8a | Benzyl | H | Methyl | 4-F | 50.0 ± 2.0 |
| 8b | Benzyl | H | Methyl | 4-Cl | 64.1 ± 1.8 |
| 8c | Methyl | Methyl | Methyl | 4-F | 11.9 ± 2.3 |
| 8d | Methyl | Methyl | Methyl | 4-Cl | 13.5 ± 5.3 |
| 8e | Methyl | Methyl | Cyclopropyl | 2-Furyl | 10.5 ± 1.7 |
| 8f | Methyl | H | 4-Tolyl | H | 23.4 ± 0.8 |
| 8g | Methyl | Methyl | 4-Tolyl | H | 12.1 ± 1.3 |
| 8h | Methyl | H | Propyl | H | 3.1 ± 0.1 |
| 8i | H | H | Propyl | H | 1.0 ± 2.8 |
Same as in Table 1.
Figure 4.

Dose–response curves of compounds 7 and 8 on the native neuronal α7 nAChR in rat brain membranes. The error bars indicate the standard deviation of the measurements. The binding assay was performed according to the previously reported method using [125I]α-BTX as the radioligand.28 IC50 of 7 = 1.6 μM; IC50 of 8 = 2.9 μM.
The functional activity of representative compounds (7, 7i, and 8) was determined by electrophysiological experiments on Xenopus oocytes expressing human α7 nAChR.29 These three compounds were found to inhibit acetylcholine-evoked receptor responses in a dose-dependent manner, suggesting the subject compounds are α7 nAChR antagonists (Fig. 5). The IC50 values of 7, 7i, and 8 are 11.9 μM, 3.7 μM, 18.9 μM, respectively. The α7 functional potency was 6–8-fold lower than the affinity estimated from human α7 receptor binding, and this difference may come from interspecies variation (rat vs human), or variability in receptors (native vs recombinant), or due to technical aspects of the functional assay in the oocyte expression system.30
Figure 5.
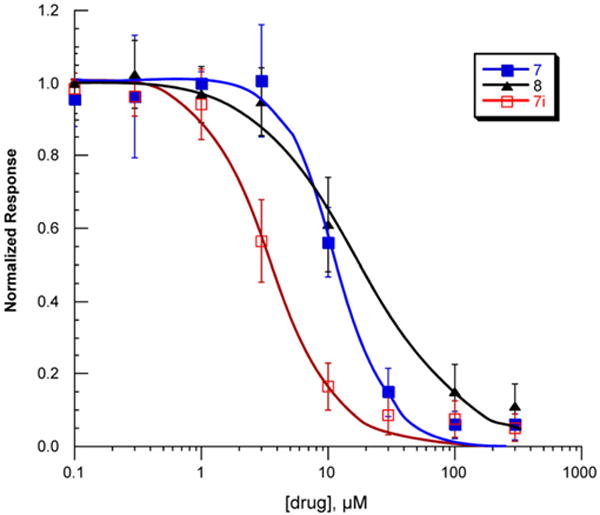
Inhibition of the human α7 AChR responses expressed in Xenopus oocytes. The error bars indicate the standard deviation of the measurements. Data shown are the averaged normalized mean of net charge responses to co-application of ACh and compounds 7, 7i, and 8 from oocytes expressing human α7 subunits. IC50 values of 7, 7i, and 8 are 11.9 μM, 3.8 μM, and 18.9 μM, respectively.
Although all training set compounds act as partial or full agonists for the α7 receptor, the resulting hits from pharmacophore-based virtual screening were shown to be α7 antagonists. Structural overlap of training compounds (e.g., 2) with hit compounds (7 and 8) revealed that all training compounds lack bulky substitutions (e.g., benzyl) at R1 (Fig. 6). Efforts were taken to investigate if the bulky R1 group contributes to the functionality of these novel α7 antagonists. Compounds with less bulky substitutions at R1 were synthesized and biologically assayed (Table 2).31 Compared with benzyl substitution (8), less bulky groups at R1 like methyl (8h) or hydrogen (8i) completely abolished the binding affinity to the α7 nAChR, suggesting the benzyl group at R1 is an important structural feature that confers receptor binding potency for compound 8 and its analogs.
