

Abstract
Therapy-induced accelerated cellular senescence (ACS) is a reversible tumor response to chemotherapy that is likely detrimental to the overall therapeutic efficacy of cancer treatment. In order to further understand the mechanism by which cancer cells can escape the sustained cell cycle arrest in ACS, we established a tissue culture model, in which the p53-null NCI-H1299 cells can be induced into senescence by an abbreviated exposure to a chemotherapeutic agent. Previously, we have reported that senescent cells over-express Cdc2/Cdk1 when they bypassed the prolonged arrest and their viability is dependent on Cdc2/Cdk1 kinase activity. In the present study, we show that human survivin is the immediate downstream effector of the Cdc2/Cdk1 mediated survival signal. Survivin cooperates with Cdc2/Cdk1 to inhibit apoptosis following chemotherapy and promote senescence escape. Using HIV-1 TAT peptides to disrupt survivin phosphorylation by Cdc2/Cdk1, we also found that phosphorylated survivin is necessary both for the escape of senescent cells and for maintenance of subsequent viability after bypassing senescence. These results further propose survivin as an important determinant of senescence reversibility and as a putative molecular target to enforce cell death in ACS.
Keywords: survivin, accelerated senescence, Cdc2/Cdk1
Introduction
In response to anti-cancer drugs, ionizing radiation, differentiating agents, oxidative stress and selective oncogenic stimuli, immortalized cancers cells can undergo terminal growth arrest despite having bypassed telomere-dependent replicative senescence 1, 2. This telomere-independent response, termed accelerated cellular senescence (AKA therapy-induced or stress-induced cellular senescence), is believed to overlap with the physiological senescence program and shares a senescence-like phenotype. This phenotype includes morphologic alterations such as enlarged and flattened shape with increased cytoplasmic granularity, presence of polyploidy, and expression of the pH-restricted, senescence-associated β-galactosidase (SA-β-gal) 1, 3. Both physiological cellular senescence and accelerated cellular senescence (ACS) appear to be modulated by p53 effector pathways not restricted to those mediated by p21waf1/cip1, and by p16 INK4A through its regulation of pRB 4, 5. However, neither pathway appears to be absolutely required in ACS 6. ACS has been demonstrated in vivo using xenograft and transgenic mouse models 7, 8. More recently, expression of the senescence marker SA-β-gal has been shown in archival tumor samples obtained from breast cancer patients treated with chemotherapy and also in resected lung cancer specimens from patients treated with neoadjuvant chemotherapy 6, 9.
ACS appears to be reversible. We have established a tissue culture model in which ACS can be induced in a p53-null, p16-deficient, NCI-H1299 human lung carcinoma cell line by a variety of chemotherapeutic agents 6. The senescent cells arrest at G2/M reinforced by low levels of cellular Cdc2/Cdk1 and cyclin B1. Rare cells can bypass or escape terminal arrest and reenter the cell cycle. These escape cells frequently over-express Cdc2/Cdk1 and rely on Cdc2/Cdk1 kinase activity for their viability. To further understand the mechanism by which Cdc2/Cdk1 promotes escape and influences cell fate in ACS, we have extended these studies to survivin, a known substrate of Cdc2/Cdk1 kinase.
Human survivin, a 16.5 kDa nuclear protein containing a single 70 amino acid BIR (Baculovirus IAP repeat) domain, is the smallest member of the human inhibitor of apoptosis protein (IAP) family 10. Survivin is expressed in a cell cycle-dependent manner, and its protein levels markedly rise during mitosis. The survivin protein associates with Cdc2/Cdk1 and is phosphorylated at the threonine-34 (T34) residue. This phosphorylation stabilizes the survivin protein and is apparently necessary for its interaction with mitotic spindle and inhibition of caspase-9 activity 11. In HeLa cells, the microtubule inhibitor taxol appears to activate a survival checkpoint through the up-regulation of Cdc2/Cdk1 kinase activity, leading to the phosphorylation and accumulation of survivin. Suppression of survivin phosphorylation with the Cdc2/Cdk1 kinase inhibitor flavopiridol enhances adriamycin-induced apoptosis 12. Additionally, survivin knockout is embryonic lethal and fibroblasts derived from these animals demonstrate catastrophic defects in microtubules, centrosomes, spindle poles, and in mitotic spindle microtubule formation 13 14. Therefore, survivin exerts a key role in the regulation of cell division.
We show in this study that survivin functions to influence cell fate in response to chemotherapy. Survivin knockdown abrogates the senescence response in H1299 lung cancer cells in favor of apoptosis. In contrast, survivin is up-regulated in senescent escape cells that are able to reenter the cell cycle. In therapy-induced senescent cells, the conditional over-expression of survivin facilitates reentry into the cell cycle and escape from terminal arrest. To examine the role of survivin phosphorylation by Cdc2/Cdk1, we disrupted the intracellular survivin phosphorylation using HIV-1 TAT-peptides with sequences derived from the Thr34 region of survivin. These peptides completely abrogate escape of senescent cells treated with chemotherapy and elicit marked apoptosis in cells that have previously bypassed senescence. Altogether, these results suggest survivin acts as a downstream effector of Cdc2/Cdk1 in therapy-induced senescence and underscore the importance of phosphorylated survivin as a determinant of cancer therapy resistance.
Materials and methods
Tissue culture
The NCI-H1299 (ATCC) non-small cell carcinoma cells were maintained in RPMI 1640 supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Biowhittaker) at 37°C and in a 5% CO2 humidified incubator.
Immunoblot analysis
Immunoblot analysis was performed using whole cell lysates extracted with WE16 buffer as described previously 6. Equivalent amount of proteins (20-40 μg) were loaded for each sample lane. Survivin and AKT2 were detected with D-8 and F-7 antibodies respectively (Santa Cruz). Phosphorylated survivin was determined with anti-phosphosurvivin (Thr34) antibody (Novus) following immunoprecipitation of cell lysates with D8 antibody. Antibodies for Cdc2/Cdk2, cyclin B1 and β-actin were reported previously 6. Antibodies against X-IAP, C-IAP1, C-IAP2, and Bcl-2 were purchased from Santa Cruz. The blot images were quantitatively analyzed by BIO-RAD Chemidoc XRT+ system (Quantity one).
Antisense oligonucleotide and siRNA methodologies
Two 20-mer, antisense phosphorothioate oligonucleotide were synthesized (Invitrogen). Antisense sequence against survivin is 5′- CCCAGCCTTCCAGCTCCTTG-3′ (nucleotides 232–251 on survivin mRNA) 15 and the control scrambled oligonucleotide sequence is 5′-GGAGCCAGGGGGGAGCAGGG-3′. At indicated concentrations, these oligonucleotides were transfected into H1299 cells with Effectene (Qiagen) according to the manufacturer's recommendation. Twenty-four hours following transfection, cells were recovered in fresh media and camptothecin was added at indicated concentrations. The control siRNA and siRNA's against Cdc2/CDk1 and AKT2 were obtained commercially (Smartpool, Upstate) and transfected with Lipofectamine following the manufacturer's protocol (Invitrogen).
