

Aromatic thioethers are valuable synthetic intermediates frequently found in biologically and pharmaceutically active molecules or in polymeric materials. In particular, diaryl-and aryl-heteroaryl thioethers are essential components of numerous drugs with potential application in the treatment of inflammation, cancer, human immunodeficiency virus (HIV), asthma, Alzheimer’s and Parkinson’s diseases.[1–6] Furthermore, diaryl thioethers are precursors to the corresponding sulfoxides and sulfones that also exhibit important biological activities and are contained in antifungal and anti-cancer agents as well as in potential drug candidates for Alzheimer’s disease or HIV.[7–11] Classical methods for the synthesis of such thioethers encompass thermal reaction of arenes with sulfur,[12,13] base-mediated reactions of activated chloroarenes with thiophenols,[14] and condensation of organolithium or Grignard reagents with chlorophenyl sulfide.[15] However, these reactions often require harsh reaction conditions, occur with low regioselectivity, and form disulfide and thiantrene side products.[12,13]
To address these limitations, cross-coupling reactions catalyzed by transition metals, including reactions catalyzed by complexes of palladium,[16–19] nickel,[20] copper,[21–23] iron,[24] and cobalt,[25] have been developed to form aromatic carbon–sulfur bonds. Although these metal-catalyzed coupling reactions often occur in high yield under milder conditions than the uncatalyzed methods, few aromatic thiols are commercially available. Such arenethiols are accessible from phenols by Newman–Kwart[26] and Schönberg[27] rearrangements, but both processes require drastic thermal conditions. In addition, many aromatic thiols are unstable to oxidation, and these thiols can decay upon storage. Thus, a process to generate diaryl thioethers from an H2S equivalent and two different aryl halides would be a marked improvement over current methods. Palladium-catalyzed reactions of hydrogen sulfide surrogates, such as isooctyl-3-mercaptopropionate[18,28] and triisopropylsilanethiol (TIPS-SH) or its corresponding alkali metal thiolates are known,[29–31] but these reactions have not been developed into a simple sequence to form unsymmetrical diaryl thioethers.
The most active and functional-group-tolerant catalyst system for the coupling of haloarenes with thiols is based on the complex containing the alkylbisphosphine ligand in Scheme 1 (1, CyPF-tBu).[32–35] Detailed mechanistic studies on these coupling reactions have also been published recently.[36] Reactions with this catalyst occur with turnover numbers and substrate scope that far surpass those of previous catalysts containing other ligands. Herein, we report reactions of aryl halides with TIPS-SH catalyzed by the palladium complex generated from the alkylbisphosphine 1,[37] and the use of this process to prepare unsymmetrical diaryl sulfides in a one-pot fashion from two different aryl bromides or in a tandem fashion with one aryl bromide and one aryl chloride. This synthesis of diaryl thioethers thus occurs with readily available reagents and avoids the need to independently prepare and isolate an intermediate arene thiol.
Scheme 1.

Palladium-catalyzed synthesis of p-tolyl phenyl sulfide from phenyl triisopropylsilyl sulfide and p-bromotoluene.
To prepare diaryl thiothers in a simple fashion, we envisioned a one-pot protocol involving the reaction of aryl halides with TIPS-SH, followed by coupling of this silylated thioether. We previously reported[34] the synthesis of p-tolyl phenyl sulfide 2a from reaction of phenyl triisopropylsilyl sulfide with p-bromotoluene in the presence of CsF and the palladium catalyst (Scheme 1). These preliminary data suggested that a one-pot method to form diaryl thioethers should be possible through the silyl thioether.