Figure 6.
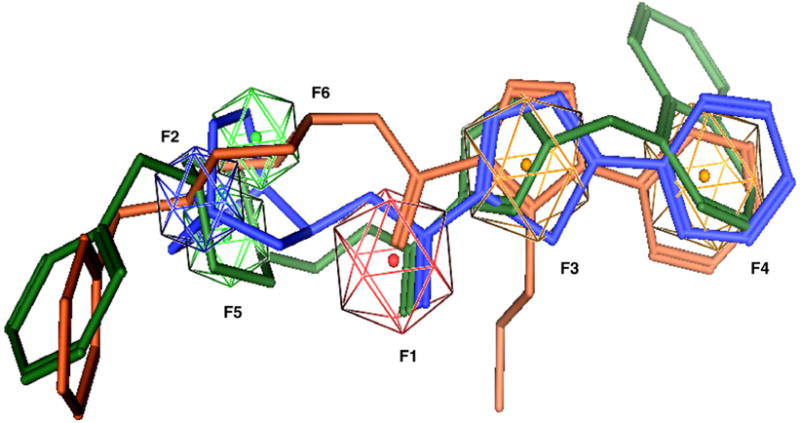
3D-Structural alignment of compound 2 (blue) and two hit compounds 7 (green) and 8 (brown). The pharmacophore features are shown in the same color coding as Figure 2. The compounds are rendered as stick-models.
The selectivity of compounds 7 and 8 were measured using previously reported methods on three other receptors: neuronal α4β2 nAChRs, muscle-type nAChRs, and 5HT3 receptors (Fig. 7).32–34 Briefly, the binding affinity for the α4β2 receptor was performed on rat cortical membranes using [3H]epibatidine as the radioligand.34 The muscle-type nAChR binding was determined using human TE671 cells with [125I]α-BTX as the radioligand.33 5HT3 binding was measured on recombinant CHO cells expressing human 5HT3 receptor using [3H]BRL 43694 as the radioligand.32 The orthosteric binding sites of the α7 nAChR and the 5HT3 receptor share a high degree of homology, therefore ligands for α7 nAChR and 5HT3 ligands frequently exhibit cross-activity. At 10 μM, compound 7 exhibited 82.5% binding to the α7 receptor and 18.8% and 8.4% binding to the neuronal α4β2 and the 5HT3 receptor, respectively. Similarly, compound 8 showed binding affinities of 82.5% to α7, 1.3% to α4β2, and 14.3% to 5HT3. Meanwhile, both compounds exhibited no detectable binding to the muscle-type nAChR at 10 μM. Taken together, these results demonstrated the selectivity of compounds 7 and 8 for the α7 receptor over α4β2, muscle-type nicotinic, and 5HT3 receptors.
Figure 7.

Binding selectivity of compounds 7 and 8 to neuronal α7 nAChR, α4β2 nAChR, and 5HT3 receptor at 10 μM. The error bars indicate the standard deviation of the measurements. The binding affinity for the α4β2 receptor was performed on rat cortical membrane cells using [3H]epibatidine as the radioligand.34 The 5HT3 binding was measured on recombinant CHO cells expressing the human 5HT3 receptor using [3H]BRL 43694 as the radioligand.32 Percent inhibition by compound 7: 82.5% (α7), 18.8% (α4β2), and 8.4% (5HT3). Percent inhibition by compound 8: 82.5% (α7), 1.3% (α4β2), and 14.3% (5HT3).
Agents useful for probing the physiological functions of neuronal nAChRs must efficiently penetrate the brain. Compounds 7i and 8 were tested in male C57Bl/6 mice (n ≥3 per time point) for blood–brain penetration after cassette dosing at 10 mg/kg (Fig. 8) via intraperitoneal (ip) administration.35 Brain and blood samples were collected at specific time points after drug administration. The area under the curve (AUC) ratios of brain to plasma were 2.8 and 3.1 for compounds 7i and 8, respectively, suggesting good brain penetration for both compounds. Compound 8 achieved high concentration in brain (9 μM). Cassette dosing of compounds can lead to incorrect estimates of plasma drug levels by drug–drug interactions such as at the level of Cytochrome P450 enzymes or by interfering with transporter systems. In order to eliminate this possible artifact we dosed compound 7i individually to mice using the same protocol and very similar levels of drug were found. The biphasic nature of the drug plasma and brain levels would suggest some type of secondary uptake mechanism as is seen for drugs that are eliminated from the blood in part by the bile system and therefore available in the intestine to be taken up into the circulation a second time.