Apoptosis assay
Where indicated, apoptosis of camptothecin-treated H1299 cells were determined quantitatively by DNA content, DNA fragmentation, and Annexin V analyses. DNA content analyses were performed with a Becton-Dickinson FACSCalibur on propidium iodide labeled cells per standard fluorescence-activated cell sorting (FACS) protocols 6. Sub-G1 fractions of the DNA content analysis were determined with the CellQuest software (Becton-Dickinson). DNA fragmentation assay was performed by agarose gel electrophoresis 16. Indicated cells were lysed in DNA lysis buffer (1% triton X-100, 10mM tris-HCl (pH 8.0), and 100 mM EDTA) and DNA was extracted by phenol/chloroform and precipitated by isopropanol. After resuspension in TE and digested with 100μg/ml of DNase free RNase I, the DNA samples were analyzed on 1.2% agarose gels and visualized with ethidium bromide staining. Annexin V analyses were also performed by FACS by incubating indicated cells with FITC-labeled Annexin and propidium iodide following manufacturer's instructions (BD Pharmingen).
Conditional and transient protein expression
cDNA's of survivin, T34A mutant survivin, Cdc2/Cdk1 and cyclin B1 were subcloned into pTRE-shuttle2 and repackaged into replication defective adenovirus in HEK293 cells (Adeno-X tet-off, Clonetech). Proliferating and senescent cells were co-infected with indicated conditional expression adenovirus and control ptTA adenovirus at MOI of 10-20 for 4-6 hours. Doxycycline was added at 300 ng/ml to repress transcription and was withdrawn from selected plates at indicated times to induce gene expression. Myristylated-AKT2 expression adenovirus was prepared according to manufacturer's recommendations (Vector Biolabs).
Colony escape assay
The senescence colony escape assay was performed as reported previously 6. Briefly, H1299 cells were plated at a known cell density and exposed to camptothecin at indicated concentrations for three days. On day four, the cells were recovered in fresh media and characterized for senescence and escape colony formation. Colonies typically emerge 14-21 days following chemotherapy and are scored from triplicate plates. In experiments involving conditional protein over-expression, adenovirus infections were carried out for 4-6 hours one day following camptothecin removal and release of cells into fresh media. Tissue culture media on these plates was replaced every 5-7 days in the presence or absence of doxycycline and/or selected inhibitors, including iso-olomoucine, olomoucine, PNU11245A, and roscovitine (Calbiochem).
TAT-peptides
TAT-survivin peptide: (d) RKKRR-Orn-RRR, b-A-(l)-TFKNWPFLEGCACTPERMAE-COOH, TAT-T34A peptide: (d)RKKRR-Orn-RRR, b-A-(l)-TFKNWPFLEGCAAAAERMAE-COOH and TAT-scramble peptide: (d)RKKRR-Orn-RRR, b-A-(l)-TERLFMAGPCWNKEACPTFG-COOH were synthesized (Biosynthesis). TAT-survivin peptide is derived from the survivin T34 region (amino acids 21 to 40). TAT-T34A peptide is identical to TAT-survivin peptide with the exception of amino acids substitution at positions 33-35 resulting in AAA from CTP. For TAT basic domain, (d)-amino acids were used for synthesis of TAT because these are slowly metabolized leading to prolongation of t1/2 of the compound. Glutamine in TAT region was substituted by ornithine to increase the cell permeation of the TAT- peptides. C-terminal of the TAT-peptides was biotinylated for visualization by immunofluorescence staining.
Generation of recombinant survivin and in vitro Cdc2/Cdk1 kinase reaction
Recombinant survivin was first expressed as a GST-fusion protein from in-frame cloning of the Survivin cDNA into pGEX-4T1 bacterial expression vector (Pharmacia). Following induction, GST was cleaved from the GST-survivin fusion proteins using thrombin per manufacturer established protocol. Either 100 ng of recombinant survivin or histone H1 (Sigma) were used in the Cdc2/Cdk1 kinase assay in presence of 20 ng of dimerized Cdk1-cyclin B (Upstate), 100 μM [32P]-γ-ATP (1μCi), and 15mM MgCl2 in 20mM MOPS, pH 7.2, 25mM β-glycerol phosphate, 5mM EGTA, 1mM sodium orthovanadate, and 1mM dithiothreitol at 37°C at indicated times. Peptides and inhibitors were included at indicated concentrations. Phosphorylated substrates were resolved on 12% TBE polyacrylamide gel and subjected to autoradiography on Cyclone Phosphorimaging System (Packard).
Immunofluorescence staining
Intracellular penetration of TAT-peptides (biotinylated) in H1299 was detected by immunofluorecence microscopy (Nikon eclipse E-800). The cells grown in multi-chamber slides were incubated at 37°C with TAT-survivin, TAT-T34A and TAT-scramble peptides at indicated concentration for 30min. The cells were then fixed in 1% paraformaldehyde, washed in PBS, permeabilized with 0.2% triton X-100, and followed by further incubation with FITC-avidin. After mounting with medium containing 0.2% DAPI and covered with a cover slip, the cells were examined under fluorescence microscope (Nikon).
Image reproduction and processing
Images of gel and digital photographs were generated with Adobe Photoshop 7.0 and Canvas 6.
Results
Survivin expression is sustained during DNA damage, and reaches a nadir during senescence, but is over-expressed in the cells that escape senescence
We have shown previously that 85-90% of H1299 cells arrest at G2/M and assume senescence phenotype following recovery from a 3-day exposure to a moderate dose of camptothecin (CPT) 6. The schema of this system is depicted in Fig. 1A. In these treated H1299 cells, the activation of the DNA damage G2 checkpoint was shown to rapidly trigger inhibitory phosphorylation of Cdc2/Cdk1 6, which is followed by a gradual decline of both Cdc2/Cdk1 and cyclin B1 protein to a nadir level in days 7 to 9 during recovery (Fig. 1B). Survivin level mirrors the protein kinetics of Cdc2/Cdk1 and cyclin B1 such that it is sustained initially during camptothecin exposure and then decreases after withdraw of chemotherapy. Thr34 (T34)-phosphorylation on survivin, previously shown to be mediated by the activated Cdc2/Cdk1 kinase11, 12, closely corresponds to the overall survivin level during this period of time. These alterations during and following camptothecin were not observed for the G1 regulatory protein Cdk2 (Fig. 1B), and were not found for other human IAP family members including X-IAP, C-IAP1 and C-IAP2, nor for Bcl-2 (Fig. 1C). Similar survivin and Cdc2/Cdk1 protein kinetics were found in other cancer cell lines, including the p53-mutated HCT116 colon cancer cell line previously demonstrated to undergo senescence in response to chemotherapy 1 (data not shown).
