

Abstract
We report a novel mutation (erlong, erl) of the cadherin 23 (Cdh23) gene in a mouse model for DFNB12 characterized by progressive hearing loss beginning from post-natal day 27 (P27). Genetic and sequencing analysis revealed a 208T>C transition causing an amino acid substitution (70S-P). Caspase expression was up-regulated in mutant inner ears. Hearing was preserved (up to 35-dB improvement) in pan-caspase inhibitor Z-VAD-FMK-treated mutants compared to untreated mutants (P < 0.05). Outer hair cell (OHC) loss in the cochleae of Z-VAD-FMK-treated mutants was significantly reduced compared to those of untreated mice. Thus, the erl mutation can lead to hearing loss through apoptosis. This is the first genetic mouse model of hearing loss shown to respond to otoprotective drug therapy. The short interval from initial hearing loss to deafness (P27-P90) makes this model ideal for screening and validating otoprotective drugs.
Keywords: mouse model, Cdh23, mutation, hearing loss, apoptosis, Z-VAD-FMK
Introduction
Cadherin 23 (CDH23) is encoded by a large Cdh23 gene containing 69 exons spanning at least 350 kb in the mouse. It is a 365 kDa transmembrane protein composed of 27 extracellular cadherin (EC) repeats, a single-pass transmembrane region, and a cytoplasmic region and is an important component of the hair cell tip-link in the organ of Corti.1–3 Mutations in the CDH23 gene in humans have been linked to age-related hearing loss (AHL),4 Usher syndrome type 1 subtype D (USH1D),5, 6 and a form of nonsyndromic autosomal recessive deafness designated as DFNB12.6 Different mutations in the CDH23 gene can cause either syndromic or nonsyndromic forms of deafness: Only missense mutations of CDH23 have been observed in families with nonsyndromic deafness, whereas nonsense, frameshift, splice-site, and missense mutations have been identified in families with Usher syndrome (syndromic).7 Age-related hearing loss (AHL) is a characteristic of the widely used C57BL/6J mouse strain.8 AHL starts with a moderate hearing impairment in 1-year-old C57BL/6J mice, and progresses to complete hearing loss with age. Linkage studies associated AHL with a locus named ahl.9 A single-nucleotide polymorphism in exon 7 of Cdh23 was significantly associated with AHL and the deafness modifier mdfw (modifier of deaf waddler). The hypomorphic Cdh23753A (synonymous with Cdh23ahl) allele causes in-frame skipping of exon 7.4 Histological analysis correlated AHL with a gradual loss of hair cells, spiral ganglion cells and degeneration of fibrocytes in the spiral ligament.10, 11 Most other mouse models of point mutation in the Cdh23 gene are characterized by congenital deafness with circling behavior.1, 2, 12, 13 Each of these mutations leads to the loss of functional domains. In four waltzer alleles (v2J, v6J vAlb, vngt), loss of functional protein has been reported to disrupt the highly organized stereocilia bundle of hair cells in the cochlea and the vestibule during late embryonic/early postnatal development.1, 13, 14 In all previously characterized Cdh23 mouse models, the mice are either deaf at birth,2 exhibit severe very early onset hearing loss15 or very late-onset hearing loss with slow progression.9 The deaf (v-df) mutant was reported by Deol MS, that v-df mice may be deaf from the beginning or may be able to hear for a few days before weaning but otherwise behave normally16. Although the final pathological outcome is consistently observed as inner ear hair cell loss2, none of these models provide an easily manageable time interval for evaluating pathophysiological changes and/or for screening and testing drug therapies. Here, we introduce a new mouse model of DFNB12 which is characterized by progressive hearing loss starting at P27 and progressing to deafness by p100. This is an ideal time window for testing otoprotective drugs. Nevertheless, we have found that apoptosis plays a major role in hair cell loss in this mouse model. Most importantly, we report here that a pan-caspase inhibitor not only preserved inner ear hair cells but also prevented hearing loss by up to 35 dB in the mutant mice.
Materials and methods
Mice, genetic linkage cross, and DNA sequencing
Mice were originally housed in The Jackson Laboratory (Bar Harbor, Maine) research facilities and all procedures were approved by the Institutional Animal Care and Use Committee (protocols 5U01NS041215 & R01DC007392). Mice were then relocated to Case Western Reserve University (CWRU, Cleveland, Ohio). Further studies were conducted in accordance with the principles set forth in the Guide for the Care and Use of Laboratory Animals, Institute of Laboratory Animal Resources, and were approved by the Case Western Reserve University of the Health Sciences Institutional Animal Use and Care Committee (R01DC009246). ENU-induced (N-ethyl-N-nitrosourea, C3H6N3O2) mutagenesis was performed as part of a large-scale screening program for new mutants at the Jackson Laboratory using predominantly the C57BL/6J (B6) mouse strain (The Jackson Laboratory Neuromutagenesis Facility website: http://nmf.jax.org). A detailed protocol is described at http://nmf.jax.org/protocols/genetics_scheme.html. As hearing impairment was the only measurable phenotype of erl mutant mice, we had to trace phenotype by testing hearing with ABR thresholds for maintaining colonies and performing all experiments. Genetic intercross generated 13 affected ((B6XC3H/HeJ)F1-erl/+)X((B6XC3H/HeJ)F1-erl/+) F2 progeny with elevated ABR thresholds as shown in Supplementary Table 1. A DNA pooling method for gene mapping was used as previously described.17, 18 Genomic DNA sequencing to identify the alteration in the erl mutant mouse was performed as follows: Genomic DNA was prepared from tail tips of mice. Briefly, 2-mm mouse tail tips were digested with 0.3 ml of 50 mM NaOH in a 0.5 ml Eppendorf tube at 95°C for 10 min. 26 μl of 1M Tris-HCl was then added to each tube. The mixtures were centrifuged at 12,000×g for 5 min and the DNA concentration in supernatants was measured using a BioPhotometer (Eppendorf AG, Hamburg, Germany). Forty-eight pairs of PCR primers spanning exons of the Cdh23 gene were designed using the Primer3 freeware (http://frodo.wi.mit.edu/primer3/) according to the exon sequence of the Cdh23 gene in the Ensemble Mouse Genome Server (www.ensembl.org), and synthesized by Integrated DNA Technologies, Inc. (San Diego, CA, USA). PCR for comparative DNA analysis between Cdh23erl/erl and Cdh23ahl/ahl mice was performed according to the Tm of the primers and the expected product sizes. PCR products were purified with the QIAquick PCR Purification kit (Qiagen, Inc. Valencia, CA, USA). DNA sequencing was performed using the same primers as for DNA amplification and then run on an ABI Applied Biosystems 3730 DNA Analyzer (Life Technologies Corp., Carlsbad, CA, USA).
To confirm the mutation and to identify potential aberrant exon splicing, Cdh23erl/erl and Cdh23ahl/ahl mice at 2 weeks of age were used for RNA extraction and RT-PCR. After mice were sacrificed under anesthesia (avertin 5 mg/10 g), the inner ears were quickly removed. Total RNA (DNA-free) was prepared using the pure-LinkTM Micro-to-Midi Total RNA Purification System (Invitrogen, Carlsbad, CA, USA). cDNA synthesis was carried out using the SuperScript™ First-Strand Synthesis System (Catalog No. 11904-018). PCR primers (CdhmF and CdhmR in table 2) were designed in exon 2 and exon 4 yielding a 228-bp PCR product containing exon 3 and its flanking regions from exons 2 and 4.
Table 2.