To enact this proposed synthesis, it was necessary to develop a general coupling of aryl bromides with TIPS-SH.[31, 37, 38] Studies of the coupling reactions of TIPS-SH with the model aryl bromide 4-bromotoluene under reaction conditions initially developed for the reaction of phenyl triisopropylsilyl sulfide[34] showed that the protected thiophenol forms in excellent yield (Table 1, entry 1) in the presence of just 500 ppm of catalyst and LiHMDS as base in toluene at 110°C. In contrast to related couplings with the same catalyst,[32] reactions of TIPS-SH with NaOtBu as base or in DME (1,2-dimethoxyethane) as solvent occurred in slightly lower yields (Table 1, entries 2 and 3). Reactions were also performed at the lower temperature of 90°C in the presence of 0.1 mol% catalyst (Table 1, entry 4). These reactions occurred with aryl chlorides, as well as aryl bromides, although longer reaction times (12 h) were required when using just 0.1 mol% catalyst (Table 1, entry 5). Reactions of bromo-naphthalene also occurred with 0.05 mol% catalyst (Table 1, entry 12).
Table 1.
Coupling of aryl halides with TIPS-SH catalyzed by Pd(OAc)2 and CyPF-tBu ligand.[a]
 | ||||
|---|---|---|---|---|
| Entry | ArX | Cat. [mol%] | Product | Yield [%] |
| 1 | 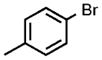 |
0.05 | 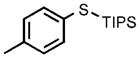 |
98 |
| 2[b] | 0.05 | 88 | ||
| 3[c] | 0.05 | 90 | ||
| 4[d] | 0.1 | 91 | ||
| 5[e] | 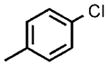 |
0.1 | 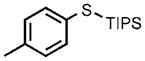 |
96 |
| 6 | 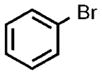 |
0.05 | 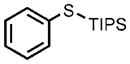 |
91 |
| 7 | 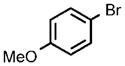 |
0.25 | 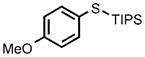 |
96 |
| 8 | 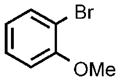 |
0.25 | 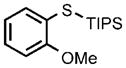 |
99 |
| 9 | 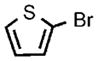 |
0.25 | 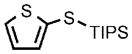 |
76 |
| 10 | 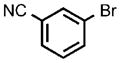 |
0.25 | 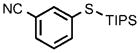 |
87 |
| 11 | 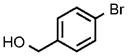 |
0.25 | 74 | |
| 12 | 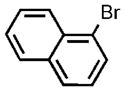 |
0.05 | 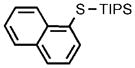 |
97 |
| 13 | 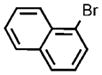 |
0.05 | 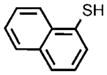 |
95[f] |
Reactions were conducted with a 1:1 ratio of metal to ligand, 1 mmol of both ArX and thiol, and 1.1 equiv of LiHMDS at 110°C in toluene (1.5 mL) requiring 2–4 h to complete.
Reaction performed with NaOtBu as base.
Reaction conducted in DME.
Reaction performed at 90°C.
Reaction required 12 h to complete.
TBAF (2 equiv) was added to the crude mixture and stirred 30 min at RT.
After establishing these reaction conditions, we explored the scope of this process with aryl bromides that often react in coupling processes in lower yields or require large amounts of catalyst. TIPS-SH successfully coupled with a variety of such aryl bromides in good to excellent yields within short reaction times (2–4 h) with relatively low catalyst loadings. For example, the coupling of bromoarenes that are electron-rich or sterically demanding or both (Table 1, entries 7 and 8), as well as the coupling of thienyl bromide (Table 1, entry 9) and aryl bromides containing a cyano or hydroxyalkyl group (Table 1, entries 10 and 11) all occurred with just 0.25 mol% catalyst. These reactions occur with turnover numbers that are one or two orders of magnitude higher than those previously reported for related couplings of TIPS-SH with other catalysts.[29–31]
To test the viability of using the developed methodology to prepare pure, unprotected aromatic thiols, we investigated conditions for removal of the silyl group. Pure naphthalene-1-thiol was obtained by deprotection with TBAF in situ after the coupling process (Table 1, entry 13). However, the analogous sequence with p-bromotoluene exclusively formed, under all conditions tested, di-p-tolyl disulfide from oxidation of the aromatic thiol. Furthermore, partial cleavage of the silicon–sulfur bond to give thiol and disulfide impurities occurred during the purification of some of the silyl thioethers. Thus, a decrease in the yield from that determined by GC/MS and NMR spectroscopy of the crude reaction was observed in some cases after isolation (Table 1, entries 9–11).[39]
These observations imply that an efficient methodology to synthesize diaryl sulfides from two aryl halides using TIPS-SH as hydrogen sulfide surrogate should avoid both the isolation and deprotection of silyl aryl sulfides. Considering that catalysts containing CyPF-tBu as ligand are able to couple aromatic silyl sulfides in the presence of CsF (Scheme 1), we investigated the potential for developing a one-pot procedure that would circumvent the isolation of protected thiols and, therefore, prevent competing undesired deprotection of the silyl thioether and subsequent oxidation of the resulting thiol.