Fig. 1. Western blot analyses of survivin and related proteins in H1299 cells during camptothecin treatment (CPT), at arrest and in escaped colonies (SE).

A. System: 1.5 × 106 H1299 (or other cell lines) cells were plated and treated with 30-90 nM camptothecin, washed and released into fresh media on day 3. Senescent cells exhibiting senescence-like phenotype (SLP) are observed between days 5-9. Media is then changed every 5-7 days (Δ) before escape colonies emerge between 14-21 days. B. Western blot analyses of survivin, T34-phoshorylated survivin, Cdc2/Cdk1, cyclin B1 and Cdk2 levels during and following withdraw of camptothecin (60nM). C. Western blot analyses of anti-apoptotic protein Bcl2, c-IAP1, c-IAP2 and X-IAP levels during and after camptothecin. D. Western blot analyses of survivin and Cdc2/Cdk1 in five unselected senescence escape (SE) colonies in comparison to the survivin level in untreated H1299 cells (H) and on day 6 (ACS). Density of survivin and Cdc2/Cdk1 protein bands by chemiluminiscence is determined by Quantity one, Chemi-DOC XRT+ normalized to the corresponding β-actin protein band. E. Western blot analyses of survivin protein level as result of si-RNA mediated knockdown of AKT2 and Cdc2/Cdk1. “U” designates untransfected control; “N” designates unrelated RNA control; “si-A” designates si-RNA against AKT-2 and “si-C” designates si-RNA against Cdc2/Cdk1.
We have reported previously that 1 in 104 to 105 H1299 cells arrested in senescence following camptothecin exposure can escape the cell cycle arrest to form colonies 6 (see Fig. 1A). In these “escaped” colonies, the aberrantly up-regulated Cdc2/Cdk1 both promotes cell cycle reentry and is necessary for its viability. We proceeded to examine the level of survivin in these senescence escape (SE) colonies and found that the survivin level is similarly over-expressed (Fig. 1D). In cells derived from five consecutive, unselected SE colonies, survivin level was found to be increased 2 to 4 folds relative to untreated H1299 cells.
Survivin level is regulated by Cdc2/Cdk1
To understand the regulation of survivin, we performed knockdown of Cdc2/Cdk1 and AKT2, since the latter has been shown to be frequently altered in human malignancy, influence chemosensitivity, and up-regulate survivin in response to growth factor stimulation 17, 18 (Fig, 1E). AKT2 therefore represents a possible regulatory pathway for survivin in this setting. Selective knockdown of either AKT2 or Cdc2/Cdk1 was carried out with siRNA to determine their effect on survivin expression. As shown in Fig. 1E, only knockdown of Cdc2/Cdk1 resulted in significant reduction of survivin protein levels both in the presence or absence of camptothecin. Neither siRNA against AKT2 nor control siRNA altered the level of survivin suggesting that survivin is selectively regulated by Cdc2/Cdk1. This regulation appears to occur at the post-transcriptional level as the overall survivin RNA level was found to be largely unaltered (data not shown).
Knockdown of survivin expression during camptothecin promotes apoptosis and reduces ACS
To determine the role of survivin in therapy-induced senescence, we performed knockdown of survivin with sequence specific antisense (AS) oligonucleotide. A twenty base-long phosphothioate oligonucleotide directed against survivin mRNA and a control oligonucleotide with a scrambled sequence (SC) were independently transfected into H1299 cells 24 hours prior to camptothecin treatment. The cells were subsequently followed for the next six days (Fig. 2). After 48 hours, the AS oligonucleotide selectively knocked down survivin in a dose dependent manner such that survivin is reduced to below 10% of its basal level by 400 nM AS oligonucleotide (Fig. 2A). The AS effect was sustained for a minimum of five days (data not shown) and is specific as shown by the unaffected levels of Cdc2/Cdk1 and Cdk2 by either oligonucleotides. We followed the growth of these cells through senescence induction (Fig. 2B). We found that AS transfected cells rapidly underwent cell death within 24 hours of camptothecin treatment, whereas SC treated cells proliferated, arrested and died such that approximately 75% of plated cells remained on day 7. Between 75-80% of the SC-transfected cells were found to exhibit senescence morphology and express SA-β-gal (Fig. 2C). This level is very similar to the 85% of H1299 cells expressing SA-β-gal when treated with camptothecin alone. None of the AS-transfected cells were found to express SA-β-gal or exhibit senescence features, but instead the AS-transfected cells displayed apoptotic morphology after treated with camptothecin (Fig. 2D). Apoptosis was confirmed by the detection of DNA-ladder on agarose gel electrophoresis after 48 hours of camptothecin treatment for AS-oligonucleotide transfected cells (Fig. 2E, right panel). However, DNA fragmentation was not detectable if cells were either not exposed to camptothecin (-CPT, left panel) or transfected with SC-oligonucleotides and treated with camptothecin (SC/+CPT). Finally, the cell cycle profiles of these cells were analyzed by flow cytometry and the apoptotic cell population was quantitated as the sub-G1 fraction (Figure 2F). The knockdown of survivin by AS oligonucleotide resulted in 11.3% apoptotic population after 48 hours of camptothecin, significantly higher than levels found for SC oligonucleotide transfection (4.95%) or camptothecin treatment alone (3.9%). These results suggest that survivin mediates cell survival during chemotherapy treatment by inhibiting apoptosis. This function is a prerequisite to establishing therapy-induced senescence.
Fig. 2. Response of H1299 cells to knockdown of endogenous survivin untreated and treated with camptothecin.

A. Western blot analyses of indicated protein levels in cells transfected either with the SC oligonucleotides (scrambled sequence) or with the AS oligonucleotides (survivin antisense) at indicated concentrations. H1299-U represents levels in untreated proliferating cells. Relative density of survivin protein bands is determined by Quantity one, Chemi-DOC XRT+ normalized to the corresponding β-actin protein level. B. Growth curve of H1299 cells either untransfected or transfected with SC or AS oligonucleotides 16 hour prior to camptothecin treatment (CPT) or DMSO. C. Percentage of cells expressing in situ SA-β-gal on day 7 of H1299 cells transfected with either oligonucleotides (SC or AS) and subsequently treated with CPT or DMSO for 3 day and recovered for 4 days. D. Morphology of cells under light microscopy is shown for cells treated under the specified conditions at day 7. E. Agarose gel electrophoresis of DNA isolated from cells at 48 hours following indicated treatment. U designates untransfected cells. F. Sub-G1 fraction is determined by FACS DNA content analyses in cells transfected with oligonucleotides (SC or AS) and subsequently treated with CPT for 48 hours. Error bars in B, C, and F indicate standard deviations of mean determined from three independent experiments.