Sequences for PCR/RT-PCR primers
| ID | Sequence | Product Size (bp) | References |
|---|---|---|---|
| ex3-F1 | CCTGACCCTCTCTGTTTTCTTTTCC | ||
| ex3-R1 | TGCTTACCCAATGCTACTCCTGGC | 406 | |
| ex3m-F | CAGGTTCTTCTGTGACCCAGC | ||
| ex3m-R | CCAAAAGATGGTGAGCCACCT | 219 | |
| GAPDH-F | AACTTTGGCATTGTGGAAGG | 351 | 41 |
| GAPDH-R | GGAGACAACCTGGTCCTCAG | ||
| Cdh23m-F | TGACACGTACCTGCTCATCA | ||
| Cdh23m-R | CCTTGGTGGTCACTGACAGA | ||
| caspase3-F | TGTCATCTCGCTCTGGTACG | 201 | 41 |
| caspase3-R | AAATGACCCCTTCATCACCA | ||
| caspase8-F | ATGGCGGAACTGTGTGACTCG | 345 | 42 |
| caspase8-R | GTCACCGTGGGATAGGATACAGCA | ||
| caspase9-F | CCTAGTGAGCGAGCTGCAAG | 232 | |
| caspase9-R | ACCGCTTTGCAAGAGTGAAG | ||
| caspase12-F | GCCCATGTGGAGACAGATTT | 206 | |
| caspase12-R | ATAGTGGGCATCTGGGTCAG | ||
| m-calpain-F | GAGGTGGTGGTGGACGACAG | 390 | 38 |
| m-calpain-R | TTTCTGCAGGCTTCCTGAAC | ||
| μ-calpain-F | CTACGGAACTGCTGTCAAAC | 727 | 38 |
| μ-calpain-R | TCCATCTTGACCCTCAGCTG | ||
| TNFα-F | CCACCACGCTCTTCTGTCTAC | 303 | |
| TNFα-R | CCTTGAAGAGAACCTGGGAGT |
Auditory-evoked brainstem response
ABR was measured at various intervals for Cdh23erl/erl and Cdh23ahl/ahl mice (at ages over P14). A computer-aided evoked potential system (Intelligent Hearing Systems, Miami, FL, USA) was used to test mice for ABR thresholds as previously described.8 Briefly, mice were anesthetized and body temperature maintained at 37–38°C by placing them on a heating pad in a sound-attenuating chamber. Subdermal needle electrodes were inserted at the vertex of (active) and ventrolaterally to (reference) the right ear and to the left ear (ground). Clicks, and 8-, 16- and 32-kHz tone-bursts were respectively channeled through plastic tubes into the animal’s ear canals. The amplified brainstem responses were averaged by a computer and displayed on a computer screen. Auditory thresholds were obtained for each stimulus by reducing the sound pressure level (SPL) at 10-dB steps and finally at 5-dB steps up and down to identify the lowest level at which an ABR pattern could be recognized. ABR threshold values above 55 (for click stimulus), 40 (for 8 kHz), 35 (for 16 kHz), or 60 (for 32 kHz) dB SPL were considered to be hearing-impaired.8
Distortion product oto-acoustic emission (DPOAE)
To test the function of outer hair cells of different mice at different time points, we used the IHS Smart EP 3.30 USBez Software (Intelligent Hearing Systems, Miami, FL, USA) for DPOAE measurement, which was conducted for pure tones from 2 to 36 KHz.19 An Etymotic 10B+ (Etymotic Research, Inc., Elk Grove Village, IL, USA) probe was inserted into the external ear canal and used with two different types of transducers depending on the range of the stimulation frequency. For frequencies ranging from 2 to 16 kHz, an Etymotic ER2 stimulator was used and for frequencies ranging from 16 to 30 kHz, an IHS high frequency transducer was used. Stimulus response signals were sampled at a rate of 128 kHz using a 16-bit D/A converter; L1 and L2 amplitudes were set to the same level. Frequencies were acquired with an F2-F1 ratio of 1.22. The stimuli were presented starting from the lowest frequencies tested and increasing to the highest frequencies tested. Five stimulation levels ranging from 65 to 25 dB SPL in 10-dB steps were used.
Histological analyses of inner ears
Histological analyses of inner ears were performed following the methods described previously.20 Briefly, anesthetized mice were perfused through the left ventricle of the heart with phosphate-buffered saline (PBS) followed by Bouin’s (for H&E staining) or 4% paraformaldehyde (for all others) fixative. For microscopic analysis of cross-sections, inner ears from Cdh23erl/erl and Cdh23ahl/ahl mice were dissected, perfused with fixative, immersed in same for 48 h, decalcified with Cal-EX solution for 6 h, and embedded in paraffin. Sections (5 μm) were cut, mounted on glass slides and counterstained in hematoxylin/eosin (H&E).
Cytocochleograms
Cytocochleograms were obtained by a modified method as described previously.20 Briefly, the organ of Corti was carefully microdissected out and mounted in glycerin on glass slides. The surface preparations were stained for F-actin with Alexa Fluor 568 conjugated to phalloidin to show hair bundles and examined with a fluorescence microscope (Leica DM4000 B, Leica Microsystems, Wetzlar, Germany). Hair cells were counted as present if V-shapes of hair bundles were intact. Inner and outer hair cell counts were made by subdividing the cochlea into 10 regions at 10% distance intervals, beginning at the apex and continuing toward the base. Individual cochleograms were constructed to show the percentage of hair cells missing as a function of distance from the apex.
Semi-Quantitative-RT-PCR for measuring mRNA accumulation levels of apoptosis-related genes
Cdh23erl/erl and Cdh23ahl/ahl mice were sacrificed under avertin anesthesia conditions at 2 weeks or 2 months of age. The inner ears and left temporal brain lobes (50 mg) were quickly isolated for total RNA and cDNA preparation as described in the previous section. One μg of total RNA from each sample was used as template for cDNA synthesis. The 20 μl reaction mixture contained 50 mM KCl, 10 mM Tris-HCl, pH 9.0 (at 25°C), 0.01% Triton X-100, 2 mM MgCl2, 250 nM of each primer (forward and reverse), 200 μM dNTPs, 1 μl of cDNA and 0.5 U of Taq DNA polymerase (New England BioLabs, Inc., Ipswich, MA, USA). PCR primers are listed in Table 2. PCR was performed in a Bio-Rad PTC-200 Peltier Thermal Cycler (Bio-Rad Laboratories, Inc. Hercules, CA, USA). Amplification conditions were 94°C for 2 min; followed by 28 cycles of 94°C for 30 s, 60°C for 40 s, and 72°C for 50 s; followed by 5 min at 72°C. Ten μl of the PCR products were subject to agarose gel electrophoresis and the gray intensity of each band was digitized using ImageJ software (rsb.info.nih.gov/nih-image/NIH, Bethesda, MD, USA) and corrected by the GAPDH mRNA accumulation level of the same sample.
Caspase-3/7 activity measurement
Caspase-3/7 activity was detected in inner ears of 9 Cdh23erl/erl and 7 Cdh23ahl/ahl mice by the Apo-ONE® Homogeneous Caspase-3/7 Assay (Promega Corp., Madison, WI, USA) according to the manufacturer’s instructions. In brief, the inner ears were isolated and immersed in 300 μl of 0.1 M phosphate buffer saline (PBS) and homogenized on ice with a Tissue Mincer (Fisher Scientific, Pittsburgh, PA, USA). After centrifugation at 12000×g for 15 min at 4°C, the supernatant was retained and the protein concentration determined by the Lowry method. For the caspase assay, the non-fluorescent caspase substrate (Z-DEVD-R110, diluted 100-fold in the provided buffer) was mixed with an equal volume (20 μl) of solution containing the same amount of total inner ear protein (20 μg) for each sample. The protein/substrate mixture was incubated at room temperature for 4 h and diluted 300-fold afterward. The fluorescent product was detected using a spectrofluorometer configured to an excitation wavelength of 499 nm and an emission wavelength of 521 nm. The relative fluorescence unit as a measure of caspase activity was calculated as the fluorescence counts divided by the total amount of protein.