We selected the combination of bromobenzene, bromotoluene, and TIPS-SH as a model system and tested several reaction conditions with 0.5 mol% Pd(OAc)2 and ligand 1. The initial reaction between TIPS-SH and bromobenzene occurred in less than 1 h in toluene at 90°C with LiHMDS (1.1 equiv) as base. Addition of p-bromotoluene and four equivalents of CsF to the resulting reaction mixture, followed by heating at 110°C for an additional 5 h, formed p-tolyl phenyl sulfide 2a in 97% yield (Table 2). Reactions conducted in DME at different temperatures produced 5–10% of undesired symmetrical diaryl sulfides as side products[33] or required higher catalyst loadings to reach full conversions. Reactions conducted with 0.25 mol% for a total time of 9 h occurred in yields that were comparable to those from reactions with 0.5 mol% palladium over 4–6 h.
Table 2.
One-pot synthesis of unsymmetrical diaryl sulfides from two aryl bromides and HSTIPS catalyzed by Pd(OAc)2 and CyPF-tBu ligand.[a]
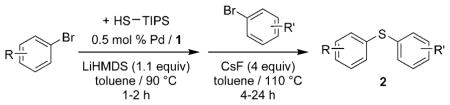 | ||
|---|---|---|
| Products (Yield [%]) | ||
|
|
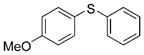 2b (97%) 2b (97%) |
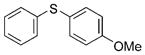 2b (95%) 2b (95%) |
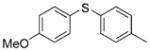 2c (98%) 2c (98%) |
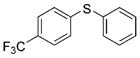 2d (98%) 2d (98%) |
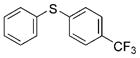 2d (92%)[c] 2d (92%)[c]
|
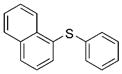 2e(91%) 2e(91%) |
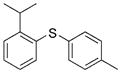 2f (85%) 2f (85%) |
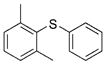 2g (95%)[d] 2g (95%)[d]
|
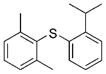 2h (94%)[e] 2h (94%)[e]
|
|
|
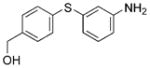 2k (69%)[f] 2k (69%)[f]
|
|
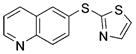 2m (85%)[f] 2m (85%)[f]
|
All reactions were conducted with a 1:1 ratio of metal to ligand and 1 mmol of each aryl bromide and TIPS-SH.
0.25 mol% catalyst loading.
<5% of symmetrical sulfides were detected.
2.0 mol% catalyst loading.
3.0 mol% catalyst loading.
1.0 mol% catalyst loading.
The identity of the base was crucial for obtaining high yields of the mixed thioether. Both LiHMDS and CsF were needed in the amounts showed in Table 2. Reactions conducted with a larger excess of LiHMDS, fewer equivalents of CsF, or the addition of CsF during the first step of the process all occurred in lower yield. Moreover, reactions conducted with alkali metal carbonates were slow and led to low yields after the second step, presumably due to significant catalyst decomposition during the second coupling. Reactions conducted with KF instead of CsF did not lead to efficient cleavage of the silyl thioether, as determined by GC/MS, and formed the diaryl thioether in <5% yield.