Survivin cooperates with Cdc2/Cdk1 to promote escape from ACS
Since both Cdc2/Cdk1 and survivin are aberrantly over-expressed in senescence escape cells, we proceeded to determine the effect of conditionally enforced expression of survivin, Cdc2/Cdk1, and cyclin B1 on the ability of senescent cells to escape growth arrest and form colonies (Fig. 3). We found that senescent H1299 cells are susceptible to adenovirus infection after their release from camptothecin treatment (data not shown). For example, greater than 85% of senescent cells express marker fluorescent protein after a 4-6 hour exposure to a marker adenovirus one day after chemotherapy. We therefore used an adenoviral delivery system to conditionally express survivin, Cdc2/Cdk1 and cyclin B1 in senescent cells during the colony formation assay. In these experiments, camptothecin treated H1299 cells were co-infected 16 hours following camptothecin removal with ptTA adenovirus encoding the tetracycline repressor and the indicated TRE (tetracycline-responsive element)-expression adenovirus encoding the indicated protein. The viral infections were carried out for 4-6 hours before the cells were recovered in fresh media containing 300ng/ml of doxycycline co-repressor or no doxycycline for protein induction. As shown in Fig. 3A, this system resulted in the conditional over-expression of survivin, Cdc2/Cdk1, and cyclin B1 proteins in senescent cells either one at a time or in combination. All the protein expressions are tightly regulated by the presence or the absence of doxycycline (Fig 3A). Ad-EGFP was used for visualization of infectivity and serve as a control for the Western blot analysis.
Fig. 3. Effect of conditional over-expression of Cdc2/Cdk1, survivin and Cyclin B1 either alone or in combination on senescence escape.

A. Western blot analyses of indicated proteins in senescent cells 48 hours following adenoviral-mediated conditional expression in the presence or the absence of doxycycline. B. Colony formation assay in senescent cells conditionally over-expressing proteins from indicated adenoviral vectors. Colonies were counted between days 19-21 following the camptothecin. Standard deviations were determined from three independent experiments. C. The effect of iso-olomucine (50 μM), olomucine (50 μM and 100 μM), roscovitine (10 μM), PNU12455A (1 μM) on escape colony formation in cells ectopically expressing survivin at indicated concentration. Inhibitors were added one day after adenoviral infection. Error bar indicates standard deviations of mean determined from three independent experiments.
The effect of conditional expression of survivin, Cdc2/Cdk1 and cyclin B1 on escape colony formation of senescent H1299 cells is depicted in Fig. 3B. Induction of either survivin or Cdc2/Cdk1 expression alone in senescent cells increased escape colony formation 2 to 2.5 fold over control levels. This finding suggests that either protein can promote the bypass of cell cycle arrest in senescent cells. When survivin and Cdc2/Cdk1 were co-expressed together, colony formation was further increased 3.5 times above control. Ectopic expression of exogenous cyclin B1 either alone or in combination with survivin modestly reduced colony formation implying that high levels of cyclin B1 may be detrimental to senescent cells. The over-expression of mutant T34A survivin, previously reported to be dominant negative, completely abrogated senescence escape. As expected, a constitutively expressed myristylated-AKT2 had no effect on colony formation. Finally, the effects of ectopic survivin expression on colony formation can be abrogated by the Cdc2/Cdk1 inhibitors, olomoucine and roscovitine, added at indicated concentrations one day following adenoviral infection, but not by the Cdk2 inhibitor PNU1124455A (Fig. 3C). These results suggest that survivin functions downstream of Cdc2/Cdk1 to promote survival in therapy-induced H1299 cells and their subsequent cell cycle reentry from senescent arrest. This role is distinct from its function during chemotherapy to suppress the induction of apoptosis.
Small TAT-peptide derived from T34 region on survivin inhibits Cdc2/Cdk1-mediated phosphorylation of survivin
To address the role of phosphorylation at the T34 residue on survivin function during chemotherapy treatment and senescence, we designed a series of peptides with amino acid sequence derived from the T34 domain of survivin. A modified leader sequence derived from the HIV-1 TAT was added to the amino terminus of these peptides to facilitate their intracellular delivery and localization 19. We found that a 20 amino acid survivin peptide with a sequence identical to amino acids 21-40 of survivin (survivin, Fig. 4A) can inhibit the in vitro phosphorylation of recombinant survivin by dimerized cyclin B1-Cdc2/Cdk1 (Fig. 4B). We tested the effect of the peptides between 1 to 10 mM and found that the peptide inhibition of survivin phosphorylation is concentration dependent. This finding along with the phosphorylation of the TAT-survivin peptide by activated Cdc2/Cdk1 kinase (not shown) suggests that the survivin peptide mediates inhibition by competing as a substrate for Cdc2/Cdk1. A second peptide with mutated sequence at amino acids 33-35 CTP→AAA (designated as T34A) also inhibits survivin phosphorylation in vitro at similar concentrations despite lacking the threonine phosphorylation target. A control peptide with scrambled survivin sequence (scramble) did not inhibit survivin phosphorylation. None of the three peptides affected the phosphorylation of histone H1 by Cdc2/Cdk1, suggesting that the peptide inhibition is substrate specific. All three peptides efficiently penetrate the cell membrane and are distributed both in the nucleus and in the cytoplasm of H1299 cells (Fig. 4C).
Fig. 4. Characterization of the TAT-peptides.

A. Sequence of TAT-peptides composing of an eight A.A. modified HIV-1 TAT derived cell penetrating leader peptide followed by a 20 A.A. effector sequence for survivin, mutated survivin (positions 33-35: CTP→AAA), and scrambled sequence. B. Effect of TAT-peptides on the in vitro phosphorylation of survivin and histone H1 by Cdc2/Cdk1. Recombinant survivin protein and histone H1 were phosphorylated by activated Cdc2/Cdk1 alone and in the presence of DMSO. Cdc2/Cdk1 inhibitor olomoucine (Olo) was used at 50 μM. The effects of scramble, survivin, and T34A peptides on the phosphorylation of recombinant survivin and histone H1 were tested at final concentrations of 1, 2.5, 5, and 10 mM. C. Intracellular penetration of the biotinylated TAT-peptides was analyzed by fluorescence microscopy using FITC conjugated strep-avidin.