Immunostaining for active caspases
A time course immunocytochemistry study of caspase expression was carried out for Cdh23erl/erl and Cdh23ahl/ahl. The following antibodies from Cell Signaling Technology, Inc. (Danvers, MA, USA) were used. Cleaved caspase-3 (Asp175) antibody detects endogenous levels of the large fragment (17/19 kDa) of activated caspase-3 resulting from cleavage adjacent to Asp175 (This antibody does not recognize full-length caspase-3 or other cleaved caspases.); caspase-8 antibody (mouse-specific) detects endogenous levels of the large 18 kDa subunit of active caspase-8; cleaved caspase-9 (Asp353) antibody (mouse-specific) detects endogenous levels of the 37 kDa subunit of mouse caspase-9 only after cleavage at aspartic acid 353. It does not cross-react with full-length caspase-9 or with other caspases at endogenous levels. Mice were subjected to ABR testing (age ≥ 14 days) under anesthetizing conditions and then sacrificed. The inner ears were removed and cryosections were made and fixed in 4% PFA (diluted in 1× PBS) for 2 h. The sections were washed in PBS at room temperature twice for 5 min and permeabilized in 0.5% Triton X-100 for 30 minutes. After being washed twice in 1× PBS for 5 min and blocked in 5% BSA for 1 h, the samples were immersed in anti-active caspase-3 or caspase-8 or caspase-9 (1:200 dilution) and incubated at 4°C overnight. After being washed twice in 1× PBS for 5 min, the samples were immersed in anti-rabbit secondary antibody Alexa 488 (1:500 dilution) for 1 h. The samples were also stained with propidium iodide (10 mg/ml in PBS) for 30 min at room temperature. The sample mounts were observed under immunofluorescent microscopy.
Intraperitoneal injection and treatment regime optimization
Cdh23erl/erl was used as a model for testing anti-apoptotic drug therapy. Sixty Cdh23erl/erl mice at the age of 7 days (a starting point based on the goal of preventing caspase increases, detected as early as P14 in untreated Cdh23erl/erl mice) were divided into 3 groups: a test group, a DMSO group and an untreated group. The following treatment regime was selected as the best treatment from three sets of preliminary regimes (data not shown). In the test group, 22 mice were injected intraperitoneally (IP) under sterile conditions with Z-VAD-FMK (1μg/μl) (Z-Val-Ala-Asp(OMe)-Fluoromethylketone, Alexis Biochemicals, Farmingdale, NY) in PBS-diluted DMSO (1:1) at the dosage21 of 1.5 μg/g mouse weight: firstly, starting at P7, 8 injections, once every other day; secondly, 4 injections, once every 3 days; then, one injection every 4 days until the time of euthanization for experimental procedures. Eighteen mice in the DMSO group received 1.5 μl of PBS-diluted DMSO (1:1) per gram of mouse weight at the same time points as the test group. ABR and DPOAE were tested at 4 weeks, 6 weeks, 8 weeks and 12 weeks for the mice in each group and 4–5 mice from each group were sacrificed at each time point for histological investigation.
Statistical methods
The ANOVA was used for all data analysis except that hair cell loss data were analyzed by the Chi-Square Test. A value of P < 0.05 was considered significant.
Results
Linkage analysis and genetic complementation tests demonstrated that erl is a new allele of the Cdh23 gene
A new hearing-impaired mouse mutant was discovered at the Jackson Laboratory on the C57BL/6J (B6) background initially based on its lack of a Preyer reflex when presented with a calibrated 20 kHz 90 dB SPL (decibel sound pressure level) tone burst from a click box at two months of age. This mutant was originally generated from the Neuroscience Mutagenesis Facility (http://nmf.jax.org/, was designated #NMF308), but here is named erlong (symbol: erl), meaning hearing impaired in Chinese language. Subsequently, hearing loss (starting at P27, Table 1) was shown to progress to deafness (at P90) by auditory brainstem response (ABR) threshold testing in the homozygous colony (data not shown). No balance defect was observed throughout the lifespan of these mice. Initially, the mutant was mated with an unrelated B6 mouse to determine the heritability pattern of the mutation. ABR testing revealed no hearing loss in (erl X B6) F1 progeny up to 3 months of age, thus indicating that erl is a recessive hearing loss mutation. B6-erl/erl mice were outcrossed with C3H/HeJ mice to generate the (B6XC3H/HeJ)F1-erl/+ mice. These F1 mice were intercrossed (F1xF1) and yielded a Mendelian proportion of a single gene recessive inheritance pattern, generating roughly ¼ (13/51) erl/erl F2 progeny with ABR thresholds above 80 dB SPL and ¾ (38/51) ?/+ (¼ +/+ and ½ erl/+) F2 progeny with ABR thresholds below 50 dB SPL at five months of age (see Supplementary Table 1).
Table 1.
ABR thresholds showing a positive complementation test that revealed allelism of the erl and v-2J loci (abnormal values are in bold)
| Parental mating | ID | Age (days) | ABR (dB SPL) | gait | genotypes | |||
|---|---|---|---|---|---|---|---|---|
| Click | 8 kHz | 16 kHz | 32 kHz | |||||
| ahl/v2J x erl/erl F1 | 1 | 26 | 50 | 50 | 70 | 80 | normal | erl/v2J (codominant) |
| ahl/v2J x erl/erl F1 | 2 | 26 | 45 | 40 | 60 | 90 | normal | erl/v2J (codominant) |
| ahl/v2J x erl/erl F1 | 3 | 26 | 40 | 40 | 40 | 70 | normal | erl/v2J (codominant) |
| ahl/v2J x erl/erl F1 | 4 | 26 | 40 | 50 | 40 | 70 | normal | erl/v2J (codominant) |
| ahl/v2J x erl/erl F1 | 5 | 26 | 40 | 40 | 20 | 40 | normal | ahl/erl |
| ahl/v2J x erl/erl F1 | 6 | 26 | 40 | 40 | 20 | 40 | normal | ahl/erl |
| ahl/v2J x erl/erl F1 | 7 | 26 | 40 | 40 | 20 | 40 | normal | ahl/erl |
| ahl/v2J x erl/erl F1 | 8 | 26 | 40 | 40 | 20 | 50 | normal | ahl/erl |
| C57BL/6J-v2J/v2J | 9 | 26 | 100 | 100 | 100 | 100 | circler | v2J/v2J |
| C57BL/6J-erl/erl | 10 | 26 | 40 | 50 | 20 | 50 | normal | erl/erl |
| C57BL/6J-erl/erl | 11 | 40 | 40 | 40 | 70 | 90 | normal | erl/erl |
| v2J/v2J X C57BL6J-ahl/ahl | 12 | 90 | 45 | 40 | 20 | 55 | normal | ahl/v2J |
Our previous publications showed that, for age-related hearing loss, many strains have a Chromosome 10 (Cdh23) effect4, 8, 20, 22–25 that causes hearing loss; therefore, we first tested this possibility by analyzing the genotype of a marker (D10Mit194, Chr location: 46MB) close to the Cdh23 gene (Chr location: 59MB). A PCR assay on pooled genomic DNA from the 13 hearing-impaired F2 progeny showed a distinctive single PCR band for the B6 allele of D10Mit194, suggesting that erl might be another AHL-related allele of the Cdh23 gene on Chr 10 on which we previously worked4, 8, 9, 22, 24, 25 or another locus in the same region. Then individual DNA samples of the 51 F2 progeny were genotyped with respect to D10Mit194, D10Mit166 (Chr location: 6MB) and D10Mit42 (Chr location: 82MB). The results showed that the candidate interval was between D10Mit194 and D10Mit42 at 29 cM on chromosome 10; whereas, the Cdh23 gene is located at 30.3 cM as shown in the Supplementary Table 1. These results prompted us to do a genetic complementation test with mice carrying the most severe Cdh23 allele, waltzer 2 Jackson (abbreviated v-2J; complete designation is C57BL/6J-Cdh23v-2J/J), the deaf and circling allele1, 2, 25. Complementation tests revealed allelism of the erl locus and the v-2J locus indicating that erl is a new allele of the Cdh23 gene. As shown in Table 1, one litter of 8 pups was born (ID 1–8). The 4 erl/v-2J compound-heterozygous mice (ID 1–4) showed a hearing-loss level intermediate (without circling behavior) between that of v-2J/v-2J (ID 9) and erl/erl (ID 10), which suggests a codominant effect. Genotypes of the v-2J and erl mice shown in Table 1 were individually confirmed afterward by sequencing. These results were corroborated by an analysis of another litter of 9 pups from a B6- erl/erl x B6-v-2J/v-2J mating that gave a deaf phenotype without any balance defect for all 9 mice at P60.