Reactions of a series of representative aryl bromides were conducted to evaluate the scope of the one-pot procedure, and the results are summarized in Table 2. Under the optimized conditions, unsymmetrical diaryl sulfides 2a–e bearing neutral, electron-rich or electron-deficient substituents were synthesized in nearly quantitative yields in short overall reaction times (5–6 h). Undesired symmetrical byproducts were formed (<5%) in the preparation of 2d when the electron-deficient bromoarene was added in the second step. However, none of these side products were detected when the more electron-deficient bromoarene was added in the first step. Reactions of hindered ortho-substituted aryl bromides also occurred to furnish the corresponding sulfides 2 f–h efficiently. The synthesis of very sterically hindered sulfides 2g and 2h with 2.0–3.0 mol% is particularly noteworthy.
We also evaluated the tolerance of the one-pot process to the presence of functional groups. In this regard, we have previously reported the extraordinary functional-group tolerance of palladium catalyst derived from ligand 1. Therefore, we focused our studies on bromoarenes that would challenge the functional group tolerance of this new method.[32,33] Reactions with aryl bromides that contain an aldehyde or an enolizable ketone produced the diaryl sulfides 2i–j in good yields. A diaryl sulfide that bears both free alcohol and free amine 2k was also formed in good yield with just 1.0 mol% catalyst. Additionally, reactions to generate aryl heteroaryl and di-heteroaryl sulfides 2l–m also occurred in high yields.
Having obtained excellent results on the one-pot synthesis of unsymmetrical diaryl sulfides by successive coupling of two aryl bromides, we decided to explore the feasibility of performing the coupling in a tandem, rather than one-pot sequential, fashion.[40] In this tandem process, both aryl halides would be added together with the catalyst at the beginning of the reaction. We have previously established that catalysts derived from CyPF-tBu are fully selective for thioetherification of aryl bromides over aryl chlorides with both aliphatic and aromatic thiols.[32] We have also shown in the present report that the coupling reactions of TIPS-SH with p-bromotoluene and p-chlorotoluene are different (Table 1, entries 1 and 5). With these data in mind, we envisioned an ideal, three-component tandem reaction in which TIPS-SH selectively couples with an aryl bromide to form a silylated thiol that subsequently reacts with the aryl chloride in the presence of the fluoride additive. In this process, the fluoride would activate or fully cleave the silyl aryl thioether to generate the thiolate for coupling, but would not cleave the silyl thiol because the thiol is present in the anionic thiolate form.
Once again we selected the formation of p-tolyl phenyl sulfide 2a as a prototype transformation, and several reaction conditions were examined. We found that the desired tandem reaction occurs to afford the unsymmetrical diaryl sulfide 2a in 80% yield in the presence of 1.0 mol% catalyst, and under conditions similar to those determined to be appropriate for the sequential one-pot procedure (Scheme 2). Apparently, the fluoride additive selectively activates the silyl aryl sulfide over the silyl thiol for the coupling process, as designed.
Scheme 2.
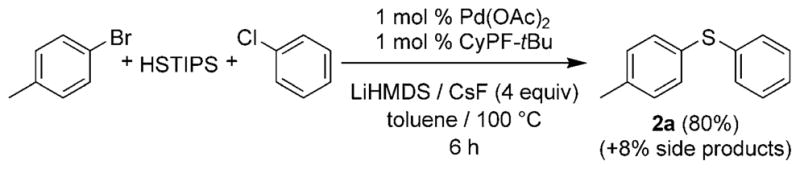
Tandem reaction between p-bromotoluene, TIPSSH, and chlorobenzene catalyzed by Pd(OAc)2 and CyPF-tBu.