TAT-survivin peptide inhibits survivin phosphorylation in H1299 cells and mediates apoptosis
Despite high TAT-survivin and TAT-T34A peptide concentrations required to inhibit Cdc2/Cdk1 kinase in vitro, both peptides surprisingly inhibited survivin phosphorylation in H1299 cells in the 1-20 μM range. At 10 μM, TAT-survivin peptide inhibited T34 phosphorylation of survivin in both untreated and camptothecin-treated H1299 cells (Fig. 5A). TAT-T34A peptide only inhibited phosphorylation of survivin in camptothecin-treated H1299 cells. The corresponding intracellular survivin levels remained relatively constant both with TAT-survivin and TAT-T34A treatment independently whether the cells have been exposed to CPT.
Fig. 5. Effect of TAT-peptides on H1299 cells.
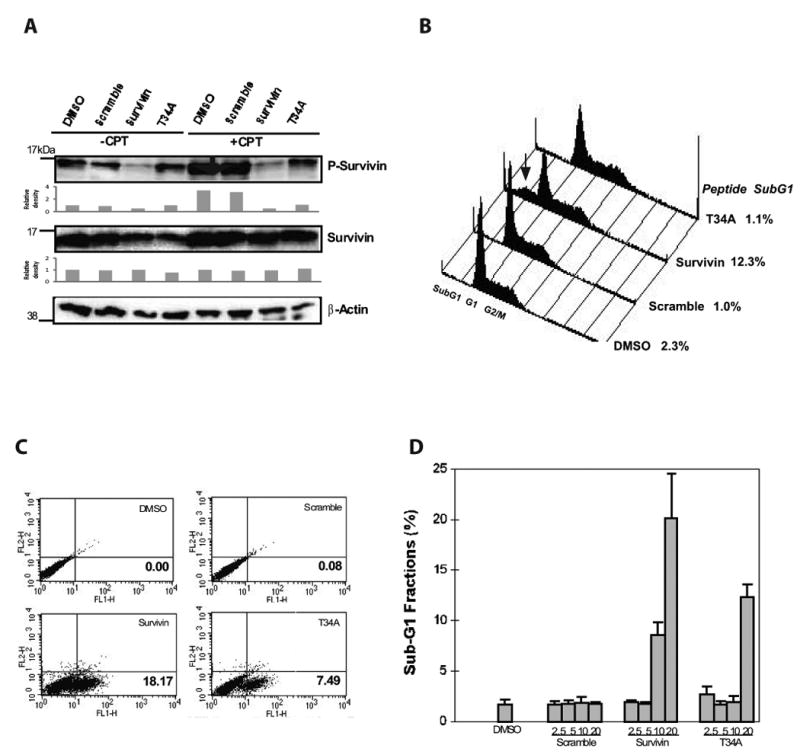
A. Levels of T34 phospho-survivin and survivin in H1299 cells treated with specified TAT-peptides by Western blot analysis. Phospho-survivin is determined following immunoprecipitation of survivin protein in lysates. Relative density of survivin protein level is determined by Quantity one, Chemi-DOC XRT+ using β-actin protein level as loading control. B. Composite FACS DNA histogram of H1299 cells treated with specified TAT-peptides. Sub-G1 fraction measured by FACS at 48 hours of peptides treatment is reported. Arrow indicates sub-G1 fraction. C. Apoptosis assessed by FITC Annexin staining. D. Effect of TAT-peptides concentration on H1299 Sub-G1 fraction. Error bar indicates standard deviation of means from three independent experiments.
We proceeded to examine the peptide effect on cell survival. FACS DNA content analysis showed that TAT-survivin peptide induces apoptosis in untreated H1299 cells (Fig. 5B and C). At 48 hours following treatment with the TAT-peptide, sub-G1 fraction was noted to be significantly increased from a baseline of 1% to 12.1% in TAT-survivin peptide treated cells, whereas TAT-T34A peptide did not appreciably promote increase of the sub-G1 fraction (Fig. 5B). This apoptosis response is verified by the annexin assay, where TAT-survivin peptide induced 18% of annexin stained cells at 48 hours whereas TAT-T34A peptide induced 7.5% annexin staining (Fig. 5C). Using sub-G1 fraction as a measure for apoptosis, we examined the concentration dependence of apoptosis induction by the TAT-peptide. We found that TAT-survivin peptide effectively promotes apoptosis at concentrations above 5 μM, whereas TAT-T34A peptide elicits apoptosis at 20 μM concentration. We have also found that these peptides exert similar effects at these concentrations in other cancer cell lines, including human colon carcinoma cell line HCT116 (data not shown). These results suggest that T34 phosphorylation of survivin is critical to the survival function of cells at baseline and during chemotherapy. Interference of T34 phosphorylation promotes apoptosis.
Interference of survivin phosphorylation is detrimental to cells escaping from senescence
We previously demonstrated that colonies which form following exposure to chemotherapy resemble senescent cells in their morphology, gene expression profile, and expression of SA-β-galactosidase marker, strongly suggesting that they have been able to bypass the senescence program 6. Moreover, the survival of the “senescence escape” cells depends on Cdc2/Cdk1, such that knockdown of Cdc2/Cdk1 or pharmacological inhibition of its kinase activity results in rapid cell death. Given our findings that survivin is up-regulated in senescence escape colonies (Fig. 1D) and survivin over-expression promotes escape colony formation (Fig. 3), we used the TAT-peptides to determine whether interference of T34 survivin phosphorylation affect the viability of senescent cells and their ability to bypass cell cycle arrest (Fig. 6). Initially, we treated cells derived from five consecutive senescence escape colonies characterized previously in Fig. 1D. As shown in Fig. 6A, cells derived from senescence escape colony 41 and senescence escape colony 42 suffered rapid cell death in response to TAT-survivin peptide and to a lesser extent with TAT-T34A peptide. This apoptotic effect can be detected as early as 16 hours (not shown). FACS analysis of senescence escape colony 41 cells demonstrates substantial induction of sub-G1 fraction by both TAT-survivin and TAT-T34A peptides at 48 hours (Fig. 6B left panel). The effect of TAT-T34A peptide is attenuated as compared with the effect of TAT-survivin peptide (34% to 74.7% in senescence escape colony 41). The magnitude of TAT-survivin peptide effect in senescence escape cells from four out of five escape colonies is two to six folds greater as compared to control H1299 cells (right panel). In senescence escape colony 43, the apoptotic response is similar to the response in H1299 cells.
Fig. 6. Effect of TAT-peptides on viability of escape colonies and colony formation.