Identification of a novel mutation in the Cdh23 gene of the hearing impaired mutant mouse
Genomic DNA screening to identify the alteration in the Cdh23 gene was performed by PCR and sequencing of all exons of the Cdh23 gene with detailed comparisons between mutants and controls using SequencherR 4.0 (genecodes.com; Gene Codes Corporation, Ann Arbor, MI, USA). A point mutation (208T>C) in the middle region of exon 3 (Genbank accession number: NM_023370) was identified by sequencing the PCR products of primer pair ex3-F1 and ex3-R1 (for three mutant and two control genomics DNA samples, see Table 2 for primer sequences) and confirmed by sequencing the cDNA of this gene (Figure 1a). No other mutations were found in any other exons of this entire gene. The RT-PCR products with primers flanking exon 3 and adjacent exons gave a clear single band (of the predicted size and identical to a normal control, data not shown) upon agarose gel electrophoresis suggesting that no aberrant splicing is caused by this mutation. The erl mutation did not quantitatively alter the expression level of Cdh23 mRNA (supplementary Figure 1a). The mutation results in an amino acid (aa) substitution of serine to proline (70S-P) in the first ectodomain (EC1, aa 38–132). The distinctive cyclic structure of the proline side chain locks its ϕ backbone dihedral angle at approximately−75°, giving proline an exceptional conformational rigidity compared to other amino acids. Proline acts as a structural disruptor in the middle of regular secondary structure element - alpha helix (aa 73–76) and to changes in the surrounding extended strands (specifically, an extended strand formed by aa 65–68 in the wild type mice shifts to that formed by aa 65–67 in the mutant, and another one formed by aa 77–80 in the wild type mice to that formed by aa 76–80 in the mutant mice) affecting the secondary structure of the protein when analyzed by the GOR IV secondary structure prediction method26. The substituted amino acid is not within but nearby a conserved calcium-binding site (aa 59–61, sequence DMD, highlighted in Figure 1c). Loss of the alpha helix and changes in the extended strands will probably affect protein function, such as formation of tip-links in the stereocilia or binding efficiency to calcium ions. Based on the mutation, a genotyping method was established to distinguish mice carrying the Cdh23erl allele from mice carrying the Cdh23ahl (C57BL/6J) allele by size differences on an agarose gel of PCR products digested with BstN I (Figure 1b). This confirms in another way that the erl mutation was accurately identified. By sequencing, we confirmed that all of the erl mice carry the ahl allele, thus this allele causes in-frame skipping of exon 7 and may contribute to conformational changes of CDH23.4
Figure 1.

Identifying the mutation in Cdh23erl/erl mice. (a) cDNA sequence traces of the Cdh23 gene from positions 205 to 213 (in the middle of exon 3) show a T>C transition mutation at position 208 (arrowhead) in Cdh23erl/erl mice (n=3) (right panel) compared to control C57BL/6J (Cdh23ahl/ahl) mice (left panel). An amino acid substitution from serine to proline (70S-P) is highlighted in red. (b) Genotyping of Cdh23 variants. Lane M, 100 bp DNA ladder; middle two lanes: an erl/erl-introduced BstN I restriction site cuts the 219 bp homozygous mutant fragment (from PCR products with primer pairs ex3m-F and ex3m-R in Table 2) into two distinct (152 bp and 68 bp) fragments. In contrast, Cdh23ahl/ahl showed one band (219 bp, left two lanes next to the ladder); and Cdh23ahl/erl showed three (219 bp, 152 bp and 68 bp, right two lanes). (c) Analysis of the conservation of serine (S, in red) and its flanking regions across species. The conserved calcium-binding site (DMD) in EC1 is highlighted in the red box. The S is conserved in mammals. However, it is changed to proline (P) in Cdh23erl/erl mice.
DPOAE and ABR testing reveal early onset, progressive hearing loss in Cdh23erl/erl mice
Distortion product otoacoustic emissions (DPOAEs) were measured to determine the mutant effect on OHC function over time in Cdh23erl/erl mice as compared to Cdh23ahl/ahl controls (Fig. 2a). Three-week-old Cdh23erl/erl mice had normal DPOAE amplitudes that were indistinguishable from those of Cdh23ahl/ahl mice which have normal hearing as determined by ABR thresholds at 3 weeks (wks). At 7 wks of age, Cdh23erl/erl mice had DPOAE amplitudes 10–20 dB lower than those of Cdh23ahl/ahl at frequencies from 10 to 18 kHz. By 22 wks of age, Cdh23erl/erl mice had no detectable DPOAE at any frequency indicating complete deafness.
Figure 2.

Phenotype and pathology of Cdh23erl/erl mice. (a) DPOAE measurement shows the progression from normal hearing (3 wks, n = 8 each), to high-frequency, partial hearing loss (7 wks, n = 10 each), to complete hearing loss (22 wks, n=6 each) in Cdh23erl/erl mice (red) compared to the Cdh23ahl/ahl control mice at the same age (blue). (b) Mean ABR thresholds (Y-axis) for Cdh23erl/erl mice (n ≥ 6) from 1 month (1M) to 9 months (9M) of age show normal hearing at 1 month progressing to severe hearing loss by 3 months. Mean ABR thresholds for Cdh23ahl/ahl mice (n = 3) undergoing a similar progression on a different time scale from 9 months to 24 months (shown on the right). Error bars represent standard deviation from the mean. (c) Surface preparations from the cochlear middle turn of the organ of Corti in control and erl mutant mice at P100 were stained for F-actin with Alexa Fluor 568 conjugated to phalloidin to show stereocilia bundles for OHC counts. Upper panel shows a full complement of OHCs of normal morphology and regular arrangement and the lower panel shows segments of OHC loss apparent as dark areas in an irregular array of OHCs. Scale bar = 50 μm. (d) Cytocochleograms showing the mean percentage of total OHCs missing (Y axis) at various time points as a function of distance from the apex (X axis) in Cdh23erl/erl mice. Error bars represent the standard deviation from the mean at each point. Vertical bars show the frequency-to-place map43 for the 8, 16 and 32 kHz test tones. Three to 7 mice (6–14 ears) were used for OHC counts at each time point.