In summary, we have shown that palladium complexes derived from the bisphosphine CyPF-tBu ligand can be used to prepare protected arenethiols by coupling aryl halides with triisopropylsilanethiol (TIPS-SH) and that this reaction can be further developed into a one-pot sequential or tandem synthesis of unsymmetrical diaryl thioethers. The reactions of aryl bromides with TIPS-SH occur with broad scope and with turnover numbers 1 or 2 orders of magnitude higher than those of related couplings of TIPS-SH by previously reported catalysts. In addition, this catalyst allows the synthesis of unsymmetrical diaryl sulfides by coupling two aryl bromides and TIPS-SH in a one-pot fashion. This protocol overcomes the limited availability and stability of arene-thiols. These reactions occur in good to excellent yield with 0.25 to 3.0 mol% catalyst. The process exhibits a substrate scope that includes hindered aryl bromides, heteroaryl bromides, as well as bromides containing an aldehyde or an enolizable ketone. Finally, we have demonstrated the viability of a tandem process using two different halides in the same vessel.
Experimental Section
General procedure for the coupling of aryl bromides with triisopropylsi-lanethiol
The appropriate quantity of catalyst system (see the Supporting Information) was added to a 4 mL vial containing the aryl bromide or chloride (1.00 mmol) and LiHMDS (184 mg, 1.10 mmol) in toluene (1.5 mL). TIPS-SH (214 μL, 1.00 mmol) was then added, and the vial sealed with a cap containing a PTFE septum. The mixture was heated at 110°C until the aryl halide was consumed, as determined by GC. Solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography on silica gel. Protected aryl thiols were isolated in the yields reported in Table 1.
General procedure for the one-pot synthesis of unsymmetrical diaryl sulfides 2 from two aryl bromides and triisopropylsilanethiol
The appropriate amount of Pd(OAc)2 and CyPF-tBu ligand was added to a 4 mL vial containing an aryl bromide (1.00 mmol) and LiHMDS (184 mg, 1.10 mmol) in toluene (1.5 mL). TIPS-SH (214 μL, 1.00 mmol) was then added, and the vial sealed with a cap containing a PTFE septum. The mixture was heated at 90°C until the aryl bromide was consumed (1–2 h), as determined by GC. The second aryl bromide (1.00 mmol) and CsF (608 mg, 4.00 mmol) were then added, and the mixture heated at 110°C until the aryl bromide was consumed (4–24 h), as determined by GC. Solvent was removed under reduced pressure and the crude mixture was purified by flash chromatography on silica gel. The corresponding diaryl sulfides were isolated in the yields reported in Table 2.
Tandem reaction between p-bromotoluene, TIPS-SH and chlorobencene
Pd(OAc)2 (2.2 mg) and CyPF-tBu (5.5 mg) were added to a 4 mL vial containing 4-bromotoluene (121 μL, 1.00 mmol), chlorobenzene (102 μL, 1.00 mmol), LiHMDS (184 mg, 1.10 mmol), and CsF (608 mg, 4.00 mmol) in toluene (1.5 mL). The vial was sealed with a cap containing a PTFE septum, and the mixture was heated at 100°C until the chlorobenzene was consumed, as determined by GC. Silica gel (0.5 g) was added, and the solvents were evaporated under reduced pressure. The crude residue was purified by column chromatography on silica gel using hexane as eluent. p-Tolyl phenyl sulfide 2a was obtained as the major product in 80% yield (160 mg). This isolated material contained 8% of the symmetrical sulfides, which could not be separated from the unsymmetrical sulfide, in this case, by column chromatography.
Supplementary Material
Acknowledgments
We thank the NIH-NIGMS (GM-55382) for support of this work. M.F.-R. thanks the Ministerio de Educación y Ciencia (Spain) for a MEC/Fulbright fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.200902313.