Figure 8.

Brain (black square) and plasma (blue triangle) concentrations of compounds 7i (left) and 8 (right) after cassette dosing at 10 mg/kg in mouse via ip administration. The error bars indicate the standard deviation of the measurements.
The acetylcholine receptors (AChRs) are activated by acetylcholine (ACh), which is hydrolyzed to choline by acetylcholineesterase (AChE). When AChE is irreversibly inhibited by organophosphorus nerve agents like DFP and sarin, the uncontrolled accumulation of ACh at peripheral and central muscarinic AChRs (mAChRs) and nAChRs causes the cholinergic syndrome. This syndrome is characterized by sweating, pupillary constriction, convulsions, tachycardia, and eventually death. The mainstay treatment for nerve agent intoxication is the mAChR antagonist atropine together with an oxime reactivator of AChE (e.g., pralidoxime). This treatment regimen does not directly target nicotinic receptors although both mAChRs and nAChRs are involved in nerve agent toxicity. In this study, the new nAChR antagonists 7i and 8 were tested in a DFP toxicity animal model to investigate their anti-seizure activity (Table 3).36,37 Compared with the DFP controls, pretreatment with compounds 7i and 8 antagonized DFP-induced seizure-like behaviors over a 2 h period post-injection by 93.4% and 91.2%, respectively. The results suggest that these compounds could provide neuroprotection against seizure-like behaviors induced by DFP and, therefore, may be useful for treatment of organophosphus nerve agent intoxication.
Table 3. Neuroprotective activities against seizure induced by DFP.
| Seizure score (mean ± SD) | Normalized % seizure | Normalized % neuroprotection | |
|---|---|---|---|
| Controla | 0 ± 0 | 0 | 100 |
| DFP controlb | 9.1 ± 6.4 | 100 | 0 |
| 7i | 0.6 ± 1.3 | 6.6 | 93.4 |
| 8 | 0.8 ± 1.3 | 8.8 | 91.2 |
Animals not treated with drugs and DFP.
Animals not pretreated with drugs.
In summary, pharmacophore-based virtual screening led to the discovery of novel α7 nAChR ligands. A battery of property and functional group filters were applied to eliminate non-drug-like molecules and to reduce false positives. Two distinct families of small molecules (e.g., 7i and 8) were identified as novel α7 nAChR antagonists with selectivity for the neuronal α7 subtype over other nAChRs and good brain penetration. Neuroprotection against the seizure-like behaviors induced by DFP were observed for these compounds in a mouse model. The compounds should be very useful in discerning the physiological roles of neuronal α7 nAChR under normal and diseased states, and in discovering potential therapies for organophosphorus nerve agent intoxication.
Acknowledgments
This work was supported, in part, by funding from the NIH (R43 MH067488-01 and RO1 GM57481) and the United States Army Medical Research and Materiel Command NETRP (DAMD 17-03-2-0019 and W81XWH-06-C-0013) to Intra-Cellular Therapies Inc.