ABR recordings yielded normal wave patterns (data not shown) but elevated thresholds at every frequency tested (click, 8, 16, 32 kHz from 30–99 dB, Figure 2b and Table 1) with high frequencies affected first, as early as P40, progressing to deafness at P90 and thereafter remaining deaf to at least 9 months of age. The Cdh23erl/erl mice exhibit hearing loss much earlier (P40) than do mice expressing other Cdh23 alleles such as Cdh23ahl/ahl mice (12-months onset, Figure 2b and Table 1). The vestibular system in these mutant mice appears to be unaffected by the erl mutation as Cdh23erl/erl mice exhibited normal swimming ability (3 Cdh23erl/erl mice at 4 months of age had similar normal swimming as B6 mice) and gait pattern as observed for all time points examined up to 9 months.
Histologic examination reveals progressive OHC loss that follows hearing loss in Cdh23erl/erl mice
To investigate cochlear pathology in Cdh23erl/erl versus Cdh23ahl/ahl control mice, cochleae were stained for F-actin with Alexa Fluor 568 conjugated to phalloidin to examine all turns and reveal the presence and morphology of OHCs (Figure 2c). The OHCs of Cdh23ahl/ahl mice at P100 (Figure 2c, upper panel) showed normal V-shaped arrangement of stereocilia. No OHC loss was observed in any of the cochlear turns of Cdh23ahl/ahl mice, as correlates with normal ABR thresholds up to 9 months (Figure 2b). In contrast, large segments of OHC loss were evident in the middle cochlear turns of Cdh23erl/erl mice at P100 (Figure 2c, lower panel). This OHC loss at P100 follows ABR threshold shifts observed at P27-P90 (Figure 2b).
To quantify OHC loss and to determine the developmental time course of OHC loss, we generated cytocochleograms from 25 Cdh23erl/erl mice ranging from one to nine months of age. For each cytocochleogram, we calculated the percentage of missing OHC and IHC by subdividing the cochlea into 10 regions at 10% distance intervals from the apex of the cochlea. Scatter plots were constructed showing the mean percentage of OHC loss as a function of age for Cdh23erl/erl mice (Figure 2d). Cdh23erl/erl mice showed no evidence of OHC loss from P0 to P28. However, OHC loss was observable at 3 months, became progressively worse over time, and moved from the cochlear base to the apex in Cdh23erl/erl mice. OHC loss in the 75–100% region increased to a nearly complete loss after 3 months of age, corresponding to a high frequency hearing loss (16 and 32 kHz, top two lines in Fig. 2b) earlier than that of low frequency hearing loss (8kHz, bottom line, Fig. 2b) as measured by ABR. Because IHC loss was not significant until after 5 months of age, IHCs were excluded from this analysis.
mRNA accumulation and activity of apoptosis-related genes was up-regulated in the inner ears of Cdh23erl/erl mice
To determine whether OHC loss might be the result of OHC apoptosis, we examined expression of apoptosis-related genes in Cdh23erl/erl mice. Gene transcription levels of apoptosis-related genes were measured in the inner ears of Cdh23erl/erl and control Cdh23ahl/ahl mice at 2 wks and 2 months of age (Figure 3). The extrinsic caspase-8 and intrinsic caspase-9 initiators, and activator caspase-3 were significantly up-regulated (P < 0.05 for each) in Cdh23erl/erl samples relative to the levels measured in control Cdh23ahl/ahl samples (Figure 3a and 3b) though the intensity for caspase-8 was higher and earlier than that of caspase-9, suggesting that the extrinsic pathway was initiated earlier than the intrinsic. At the two-month time point, the expression level of caspase-9 was also up-regulated (P < 0.05) in Cdh23erl/erl samples. However, there was no significant difference in mRNA accumulation levels of caspase-3, caspase-8 and caspase-9 genes in brain tissue between Cdh23erl/erl and Cdh23ahl/ahl mice at the ages of 2 weeks or 2 months (P > 0.05, data not shown), indicating that the apoptotic gene expression measured in ear tissue was not a generalized phenomenon.
Figure 3.

Increased apoptotic activity in erlong mice precedes hearing loss and hair cell loss. GAPDH-corrected mean mRNA expression levels of apoptosis-related genes caspase-3, −8, and −9 in inner ear tissue from Cdh23erl/erl (n = 4 mice, 8 ears) were higher than those from Cdh23ahl/ahl ( n = 4 mice, 8 ears) mice at 2 weeks (a) and 2 months (b) of age. Caspase-8 was expressed earlier and at a higher level than others. Caspase 3/7 activity measured by cleavage of the fluorescent caspase 3/7 substrate Z-DEVD-R110 and indicated as relative fluorescence units (RFLU) in the inner ear homogenates from mice at ages of 2 weeks (n = 9 mice for each group) (c) and 2 months (n = 7 mice for each group) (d). Error bars represent the standard error from the mean. *P < 0.05, **P < 0.01.
To confirm whether increased mRNA levels were correlated with changes at the protein level, the Apo-ONE® homogeneous caspase-3/7 assay (Promega) was used to measure caspase-3 and −7 activities in inner ears of erl mutants and control mice. The relative fluorescence unit is a measure of caspase activity in the homogenized inner ear tissue from the mice in each group, upon addition of a caspase substrate to the homogenate. At P14 (2 weeks) and P60 (2 months), the relative caspase-3/7 activity was significantly higher in Cdh23erl/erl mice than in Cdh23ahl/ahl mice (Figure 3c and 3d).
Immunostaining reveals signs of apoptosis early, increasing toward the 3-month time point in the organ of Corti of Cdh23erl/erl mice
To investigate inner ear apoptosis in situ in the Cdh23erl/erl mouse model and determine whether the observed OHC loss can be explained by apoptosis of the OHCs, we stained tissue sections with fluorescein-labeled antibodies to detect cells that express caspases. The antibodies used specifically detect only the active forms of caspases-3, −8 and −9. Cdh23ahl/ahl mice were used as controls. We investigated three time points leading up to the time of complete hearing loss at 3 months, and beginning at the P14 time point (Figure 4). At P14, caspases-8 and −9 were expressed in the middle turns of cochleae (mainly in the outer hair cells (OHC) and spiral ganglion cells (SG) for caspase-8 and caspase-9) of the Cdh23erl/erl mice (Figure 4d, 4e) compared to the same areas of the Cdh23ahl/ahl mice (Figure 4a, 4b). Primary antibody-omitted sham sections of the cochleae of both mouse groups demonstrated that this staining was specific and not an artifact of non-specific secondary antibody staining (Figure 4c, 4f). Notably, caspase-8 stained OHC stronger than SG (Figure 4d), whereas caspase-9 stained OHC weaker than SG (Figure 4e).
Figure 4.

FITC(Green)-anti- caspase antibody staining. Sections of the middle turns of cochleae from Cdh23ahl/ahl (a, b, c) and Cdh23erl/erl ( d, e, f) mice at P14, stained with FITC-anti-active caspase-8 (a, d) and caspase-9 (b, e). The signals for caspase-8 and caspase-9 staining in Cdh23erl/erl mice were more intense in the outer hair cell (OHC) regions, and spiral ganglion cells (SG) than in the corresponding positions of the sections from Cdh23ahl/ahl mice. There were no detectable signals when primary antibody was omitted (c, f). StV, Stria vascularis; RM, Reissner’s membrane. Scale bars = 50μm.