A. Phase-contrast microscopy (20×) of untreated cells (H1299) and cells derived from two independent senescence escape colonies (SE41 and SE43) treated with indicated TAT-peptides after 48 h. B. (left) Representative composite FACS DNA histogram of cells derived from senescence escape colonies (SE 41) treated with specified TAT-peptides. (right) Effect of TAT-peptides on sub-G1 fraction of H1299 cells and cells derived from indicated escape colonies. C. Effect of TAT-peptides at indicated concentrations on colony formation of H1299 cells following CPT. Colony formation is reported as percentage of colonies derived from H1299 cells treated with DMSO (20 μl) alone. Olomoucine (Olo) is added to 100 μM final concentration. Error bars indicate standard deviations of mean calculated based on three independent experiments.
Finally, we tested the TAT-peptides for the ability to interfere with senescence escape colony formation (Fig. 6C). In this assay, TAT-peptides were added at indicated concentrations 24 hour following withdraw of chemotherapy. Not surprisingly, TAT-survivin peptide effectively inhibits escape colony formation in a concentration dependent manner. At 10 μM, the TAT-survivin peptide completely blocked escape colony formation. This effect is significantly more pronounced in comparison to 100 μM of Cdc2/Cdk1 inhibitor olomoucine, previously shown to block escape colony formation in H1299 cells. TAT-T34A peptide also reduced colony formation by approximately 50%, which is consistent with the data that shows its reduced ability to inhibit survivin phosphorylation in vitro and in cells (Figs. 5A and 6). TAT-survivin peptide also effectively blocks escape in therapy-induced HCT116 cells (data not shown). Collectively, these results suggest that survivin functions downstream of Cdc2/Cdk1 to mediate survival in cells that have bypassed therapy-induced senescence. This function of survivin is impacted by the phosphorylation of the T34 residue. Interference of T34A phosphorylation on survivin can therefore block senescence escape.
Discussion
The pleiotropic response of tumors in vivo to chemotherapy includes accelerated cellular senescence, a state of prolonged cell cycle arrest associated with features of physiological senescence. Reversibility of ACS fundamentally distinguishes this response from programmed cell death (apoptosis and autophagy) and mitotic catastrophe as cells enter a sustained period of viability and replicative arrest. In this state, cells are either destined for cell death or the eventual bypass of senescence and cell cycle reentry. Cell fate in ACS is therefore likely an important determinant of cancer treatment outcome. To define mechanisms underlying senescence reversibility, our studies in H1299 cells have previously focused on the role of Cdc2/Cdk1 in senescence and escape following chemotherapy. We previously showed that escaped H1299 cells from chemotherapy-induced ACS frequently over-express Cdc2/Cdk1, and that elevated Cdc2/Cdk1 level can both promote cell cycle reentry and is essential to the viability of senescent cells 6. To further understand how Cdc2/Cdk1 mediates these biological effects, we have extended our study to survivin, a known substrate of Cdc2/Cdk1 kinase. In this present report, we show that survivin functions immediately downstream as a mediator of the Cdc2/Cdk1 survival signal and that survivin cooperates with Cdc2/Cdk1 to determine the fate and reversibility of cells in senescence.
Apart from its principal function as the mitotic kinase, Cdc2/Cdk1 has been implicated in a complex relationship of cell survival and death signaling 20. Cdc2/Cdk1 activity may be pro-apoptotic. For example, aberrant expression and activation of Cdc2/Cdk1 in apoptosis has been described for breast cancer cell line and hematopoietic cell lines 21 22. In these instances, genetic and pharmacological modulation of Cdc2/Cdk1 inhibits cell death. Unscheduled Cdc2/Cdk1 activation also appears to activate caspases and induce apoptosis 23. Yet, genetic evidence from disruption of Drosophila lats gene resulting in deregulated p34cdc2 kinase activity suggests Cdc2/Cdk1 may repress apoptosis as well 24. In this context LATS may function as a tumor suppressor. In its absence, increased cell proliferation and tumor formation was found, whereas enforced expression of human LATS1 inhibited Cdc2/Cdk1 kinase activity leading to mitotic arrest and apoptosis. Additionally, Cdc2/Cdk1 appeared to be involved in cell survival during the mitotic checkpoint where the protein physically associates with and phosphorylates survivin thereby enhancing survivin cytoprotection and stability11. Survivin elevation in cells treated with spindle microtubule poison paclitaxel is necessary for cell viability, as the disruption of survivin phosphorylation results in massive apoptosis. In each of these two instances, Cdc2/Cdk1 survival and apoptotic function appear to be influenced by a co-regulatory or an effector protein.
As shown here in H1299 cells, survivin expression corresponds to the expression of Cdc2/Cdk1 and cyclin B1 during and following release from camptothecin and its level is regulated by Cdc2/Cdk1 post- transcriptionally. The interplay of Cdc2/Cdk1 and survivin defines cell fate immediately following DNA damage and during senescence. Initially, chemotherapy elicits a DNA damage leading to Cdc2/Cdk1 inactivation through inhibitory phosphorylation 6; yet both Cdc2/Cdk1 and survivin levels remained sustained throughout the exposure to chemotherapy. During this time, knockdown of survivin causes rapid apoptosis and a complete elimination of the senescence response. These results confirm the pro-survival function of the protein and are consistent with the findings reported by others, in which antisense targeting of survivin has been invariably associated with spontaneous apoptosis 25, enhancement of chemotherapy-induced cell death 26, and antitumor activity in vivo 27. Survivin function is also regulated by Cdc2/Cdk1 through phosphorylation at Thr34 as interference with this event even in the absence of DNA damage induces apoptosis. Survivin therefore inhibits baseline apoptosis during normal proliferation of H1299 cells and its function is a prerequisite for therapy-induced senescence.
In senescence, the cell cycle arrest is reinforced by a low level of Cdc2/Cdk1 and cyclin B1. The low levels of survivin imply that it is not required for survival of the senescent cells and the overwhelming majority of these are destined for cell death. Rare cells however retain cell cycle reversibility and their escape from proliferative arrest coincides with high levels of survivin and Cdc2/Cdk1 found in cells derived from the escaped colonies. In these cells, survivin cooperates with Cdc2/Cdk1 to promote escape from ACS. This conclusion is supported by augmentation of escape when survivin is conditionally over-expressed either alone or in conjunction Cdc2/Cdk1 and by the abrogation of escape when Cdc2/Cdk1 mediated survivin phosphorylation is inhibited by the TAT-survivin peptide. Importantly, cells derived from escaped colonies demonstrate several-fold greater sensitivity to TAT-survivin peptide. This finding suggests that the viability of escape cells is dependent on the function of phosphorylated survivin. This observation further explains the exquisite dependence of escape cells on Cdc2/Cdk1 that we have reported previously 6. In this context Cdc2/Cdk1 appears to dominantly promote survival by mediating survivin phosphorylation.