References and notes
- 1.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB, Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S. Pharmacol Rev. 1999;51:397. [PubMed] [Google Scholar]
- 2.Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. Mol Pharmacol. 1992;41:31. [PubMed] [Google Scholar]
- 3.Lindstrom J, Anand R, Gerzanich V, Peng X, Wang F, Wells G. Prog Brain Res. 1996;109:125. doi: 10.1016/s0079-6123(08)62094-4. [DOI] [PubMed] [Google Scholar]
- 4.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. J Med Chem. 2005;48:4705. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 5.Newhouse P, Singh A, Potter A. Curr Top Med Chem. 2004;4:267. doi: 10.2174/1568026043451401. [DOI] [PubMed] [Google Scholar]
- 6.Paterson D, Nordberg A. Prog Neurobiol. 2000;61:75. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- 7.Ripoll N, Bronnec M, Bourin M. Curr Med Res Opin. 2004;20:1057. doi: 10.1185/030079904125004060. [DOI] [PubMed] [Google Scholar]
- 8.Shytle RD, Silver AA, Lukas RJ, Newman MB, Sheehan DV, Sanberg PR. Mol Psychiatry. 2002;7:525. doi: 10.1038/sj.mp.4001035. [DOI] [PubMed] [Google Scholar]
- 9.Arneric SP, Holladay M, Williams M. Biochem Pharmacol. 2007;74:1092. doi: 10.1016/j.bcp.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 10.Mazurov A, Hauser T, Miller CH. Curr Med Chem. 2006;13:1567. doi: 10.2174/092986706777442011. [DOI] [PubMed] [Google Scholar]
- 11.Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, Ellis J, Zerbe GO, Leonard S, Stevens KE, Stevens JO, Martin L, Adler LE, Soti F, Kem WR, Freedman R. Arch Gen Psychiatry. 2006;63:630. doi: 10.1001/archpsyc.63.6.630. [DOI] [PubMed] [Google Scholar]
- 12.Chang CC. J Biomed Sci. 1999;6:368. doi: 10.1007/BF02253668. [DOI] [PubMed] [Google Scholar]
- 13.Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S. Neuropharmacology. 1999;38:679. doi: 10.1016/s0028-3908(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 14.Lopez-Hernandez GY, Thinschmidt JS, Zheng G, Zhang Z, Crooks PA, Dwoskin LP, Papke RL. Mol Pharmacol. 2009 doi: 10.1124/mol.109.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Paleari L, Cesario A, Fini M, Russo P. Drug Discovery Today. 2009;14:822. doi: 10.1016/j.drudis.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 16.Sheridan RD, Smith AP, Turner SR, Tattersall JE. J R Soc Med. 2005;98:114. doi: 10.1258/jrsm.98.3.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Astles PC, Baker SR, Boot JR, Broad LM, Dell CP, Keenan M. Curr Drug Targets CNS Neurol Disord. 2002;1:337. doi: 10.2174/1568007023339256. [DOI] [PubMed] [Google Scholar]
- 18.Broad LM, Felthouse C, Zwart R, McPhie GI, Pearson KH, Craig PJ, Wallace L, Broadmore RJ, Boot JR, Keenan M, Baker SR, Sher E. Eur J Pharmacol. 2002;452:137. doi: 10.1016/s0014-2999(02)02273-2. [DOI] [PubMed] [Google Scholar]
- 19.Bodnar AL, Cortes-Burgos LA, Cook KK, Dinh DM, Groppi VE, Hajos M, Higdon NR, Hoffmann WE, Hurst RS, Myers JK, Rogers BN, Wall TM, Wolfe ML, Wong E. J Med Chem. 2005;48:905. doi: 10.1021/jm049363q. [DOI] [PubMed] [Google Scholar]
- 20.Boess FG, De Vry J, Erb C, Flessner T, Hendrix M, Luithle J, Methfessel C, Riedl B, Schnizler K, van der Staay FJ, van Kampen M, Wiese WB, Koenig G. J Pharmacol Exp Ther. 2007;321:716. doi: 10.1124/jpet.106.118976. [DOI] [PubMed] [Google Scholar]
- 21.MOE, Chemical Computing Group Inc.: Montreal, Quebec, Canada.