References
- 1.Pasquini S, Mugnaini C, Tintori C, Botta M, Trejos A, Arvela RK, Larhed M, Witvrouw M, Michiels M, Christ F, Debyser Z, Corelli F. J Med Chem. 2008;51:5125–5129. doi: 10.1021/jm8003784. [DOI] [PubMed] [Google Scholar]
- 2.Gangjee A, Zeng Y, Talreja T, McGuire JJ, Kisliuk RL, Queener SF. J Med Chem. 2007;50:3046–3053. doi: 10.1021/jm070165j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alcaraz ML, Atkinson S, Cornwall P, Foster AC, Gill DM, Humphries LA, Keegan PS, Kemp R, Merifield E, Nixon RA, Noble AJ, O’Beirne D, Patel ZM, Perkins J, Rowan P, Sadler P, Singleton JT, Tornos J, Watts AJ, Woodland IA. Org Process Res Dev. 2005;9:555–569. [Google Scholar]
- 4.Clader JW, Billard W, Binch H, III, Chen L-Y, Crosby G, Jr, Duffy RA, Ford J, Kozlowski JA, Lachowicz JE, Li S, Liu C, McCombie SW, Vice S, Zhou G, Greenlee WJ. Bioorg Med Chem. 2004;12:319–326. doi: 10.1016/j.bmc.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 5.Liu G, Huth JR, Olejniczak ET, Mendoza R, DeVries P, Leitza S, Reilly EB, Okasinski GF, Fesik SW, von Geldern TW. J Med Chem. 2001;44:1202–1210. doi: 10.1021/jm000503f. [DOI] [PubMed] [Google Scholar]
- 6.Nielsen SF, Nielsen EO, Olsen GM, Liljefors T, Peters D. J Med Chem. 2000;43:2217–2226. doi: 10.1021/jm990973d. [DOI] [PubMed] [Google Scholar]
- 7.Sciabola S, Carosati E, Baroni M, Mannhol R. J Med Chem. 2005;48:3756–3767. doi: 10.1021/jm049162m. [DOI] [PubMed] [Google Scholar]
- 8.Llauger L, He H, Kim J, Aguirre J, Rosen N, Peters U, Davies P, Chiosis G. J Med Chem. 2005;48:2892–2905. doi: 10.1021/jm049012b. [DOI] [PubMed] [Google Scholar]
- 9.Otzen T, Wempe EG, Kunz B, Bartels R, Lehwark-Yvetot G, Hänsel W, Schaper KJ, Seydel JK. J Med Chem. 2004;47:240–253. doi: 10.1021/jm030931w. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Chackalamannil S, Hu Z, Clader JW, Greenlee W, Billard W, Binch G, III, Crosby G, Jr, Ruperto V, Duffy RA, McQuade R, Lachowicz JE. Bioorg Med Chem Lett. 2000;10:2247–2250. doi: 10.1016/s0960-894x(00)00457-1. [DOI] [PubMed] [Google Scholar]
- 11.Sun ZY, Botros E, Su AD, Kim Y, Wang E, Baturay NZ, Kwon CH. J Med Chem. 2000;43:4160–4168. doi: 10.1021/jm9904957. [DOI] [PubMed] [Google Scholar]
- 12.Dougherty G, Hammond PD. J Am Chem Soc. 1935;57:117–118. [Google Scholar]
- 13.Glass HB, Reid EE. J Am Chem Soc. 1929;51:3428–3430. [Google Scholar]
- 14.Campbell JR. J Org Chem. 1964;29:1830–1833. [Google Scholar]
- 15.Kharasch N, Potempa SJ, Wehrmeister HL. Chem Rev. 1946;39:269–332. doi: 10.1021/cr60123a004. [DOI] [PubMed] [Google Scholar]
- 16.Cai L, Cuevas J, Peng YY, Pike VW. Tetrahedron Lett. 2006;47:4449–4452. [Google Scholar]
- 17.Mispelaere-Canivet C, Spindler JF, Perrio S, Beslin P. Tetrahedron. 2005;61:5253–5259. [Google Scholar]
- 18.Itoh T, Mase T. Org Lett. 2004;6:4587–4590. doi: 10.1021/ol047996t. [DOI] [PubMed] [Google Scholar]
- 19.Murata M, Buchwald SL. Tetrahedron. 2004;60:7397–7403. [Google Scholar]
- 20.Zhang Y, Ngeow KC, Ying JY. Org Lett. 2007;9:3495–3498. doi: 10.1021/ol071248x. [DOI] [PubMed] [Google Scholar]
- 21.Herrero MT, SanMartin R, Domínguez E. Tetrahedron. 2009;65:1500–1503. [Google Scholar]
- 22.Carril M, SanMartin R, Domínguez E, Tellitu I. Chem Eur J. 2007;13:5100–5105. doi: 10.1002/chem.200601737. [DOI] [PubMed] [Google Scholar]
- 23.For a review on cooper-catalyzed coupling, see: Ley SV, Thomas AW. Angew Chem. 2003;115:5558–5607. doi: 10.1002/anie.200300594.Angew Chem Int Ed. 2003;42:5400–5449. doi: 10.1002/anie.200300594.