Increased survivin levels have been described in high frequencies in many types of human cancers. Survivin over-expression has been reported in 85-96% of lung cancer specimens, 100% of colon adenocarcinomas, 71% of prostate adenocarcinomas, 80% of glioblastomas and nearly 100% of laryngeal carcinomas 28-31. Its expression, particularly in the nuclei of these tumors, appears to be associated with unfavorable clinical outcome 32-34. Survivin level has also been shown to correlate with therapy resistance in some clinical settings. Survivin has therefore become a popular target for several novel anticancer strategies 35.
Pharmacological approaches using agents such as histone deacetylase inhibitors, mitogen-activated protein kinase, and cyclin-dependent kinase lack target specificity as survivin level and function are indirectly modulated 12, 36, 37. Their use may be complicated by off-target cellular and physiological effects. Gene targeting based on small interfering RNA and antisense oligonucleotides against survivin is target specific. However, their use is limited by short duration of action and by the persistent challenge of effective drug delivery in clinical settings 15, 38, 39. Survivin proteins, either wild-type or T34A, have been used in proof of principle demonstrations. For example, survivin proteins tagged with HIV-derived TAT delivery sequence have been shown to induce apoptosis in YUSAC2 cells and to result in tumor response when it is given as intratumoral injection in mice implanted with YUSAC2 xenografts 40. Here we show that a short TAT-survivin peptide can specifically interfere with Cdc2/Cdk1 mediated survivin phosphorylation and the peptide induces apoptosis in both untreated and senescence escape p53-deficient cancer cells. Phosphorylation interference may be a more potent and direct mechanism of disrupting survivin function in cells since TAT-survivin peptide was found to induce apoptosis in both untreated and camptothecin treated H1299 cells while survivin antisense oligonucleotide only induces apoptosis in camptothecin treated cells. The advantage of using TAT-survivin peptide in antitumor approaches is that the peptide is highly permeable, target specific and active at relatively low concentrations. The peptide used here is slowly metabolized owing to the (d)-amino acids modification in the synthesis of TAT basic domain. TAT-survivin peptide could be used during chemotherapy to favor apoptosis as the dominant response. The peptide could also be considered in the adjuvant setting to enforce irreversibility of therapy-induced senescence and to target senescence escape as a mechanism of therapy resistance. These approaches to manipulate survivin function in chemotherapy response warrant further in vivo testing.
Acknowledgments
We thank Dr. Liu Yang for his critical reading of our manuscript and his insightful suggestions. This work is supported by the National Cancer Institute (R01- CA113892-01 Wu).
Abbreviations
- ACS
accelerated cellular senescence
- SA-β-gal
senescence associated β-galactosidase
- IAP
inhibitor of apoptosis protein
- FACS
fluorescence-activated cell sorting
- CPT
camptothecin
References
- 1.Chang B, Broude E, Dokmanovic M, Zhu H, Ruth A, Xuan Y, Kandel E, Lausch E, Christov K, Roninson I. A senescence-like phenotype distinguishes tumor cells that undergo terminal proliferation arrest after exposure to anticancer agents. Cancer Res. 1999;59:3761–7. [PubMed] [Google Scholar]
- 2.Mooi W, Peeper D. Oncogene-induced cell senescence--halting on the road to cancer. N Engl J Med. 2006;355:1037–46. doi: 10.1056/NEJMra062285. [DOI] [PubMed] [Google Scholar]
- 3.Dimri G, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano E, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Elmore L, Rehder C, Di X, McChesney P, Jackson-Cook C, Gewirtz D, Holt S. Adriamycin-induced senescence in breast tumor cells involves functional p53 and telomere dysfunction. J Biol Chem. 2002;277:35509–15. doi: 10.1074/jbc.M205477200. [DOI] [PubMed] [Google Scholar]
- 5.Chang B, Xuan Y, Broude E, Zhu H, Schott B, Fang J, Roninson I. Role of p53 and p21waf1/cip1 in senescence-like terminal proliferation arrest induced in human tumor cells by chemotherapeutic drugs. Oncogene. 1999;18:4808–18. doi: 10.1038/sj.onc.1203078. [DOI] [PubMed] [Google Scholar]
- 6.Roberson R, Kussick S, Vallieres E, Chen S, Wu D. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005;65:2795–803. doi: 10.1158/0008-5472.CAN-04-1270. [DOI] [PubMed] [Google Scholar]
- 7.Christov K, Shilkaitis A, Kim E, Steele V, Lubet R. Chemopreventive agents induce a senescence-like phenotype in rat mammary tumours. Eur J Cancer. 2003;39:230–9. doi: 10.1016/s0959-8049(02)00497-5. [DOI] [PubMed] [Google Scholar]
- 8.Roninson I, Broude E, Chang B. If not apoptosis, then what? Treatment-induced senescence and mitotic catastrophe in tumor cells. Drug Resist Updat. 2001;4:303–13. doi: 10.1054/drup.2001.0213. [DOI] [PubMed] [Google Scholar]
- 9.te Poele R, Okorokov A, Jardine L, Cummings J, Joel S. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res. 2002;62:1876–83. [PubMed] [Google Scholar]
- 10.Li F. Survivin study: what is the next wave? J Cell Physiol. 2003;197:8–29. doi: 10.1002/jcp.10327. [DOI] [PubMed] [Google Scholar]
- 11.O'Connor D, Grossman D, Plescia J, Li F, Zhang H, Villa A, Tognin S, Marchisio P, Altieri D. Regulation of apoptosis at cell division by p34cdc2 phosphorylation of survivin. Proc Natl Acad Sci U S A. 2000;97:13103–7. doi: 10.1073/pnas.240390697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wall N, O'Connor D, Plescia J, Pommier Y, Altieri D. Suppression of survivin phosphorylation on Thr34 by flavopiridol enhances tumor cell apoptosis. Cancer Res. 2003;63:230–5. [PubMed] [Google Scholar]
- 13.Uren A, Wong L, Pakusch M, Fowler K, Burrows F, Vaux D, Choo K. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–28. doi: 10.1016/s0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 14.Wheatley S, Henzing A, Dodson H, Khaled W, Earnshaw W. Aurora-B phosphorylation in vitro identifies a residue of survivin that is essential for its localization and binding to inner centromere protein (INCENP) in vivo. J Biol Chem. 2004;279:5655–60. doi: 10.1074/jbc.M311299200. [DOI] [PubMed] [Google Scholar]