- 22.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 23.Oprea TI. J Comput Aided Mol Des. 2000;14:251. doi: 10.1023/a:1008130001697. [DOI] [PubMed] [Google Scholar]
- 24.Olah MM, Bologa CG, Oprea TI. Curr Drug Discov Technol. 2004;1:211. doi: 10.2174/1570163043334965. [DOI] [PubMed] [Google Scholar]
- 25.Celie PH, van Rossum-Fikkert SE, van Dijk WJ, Brejc K, Smit AB, Sixma TK. Neuron. 2004;41:907. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 26.Zhong W, Gallivan JP, Zhang Y, Li L, Lester HA, Dougherty DA. Proc Natl Acad Sci USA. 1998;95:12088. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen IJ, Foloppe N. J Chem Inf Model. 2008;48:1773. doi: 10.1021/ci800130k. [DOI] [PubMed] [Google Scholar]
- 28.Meyer EM, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL. J Pharmacol Exp Ther. 1998;287:918. [PubMed] [Google Scholar]
- 29.nAChR expression in Xenopus oocytes: mature (>9 cm) female X. laevis African frogs (Nasco, Ft. Atkinson, WI) were used as a source of oocytes. Prior to surgery, frogs were anesthetized by placing the animal in a 1.5 g/l solution of 3-aminobenzoic acid ethyl ester for 30 min. Oocytes were removed from an incision made in the abdomen. All procedures involving frogs were approved by the University of Florida Institutional Animal Care and Use Committee (IACUC). To remove the follicular cell layer, harvested oocytes were treated with 1.25 mg/ml Type 1 collagenase (Worthington Biochemicals, Freehold, NJ) for 2 h at room temperature in calcium-free Barth's solution with a composition in mM of: 88 NaCl, 1 KCl, 0.8 MgSO4, 2.4 NaHCO3, 15 HEPES (pH 7.6), and 12 mg/l tetracycline. Stage 5 oocytes were then isolated and injected with 50 nl (5–20 ng) each of the appropriate subunit cRNAs. The human α7 nAChR receptor clone was provided by from Dr. Jon Lindstrom (University of Pennsylvania, Philadelphia, PA). To improve the expression of α7 nAChR, α7 RNA was routinely co-expressed with human RIC-3, a gift from Dr. Millet Treinin (Hebrew University, Jerusalem, Israel). After linearization and purification of cloned cDNAs, RNA transcripts were prepared in vitro using the appropriate mMessage mMachine kit from Ambion (Austin, TX).Voltage-clamp recording in Xenopus oocytes expressing nAChRs: Experiments were conducted using OpusXpress 6000A (Molecular Devices, Union City, CA). Each oocyte received initial control applications of acetylcholine (60 μM ACh), co-applications of ACh and the experimental drugs, and then a follow-up control application of ACh. Both peak amplitude and net charge of the responses were measured for each drug application and calculated relative to the preceding ACh control responses to normalize the data, compensating for the varying levels of channel expression among the oocytes. Net charge values were used to report inhibitory effects. Means and standard errors (SD) were calculated from the normalized responses of at least four oocytes for each experimental concentration. Concentration–response data were fit to the Hill equation, assuming negative Hill slopes.
- 30.Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, de Fiebre CM. Brain Res. 1997;768:49. doi: 10.1016/s0006-8993(97)00536-2. [DOI] [PubMed] [Google Scholar]
- 31.Synthesis of 8h: 5.0 mg of compound 8 (0.012 mmol) was dissolved in 5 ml methanol and 0.5 mg 10% Pd/C was added. The mixture was hydrogenated with hydrogen balloon at 60 °C for 8 h. After LC–HRMS indicated the reaction was finished, product was purified by preparative HPLC to give 3.8 mg 8h as white powder, yield 95.0%. HRMS (E+) 327.4359, found 327.1987. 1H NMR (DMSO-d6): 8.11 (s, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.58−7.49 (m, 3H), 7.45 (d, J = 8.0 Hz, 2H), 3.70 (br, 1H), 2.90 (t, J = 8.0 Hz, 2H), 2.77 (d, J = 4.0 Hz, 2H), 2.17 (s, 3H), 1.93 (t, J = 12.0 Hz, 2H), 1.75 (d, J = 4.0 Hz, 2H), 1.58−1.50 (m, 2H), 1.44−1.35 (m, 2H), 0.72 (t, J = 4.0 Hz, 3H).Synthesis of 8i: 5.0 mg of compound 8 (0.012 mmol) was dissolved in 5 ml glacial acetic acid and 0.5 mg 10% Pd/C was added. The mixture was hydrogenated with hydrogen balloon at 60 °C for 8 h. After LC–HRMS indicated the reaction was finished, product was purified by preparative HPLC to give 3.4 mg 8i as white powder, yield 94.5%. HRMS (E+) 313.4093, found 313.1877. 1H NMR (DMSO-d6): 8.11 (s, 1H), 7.82 (d, J = 4.0 Hz, 1H), 7.58−7.49 (m, 3H), 7.45 (d, J = 2.0 Hz, 2H), 4.10 (br, 2H), 3.78 (br, 2H), 2.90 (t, J = 8.0 Hz, 2H), 1.64 (br, 2H), 1.57 (s, 1H), 1.45−1.19 (m, 5H), 0.72 (t, J = 8.0 Hz, 3H).