- 24.Correa A, Carril M, Bolm C. Angew Chem. 2008;120:2922–2925. [Google Scholar]; Angew Chem Int Ed. 2008;47:2880–2883. doi: 10.1002/anie.200705668. [DOI] [PubMed] [Google Scholar]
- 25.Wong YC, Jayanth TT, Cheng CH. Org Lett. 2006;8:5613–5616. doi: 10.1021/ol062344l. [DOI] [PubMed] [Google Scholar]
- 26.For a recent review, see: Lloyd-Jones GC, Moseley JD, Renny JS. Synthesis. 2008:661–689.
- 27.Al-Kazimi HR, Tarbell DS, Plant D. J Am Chem Soc. 1955;77:2479–2482. [Google Scholar]
- 28.Itoh T, Mase T. J Org Chem. 2006;71:2203–2206. doi: 10.1021/jo052624z. [DOI] [PubMed] [Google Scholar]
- 29.Kreis M, Bräse S. Adv Synth Catal. 2005;347:313–319. [Google Scholar]
- 30.Arnould JC, Didelot M, Cadilhac C, Pasquet MJ. Tetrahedron Lett. 1996;37:4523–4524. [Google Scholar]
- 31.Rane AM, Miranda EI, Soderquist JA. Tetrahedron Lett. 1994;35:3225–3226. [Google Scholar]
- 32.Fernández-Rodríguez MA, Hartwig JF. J Org Chem. 2009;74:1663–1672. doi: 10.1021/jo802594d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fernández-Rodríguez MA, Shen Q, Hartwig JF. Chem Eur J. 2006;12:7782–7796. doi: 10.1002/chem.200600949. [DOI] [PubMed] [Google Scholar]
- 34.Fernández-Rodríguez MA, Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:2180–2181. doi: 10.1021/ja0580340. [DOI] [PubMed] [Google Scholar]
- 35.Hartwig JF. Acc Chem Res. 2008;41:1534–1544. doi: 10.1021/ar800098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alvaro E, Hartwig JF. J Am Chem Soc. 2009;131:7858–7868. doi: 10.1021/ja901793w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.For the introduction of triisopropylsilylthiol as a reagent for the synthesis of thiols, see: de Pomar JCJ, Soderquist JA. Tetrahedron Lett. 1998;39:4409.Miranda EI, Diaz MJ, Rosado I, Soderquist JA. Tetrahedron Lett. 1994;35:3221.
- 38.For reactions with other thiol surrogates tested, such as NaSH and AcSH either no conversion was observed or complex mixtures were obtained. On the other hand, reactions with thiourea afforded symmetrical diarylsulfides as main products as a result of in situ deprotection and the subsequent reaction with a second molecule of aryl bromide.
- 39.The cleavage of the Si–S bond during chromatography purification of aryl triisopropylsilyl sulfides was previously observed: see reference [22,23].
- 40.Fogg DE, dos Santos EN. Coord Chem Rev. 2004;248:2365–2379. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.