A time course observation of caspase-3 staining in the middle turns of cochleae of Cdh23erl/erl mice is shown in Figure 5. Anti-caspase-3 (the activated, downstream, effector caspase) staining was not visible in any areas of the cochleae at P14 (Figure 5a); however, it was evident in the cytoplasm of OHCs of Cdh23erl/erl mice by P23 (Figure 5b) and became stronger by P57 (Figure 5c). Active caspase-3 was also positive in the spiral ligament (SL) at both time points at P57. An enlarged image of OHCs from Figure 5c showed the overlapping staining (Figure 5f) of PI-stained nuclei (Figure 5e) and anti-caspase-3-stained cytoplasm (Figure 5d). Anti-caspase-3 signals were not detected in any cochlear turns from Cdh23ahl/ahl mice.
Figure 5.

Caspase-3 detection in Cdh23erl/erl mice in situ in a time course observation. Representative sections from the mid-turn of cochleae of Cdh23erl/erl mice were stained for active caspase-3 expression (FITC-anti-active-caspase-3, green) at P14 (a), P23 (b) and P57 (c). Caspase-3 expression in situ is evident at P23 and P57. (d–f) Enlarged area from panel c to show individual OHCs in the organ of Corti, with propidium iodide (PI) staining (red) revealing OHC nuclei and tissue ultrastructure in panel e. (f) Merged images of d and e. SL, spiral ligament, OHC, outer hair cell. For a–c, scale bars = 50 μm. d–f, scale bars = 20 μm.
Upregulation of gene expression in the extrinsic (TNF-α) and intrinsic (Calpain) apoptosis pathways in Cdh23erl/erl mice
The relative mRNA levels of TNF-α, caspase-12, m-calpain and μ-calpain were significantly up-regulated in the Cdh23erl/erl mice at both time points of 2 weeks (Supplementary Figure 1a, c) and 2 months (Supplementary Figure 1b, d), indicating multiple apoptosis pathways are involved in the hearing loss of these mutant mice. But TNF-α (Supplementary Fig. 1a) was up-regulated at the younger age (2 weeks) more significantly than other genes (c), suggesting the extrinsic pathway may play an important role for initiation of apoptosis. The fact that caspase-8 was up-regulated at the younger age (2 weeks) (Figure 3a) more significantly (P<0.01) than other genes in the Cdh23erl/erl mice also supports this point becauseTNF-α and caspase-8 are both important indicators of the extrinsic apoptotic pathway. Calpains, caspase-12 (Supplementary Figure 1b, d, P<0.05) and caspase-9 (Figure 3b, P<0.05) were also up-regulated slightly, but significantly, as indicators of intrinsic apoptosis.
OHC protection in Cdh23erl/erl mice
Cdh23erl/erl was used as a model for testing anti-apoptotic drug therapy. Sixty Cdh23erl/erl mice were divided into a test group, a diluent (DMSO) control group and an untreated control group. Each mouse in the test group received Z-VAD-FMK (in dimethyl sulfoxide, DMSO, 1:1 diluted with PBS) intraperitoneally (ip) at a dosage of 1.5 μg/g body weight at different time points: firstly, eight injections, on a schedule of one injection every other day, beginning at age P7; then 4 injections, on a schedule of one injection every 3 days; and lastly, one injection every 4 days until the time of euthanization for analysis at various time points. Each mouse in the DMSO group received an equivalent volume of DMSO following an identical injection schedule to its cohort test group. ABR and DPOAE were tested at 4 wks, 6 wks, 8 wks and 12 wks of age. ABR thresholds were significantly lower in the DMSO+Z-VAD-FMK-treated mice than in the DMSO-treated or the untreated Cdh23erl/erl mice at stimulus frequencies of click, 8 kHz, 16 kHz and 32 kHz and at all time points (P < 0.05 at 4 wks; P < 0.01, for the time points of 6 wks, 8 wks and 12 wks) (Figure 6). There were also significant differences in ABR thresholds between the untreated and DMSO-treated mice at 12 wks for the stimulus frequency of 16 kHz (P < 0.05) (Figure 6c). The mean amplitude of DPOAE at 16 kHz was significantly higher in the DMSO+Z-VAD-FMK-treated mice than that of the untreated or DMSO-treated group at the age of 8 wks (P < 0.05, respectively, Figure 7). Loss of outer hair cells (OHC) was evident in the middle turns of cochleae of the untreated or DMSO-treated mouse groups at 2 months of age; whereas, no OHC loss was seen in the corresponding areas of the Z-VAD-FMK+DMSO-treated mice (Figure 8). OHC and inner hair cell (IHC) loss was counted and averaged from apical to basal turn of cochlear surface preparations in the three mouse groups (n=4 mice per group) at 2 months of age (Figure 9). The mean percentage of OHC loss was significantly less in Z-VAD-FMK+DMSO-treated Cdh23erl/erl mice than in the untreated or DMSO-treated mice at the apex, middle or basal cochlear regions. There was also significantly less OHC loss in the DMSO-treated mice than in the untreated mice at the basal turn (P <0.05). IHC loss was not substantially altered by the treatment. Additionally, we have not observed any adverse effects of Z-VAD-FMK treatment (such as tumor development in the intestine, colon, liver or lungs or i.p injection site) on the treated mice during the experimental period up to 3–4 months of ages (data not shown).
Figure 6.

ABR thresholds to evaluate hearing in Cdh23erl/erl mice with no treatment or after treatment with diluents of DMSO or DMSO+Z-VAD-FMK, over a period of 11 weeks. Treatments were initiated at mouse age P7 (1 wk). The number of mice tested was 14, 14, and 19 at 4 wks; 10, 11 and 14 at 6 wks; 10, 7 and 9 at 8 wks; and 6, 4 and 6 at 12 wks, for no treatment, DMSO and DMSO+Z-VAD-FMK groups, respectively. ABR thresholds were measured at each time point (indicating mouse age) on the X axis. (a–d) ABR thresholds were measured at the frequencies indicated on the Y axis of each plot. Each point represents the mean ABR threshold for a group, with error bars indicating standard error from the mean. The results show that ABR thresholds were significantly lower in the DMSO+Z-VAD-FMK-treated mice than those of the DMSO-treated or the untreated mice at stimulus frequencies of click, 8 kHz, 16 kHz and 32 kHz at all time points ( *P < 0.05 at 4 wks; **P < 0.01, respectively, for 6 wks, 8 wks and 12 wks). There were also significant differences for the ABR thresholds between the untreated and DMSO-treated mice at 12 wks at the stimulus frequency of 16 kHz (P < 0.05).
Figure 7.

A time course observation in the three treatment-based Cdh23erl/erl mouse groups of DPOAEs to stimulus frequency of 16 kHz (f2) at 4 weeks (wks), 6 wks, 8 wks and 12 wks. DPOAE amplitudes (dB SPL) gradually decreased with time in the untreated group (n = 8, 10, 10 and 8, respectively, at each time point), the DMSO-treated group (n = 11, 7, 7 and 6) and DMSO+Z-VAD-FMK-treated group (n = 14, 9, 9 and 6). The mean amplitude was significantly higher in the DMSO+Z-VAD-FMK-treated mice than in the untreated (P = 0.03294) or DMSO-treated (P = 0.03955) group at the age of 8 wks. There were no significant differences for the DPOAE amplitudes between the untreated group and DMSO-treated group at any time points. * P < 0.05. Error bars indicate standard error from the mean.