- 32.Hope AG, Peters JA, Brown AM, Lambert JJ, Blackburn TP. Br J Pharmacol. 1996;118:1237. doi: 10.1111/j.1476-5381.1996.tb15529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lukas RJ. J Neurochem. 1986;46:1936. doi: 10.1111/j.1471-4159.1986.tb08516.x. [DOI] [PubMed] [Google Scholar]
- 34.Perry DC, Kellar KJ. J Pharmacol Exp Ther. 1995;275:1030. [PubMed] [Google Scholar]
- 35.Male, 8–10 weeks-old C57/BLC mice (Jackson Laboratory, Bar Harbor, ME) were used in this experiment. All handling and use followed a protocol of Institutional Animal Care and Use Committee of Columbia University, in accordance with NIH guidelines. Compounds 7i and 8 were co-injected to the animals (N ≥ 3) at 10 mg/kg dose via an intraperitoneal (ip) administration. After various time periods (0.25, 0.5, 1, 2, 4 h), animals were sacrificed and blood and brain collected. Whole brains were collected and sonicated with PBS buffer (137 mM NaCl, 2.7 mM KCl, 10 mM phosphate buffer) at pH 7.4, using 2 ml/g (v/w) homogenate. The brain homogenates were frozen in pre-weighed Eppendorf tubes at −80 °C. Blood samples were collected from the retro-orbital veins using VWR pasteur pipettes (VWR, West Chester, PA). Blood samples were centrifuged at 60 rpm for 40 min at 4 °C, and the plasma fractions were separated and stored at −80 °C. Brain homogenates and plasma were extracted with two volumes of acetonitrile and clarified by centrifugation at 12,000 g for 20 min and then analyzed by HPLC/MS/MS. The HPLC/MS/MS system included a Waters Alliance 2695 separations module and a Micromass Quattro-Micro Mass Spectrometer (Waters, Milford, MA). Separation was achieved on the column of Synergi 4 μ Fusion-RP (Phenomenex, Torrance, CA) with a gradient of methanol over 30 min in a mobile phase of 0.1% formic acid. Sample injection volume was 10 ml and flow rate was 0.6 ml/min. Control experiments were performed to determine extraction efficiencies.
- 36.Male FVB mice (Jackson Laboratories, Bar Harbor, ME) aged 6 months were housed in a temperature (22 ± 2 °C) and humidity-controlled (30–40%) colony room maintained on a 12-h light/12-h dark cycle. Animals were allowed ad libitum access to chow and water. No cage enrichment was employed. All procedures were performed under an approved Animal Care and Use Committee of the Centers for Disease Control and Prevention-National Institute for Occupational Safety and Health. Groups of mice (N = 5 mice each) were administered a single dose of diisopropylfluorophosphonate (DFP), 4.0 mg/kg, ip, alone or 30 min after pretreatment with compounds 7i and 8, at a dosage of 25 mg/kg, sc in the flank. Saline was used as the vehicle for DFP and 50%DMSO/50% saline was used as a vehicle for the pretreatments. Behavior was rated for seizure-like behaviors at specified times (15, 30, 45, 60, 75, 90, 105, and 120 min) over a 2 h period post-injection using a modified Racine seizure rating scale, as follows: stage 1: immobility/rigid posture; stage 2: immobility/rigid posture and occasional rearing and falling; stage 3: immobility/rigid posture and frequent rearing and falling and forelimb clonus; stage 4: immobility/rigid posture and severe tonic and clonic seizures.
- 37.Racine RJ. Electroencephalogr Clin Neurophysiol. 1972;32:269. doi: 10.1016/0013-4694(72)90176-9. [DOI] [PubMed] [Google Scholar]