- 15.Kuo P, Liu H, Chao J. Survivin and p53 modulate quercetin-induced cell growth inhibition and apoptosis in human lung carcinoma cells. J Biol Chem. 2004;279:55875–85. doi: 10.1074/jbc.M407985200. [DOI] [PubMed] [Google Scholar]
- 16.Gong J, Traganos F, Darzynkiewicz Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal Biochem. 1994;218:314–9. doi: 10.1006/abio.1994.1184. [DOI] [PubMed] [Google Scholar]
- 17.Dan H, Jiang K, Coppola D, Hamilton A, Nicosia S, Sebti S, Cheng J. Phosphatidylinositol-3-OH kinase/AKT and survivin pathways as critical targets for geranylgeranyltransferase I inhibitor-induced apoptosis. Oncogene. 2004;23:706–15. doi: 10.1038/sj.onc.1207171. [DOI] [PubMed] [Google Scholar]
- 18.Ohashi H, Takagi H, Oh H, Suzuma K, Suzuma I, Miyamoto N, Uemura A, Watanabe D, Murakami T, Sugaya T, Fukamizu A, Honda Y. Phosphatidylinositol 3-kinase/Akt regulates angiotensin II-induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ Res. 2004;94:785–93. doi: 10.1161/01.RES.0000121103.03275.EC. [DOI] [PubMed] [Google Scholar]
- 19.Dietz G, Bähr M. Delivery of bioactive molecules into the cell: the Trojan horse approach. Mol Cell Neurosci. 2004;27:85–131. doi: 10.1016/j.mcn.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 20.Castedo M, Perfettini J, Roumier T, Kroemer G. Cyclin-dependent kinase-1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287–93. doi: 10.1038/sj.cdd.4401130. [DOI] [PubMed] [Google Scholar]
- 21.Shen S, Huang T, Jee S, Kuo M. Taxol-induced p34cdc2 kinase activation and apoptosis inhibited by 12-O-tetradecanoylphorbol-13-acetate in human breast MCF-7 carcinoma cells. Cell Growth Differ. 1998;9:23–9. [PubMed] [Google Scholar]
- 22.Meikrantz W, Schlegel R. Suppression of apoptosis by dominant negative mutants of cyclin-dependent protein kinases. J Biol Chem. 1996;271:10205–9. doi: 10.1074/jbc.271.17.10205. [DOI] [PubMed] [Google Scholar]
- 23.Harvey K, Lukovic D, Ucker D. Caspase-dependent Cdk activity is a requisite effector of apoptotic death events. J Cell Biol. 2000;148:59–72. doi: 10.1083/jcb.148.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tao W, Zhang S, Turenchalk G, Stewart R, St John M, Chen W, Xu T. Human homologue of the Drosophila melanogaster lats tumour suppressor modulates CDC2 activity. Nat Genet. 1999;21:177–81. doi: 10.1038/5960. [DOI] [PubMed] [Google Scholar]
- 25.Li F, Altieri D. The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res. 1999;59:3143–51. [PubMed] [Google Scholar]
- 26.Olie R, Simões-Wüst A, Baumann B, Leech S, Fabbro D, Stahel R, Zangemeister-Wittke U. A novel antisense oligonucleotide targeting survivin expression induces apoptosis and sensitizes lung cancer cells to chemotherapy. Cancer Res. 2000;60:2805–9. [PubMed] [Google Scholar]
- 27.Kanwar J, Shen W, Kanwar R, Berg R, Krissansen G. Effects of survivin antagonists on growth of established tumors and B7-1 immunogene therapy. J Natl Cancer Inst. 2001;93:1541–52. doi: 10.1093/jnci/93.20.1541. [DOI] [PubMed] [Google Scholar]
- 28.Shimizu T, O'Connor P, Kohn K, Pommier Y. Unscheduled activation of cyclin B1/Cdc2 kinase in human promyelocytic leukemia cell line HL60 cells undergoing apoptosis induced by DNA damage. Cancer Res. 1995;55:228–31. [PubMed] [Google Scholar]
- 29.Peterson Y, Kelly P, Weinbaum C, Casey P. A novel protein geranylgeranyltransferase-I inhibitor with high potency, selectivity, and cellular activity. J Biol Chem. 2006;281:12445–50. doi: 10.1074/jbc.M600168200. [DOI] [PubMed] [Google Scholar]
- 30.Fritz G, Kaina B. Rho GTPases: promising cellular targets for novel anticancer drugs. Curr Cancer Drug Targets. 2006;6:1–14. [PubMed] [Google Scholar]
- 31.Datta S, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg M. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–41. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 32.Cardone M, Roy N, Stennicke H, Salvesen G, Franke T, Stanbridge E, Frisch S, Reed J. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–21. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 33.Mayo L, Donner D. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Graaf M, Richel D, van Noorden C, Guchelaar H. Effects of statins and farnesyltransferase inhibitors on the development and progression of cancer. Cancer Treat Rev. 2004;30:609–41. doi: 10.1016/j.ctrv.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 35.Kawata S, Yamasaki E, Nagase T, Inui Y, Ito N, Matsuda Y, Inada M, Tamura S, Noda S, Imai Y, Matsuzawa Y. Effect of pravastatin on survival in patients with advanced hepatocellular carcinoma. A randomized controlled trial Br J Cancer. 2001;84:886–91. doi: 10.1054/bjoc.2000.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Schepper S, Bruwiere H, Verhulst T, Steller U, Andries L, Wouters W, Janicot M, Arts J, Van Heusden J. Inhibition of histone deacetylases by chlamydocin induces apoptosis and proteasome-mediated degradation of survivin. J Pharmacol Exp Ther. 2003;304:881–8. doi: 10.1124/jpet.102.042903. [DOI] [PubMed] [Google Scholar]
- 37.O'Connor D, Wall N, Porter A, Altieri D. A p34(cdc2) survival checkpoint in cancer. Cancer Cell. 2002;2:43–54. doi: 10.1016/s1535-6108(02)00084-3. [DOI] [PubMed] [Google Scholar]
- 38.Grossman D, McNiff J, Li F, Altieri D. Expression and targeting of the apoptosis inhibitor, survivin, in human melanoma. J Invest Dermatol. 1999;113:1076–81. doi: 10.1046/j.1523-1747.1999.00776.x. [DOI] [PubMed] [Google Scholar]
- 39.Ling X, Li F. Silencing of antiapoptotic survivin gene by multiple approaches of RNA interference technology. Biotechniques. 2004;36:450–4. 6–60. doi: 10.2144/04363RR01. [DOI] [PubMed] [Google Scholar]
- 40.Yan H, Thomas J, Liu T, Raj D, London N, Tandeski T, Leachman S, Lee R, Grossman D. Induction of melanoma cell apoptosis and inhibition of tumor growth using a cell-permeable Survivin antagonist. Oncogene. 2006;25:6968–74. doi: 10.1038/sj.onc.1209676. [DOI] [PMC free article] [PubMed] [Google Scholar]