Figure 8.
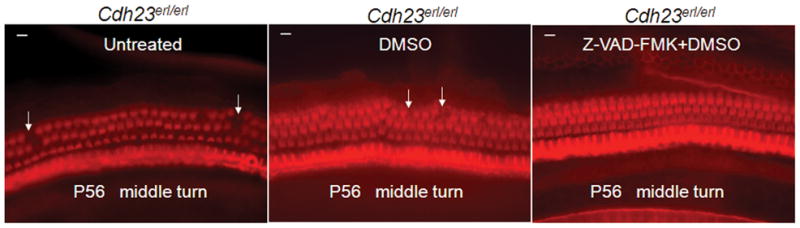
Whole-mount phalloidin-stained preparations from the mid-turn of cochleae reveal normal appearance of OHC morphology and arrangement in Z-VAD-FMK-treated Cdh23erl/erl mice at P56. Left panel, untreated mice; middle panel, DMSO-diluent-treated mice; right panel, Z-VAD-FMK-treated mice. White arrows indicate areas of OHC loss, apparent in both controls. Scale bars = 10 μm.
Figure 9.

Hair cell losses in the cochleae of the Z-VAD-FMK-treated Cdh23erl/erl mice at the age of 2 months. Surface preparations of 4 cochleae from 4 mice in each mouse group were examined. The mean percentages of outer hair cell (OHC) loss in the apex, middle and basal regions of the cochleae of the Z-VAD-FMK-treated mice (black bars) were significantly lower than that of the untreated (white bars) and DMSO-treated (gray bars) mice. There were also significant differences in OHC loss between the untreated and DMSO-treated mice at the basal regions (P < 0.05). No significant difference in inner hair cell loss occurred between the three mouse groups.* P < 0.05; ** P < 0.01. Error bars represent standard error from the mean.
Z-VAD-FMK treatment prevents OHC loss in Cdh23v-2J/v-2J mice
As represented in Supplementary Figure 2, serial sections demonstrated that Z-VAD-FMK treatment could protect OHCs from loss in Cdh23v-2J/v-2J mice, but failed to improve the hearing of the mice at any level. The survival of inner ear hair cells may still be important to release neurotrophic factors for keeping spiral ganglion neurons viable and for responding to electrical stimuli in cochlear-implanted USH1D patients, despite the inability of rescued hair cells to function in hearing. Thus anti-apoptosis treatment in USH1D patients might be useful in the future, but will require further studies.
Discussion
The Cdh23erl/erl mice introduced in this study are of the C57BL/6J background and proved to be carrying a new allele of Cdh23. Cdh23erl/erl is characterized by progressive hearing loss starting about 1 month after birth and becoming deaf at 3 months of age. Hair cell loss began near the base of the cochlea and spread toward the apical turn, and lesions of the spiral ganglion were also involved at a later stage (P149, data not shown). The Cdh23erl/erl mouse, therefore, provides a promising model for investigating the pathogenesis of DNFB12. It is also an ideal mouse model for otoprotection drug therapy and discovery.
Given that the hearing loss in Cdh23erl/erl mice happens before hair cell loss (P60 see Fig. 2d) but after caspase signals increase (P14), we hypothesized that apoptosis might be the mechanism for causing a physiological deficit to occur before hair cell loss. There are mainly two pathways of cellular apoptosis as shown in Figure 10.27, 28 In response to extra-cellular stimuli, the initiator of the extrinsic pathway (caspase-8) undergoes self-processing, releasing active enzyme molecules into the cytosol and activating caspase-3, caspase-6, and caspase-7. These activated caspases proteolytically cleave and activate other caspases, as well as relevant targets in cells. Alternatively, the intrinsic pathway involves release from the mitochondrion of pro-apoptotic proteins that activate caspase enzymes, mainly caspase-9, which ultimately trigger apoptosis by activating caspase-3, caspase-6, and caspase-7. Therefore, we attempted to determine the role of apoptosis in the pathogenesis of the Cdh23erl/erl mouse model by examining expression of apoptosis-related proteins in the inner ears of these mice. Our results showed significantly up-regulated transcriptional levels of caspase-3, caspase-8 and caspase-9 in the inner ears of Cdh23erl/erl mice at 2 weeks and/or 2 months of age, indicating both extrinsic and intrinsic pathway activation. Though the extrinsic pathway was activated slightly earlier and more strongly than the intrinsic pathway, this presumably was the result of increased TNFα signalling triggered by debris from broken stereocilia composed of mis-folded CDH23erl/erl protein, due to the ϕ backbone dihedral angle (−75°) caused by the proline from the aa substitution (70S-P). A detailed mechanistic explanation of why mutated CDH23 increases TNF-α signalling is yet to be determined. Another possibility is that the 70S-P introduced conformational changes in the first ectodomain, ultimately leading to intracellular domain and catenin complex signal changes that modulate apoptosis.29, 30
Figure 10.

Conceptual diagram of possible pathways of apoptosis involved in hair cell loss in the Cdh23erl/erl mice and the sites of Z-VAD-FMK blockage (indicated by circled minus signs). The extrinsic caspase-8 and intrinsic caspase-9 initiators, and activator caspase-3 are significantly up-regulated by the Cdh23erl/erl mutation. TNF-a, caspase-12 and calpains including m-calpain and μ-calpain were also up-regulated in this study. The interactions between Cdh23 and Pcdh15 (Zheng et al 2005) or between Cdh23 and PMCA2 (Zheng et al 2001) were demonstrated in our previous reports. Solid lines show pathways for which there is direct evidence. Dash lines are proposed links.
Immunostaining showed that caspase-8 was activated in the organ of Corti at P14, that caspase-3 was activated by P23 and that the staining for both became more intense at P57. Additionally, there were signs of apoptosis in the spiral ganglion and the stria vascularis. In contrast to a previous report in rat of a strong active caspase-3 signal in the lateral wall of the cochlear basal turn with drug-induced damage,21 our mouse experiments showed that caspase immunoreactivities were first present and strong in the basal turn and middle turns of the organ of Corti. Based on these results, we conclude that apoptosis plays a major role in Cdh23erl/erl mice in the death of hair cells, spiral ganglion cells and cells of the spiral ligament and stria vascularis in the inner ear, and that the cell death mechanism of drug-induced damage in rat may differ from the mechanism in a genetic mutant such as the Cdh23erl/erl mouse. The up-regulated apoptosis is probably localized to the inner ear in Cdh23erl/erl mice, as there was no significant difference in mRNA accumulation levels for caspase-3, caspase-8 or caspase-9 genes in brain tissue between Cdh23erl/erl and Cdh23ahl/ahl mice. We acknowledge that other mechanisms may apply for possible causes of hearing loss in other mutations.
Cadherins are glycoproteins involved in Ca2+-mediated cell-cell adhesion, migration and compaction.2 CDH23 is an important component of the Ush1 interactome and of the tip-links in the mechanotransduction (MET) system of the organ of Corti.15, 31–33 It is involved in regulating the activity of mechanically gated ion channels in hair cells.3, 34, 35 It is reported that, of the 21 missense mutations in CDH23 that have been observed in patients with nonsyndromic hearing loss, 10 occurred within the DXNDN, LDRE and DXD calcium-binding motifs. These motifs are highly conserved in sequence and spacing and are required for cadherin dimerization and Ca2+-binding, suggesting that deficiency of the calcium-dependent cell adhesion has a major impact on cochlear cell integrity36. Because calcium provides rigidity to the elongated structure of cadherin molecules thus enabling homophilic lateral interaction, mutations in the calcium-binding motifs of CDH23 are likely to impair homophilic interactions of CDH23 molecules and possibly also interactions with other proteins.37 Though the change in structure in CDH23 protein in the Cdh23erl/erl mice is not located precisely at the Ca2+-binding site, it is nearby. Thus, we speculate that it may influence the efficiency of CDH23 interactions with proteins, Ca2+ and ultimately may affect calcium-dependent cell adhesion, leading to apoptosis in the Cdh23erl/erl mutant mice by multiple pathways (Figure 10). (1) The intricate relationship between CDH23 and other proteins of the USH complex could be disrupted by the erl mutation, causing faulty OHC hair bundle formation and extrinsic apoptosis, as the mRNA level of TNF-α was significantly higher in the ears of the Cdh23erl/erl mice (Fig s1a,b). This pathway may be the earliest and strongest-affected pathway in our mouse model among all possible involved pathways. (2) We did find that calpains and caspase 12 were up-regulated at the transcriptional level in Cdh23erl/erl mutant mice (Fig s1c,d). Calpains are activated by Ca2+ ions 38 and can further activate caspase-12, which subsequently activates caspase-9 (intrinsic pathway). (3) As apoptosis was also evident in spiral ganglion, stria vascularis and supporting cells in the mutant mice, disorder in Ca2+ regulation and in calcium-dependent cell adhesion in the cochlea may be responsible for triggering apoptosis. (4) CDH23 is a bona fide type I intrinsic membrane protein and is most likely translated on rough ER membranes; therefore, the 70S-P substitution may cause CDH23 misfolding resulting in accumulation of CDH23 in the endoplasmic reticulum (ER), with subsequent ER stress followed by apoptosis (Figure 10). (5) It has been reported that a cyclic peptide can perturb cadherin-mediated endothelial cell interactions, resulting in a progressive apoptotic cell death. This effect depends on cell density, as it is only observed when dense cultures are treated with the peptide.39 CDH23 is a member of the cadherin family and could have a similar mechanism of apoptosis. This may also explain why the densely organized OHCs (three rows) were more affected than the more loosely organized IHC (one single row).
It has been documented in drug-induced hearing loss models that hair cell loss can occur by apoptosis, and that such loss can be reversed through treatment with the anti-apoptotic drug, Z-VAD-FMK (a powerful, irreversible and cell-permeable inhibitor for caspases).21 We therefore investigated Z-VAD-FMK as a candidate drug for maintenance of hearing and hair cell viability in Cdh23erl/erl mice, though the mechanism of cell death in Cdh23erl/erl mice may differ from that in drug-induced damage. Following Z-VAD-FMK treatment of the Cdh23erl/erl mice, there was a significantly reduced amount of OHC loss compared to that of the untreated or DMSO-treated mice in all regions of the cochlea. The protective effect of Z-VAD-FMK on OHC was further confirmed by significantly higher DPOAE amplitudes in the DMSO+Z-VAD-FMK-treated mice than in the untreated or DMSO-treated mice at the age of 8 weeks. Furthermore, ABR thresholds were preserved at levels approximately 15–40 dB better in the Z-VAD-FMK-treated group than in the untreated or DMSO-treated groups of littermate Cdh23erl/erl mice from 6 weeks to 12 weeks of age. The involvement of apoptosis in the pathology of Cdh23erl/erl inner ears was thus confirmed by anti-apoptotic drug therapy. Z-VAD-FMK can preserve cells (hair cells, for example) in the inner ears by stopping apoptosis. These results offer the potential that DNFB12 could be preventable in the future by anti-apoptotic intervention. However, the hearing loss of the Cdh23erl/erl mice could not be completely prevented by Z-VAD-FMK treatment in the time course observation, especially at 12 weeks of age, indicating that other factors related to alteration in CDH23 structure may be also involved in the pathology of these mouse inner ears. Additionally, we noticed that DMSO has a minor, but slightly significant, rescue effect on hearing (Figure 6c and Figure 9). This is consistent with another recent report.40
A mouse model of DFNB12 was reported recently, in which the mutation salsa was predicted to affect Ca2+ binding by altering the extracellular CDH23 domain, resulting in severe hearing loss at P21.15 The early onset and rapid progression of hearing loss in salsa mice may make it difficult to find a time period for drug intervention. Therefore, the Cdh23erl mouse model may provide a better platform for further anti-apoptotic drug screening and testing.
In summary, a point mutation in the Cdh23 gene (208T>C) of C57BL/6J mice results in hearing loss around 1 month after birth. Given the strain background on which the erl mutation arose, the Cdh23erl mice also carries the ahl mutation; thus, the combination of mutations on the C57BL/6J background produces an accelerated hearing loss phenotype that is well-suited for an experimental model. The erl mutation influences cell fate by increasing cellular apoptosis which may be attenuated by anti-apoptotic drug therapy. These results may provide avenues for improving hearing in patients with DFNB12 by an anti-apoptosis approach in the near future. Anti-apoptosis drugs could be adapted for preventive pre-clinical application after detailed evaluation of the primary effects and the side effects under various treatment regimens in this mouse model. Because this is a preventive drug, we suggest that only after a patient is clearly genetically diagnosed as DFNB12 younger than a determined age of hearing loss should a patient be included for trial of this drug.
Supplementary Material
Table. S1. Mapping of the mutation in erlong mice. (A) DNA from affected mice and unaffected mice was analyzed for MIT markers (D10Mit166, D10Mit194 and D10Mit42) on chromosome 10, which are listed in the top row and indicate the base pair position (NCBI Build 37). The mouse identification numbers (ped#) (column K) and phenotypes (ABR thresholds, db SPL, column E-H at five month of age column B) are indicated in the 2 top rows. The genotype for each marker is given as: b, homozygous C57BL/6J genotype; c, homozygous C3H/HeJ; and h, heterozygous. The chromosomal intervals that segregate with the mutant phenotype are highlighted with boxes.
Four Cdh23erl/erl and 4 Cdh23ahl/ahl mice at the ages of 2 weeks and 2 months, respectively, were used. The primers for GAPDH and Cdh23 are described in the methods and the other primers are listed in Table 2. The relative mRNA levels of Cdh23, TNF-α, caspase-12, m-Calpain and μ-Calpain were averaged for each genotype and plotted, with error bars indicating standard error from the mean. The results indicated that there was no significant difference for the relative mRNA levels of Cdh23 between the Cdh23ahlahl and Cdh23erl/erl mouse groups (P > 0.05). However, the relative mRNA levels of TNF-α, caspase-12, m-Calpain and μ-Calpain were significantly up-regulated in the Cdh23erl/erl mice at both time points of 2 weeks (a, c) and 2 months (b, d), indicating that multiple apoptosis pathways are involved in the hearing loss of the mutant mice. *P < 0.05; **P < 0.01
Six littermate Cdh23v-2J/v-2J mice and 3 Cdh23v-2J/+ mice at the age of P8 were used. Three Cdh23v-2J/v-2J mice received 4 doses of Z-VAD-FMK (1.5mg/kg mouse weight, IP injection in DMSO solvent) at P8, P9, P10 and P13. Three Cdh23v-2J/v-2J and three Cdh23v-2J/+ mice were simultaneously injected with an equivalent dose of DMSO without Z-VAD-FMK. Click and pure tone (8 kHz, 16 kHz, and 32 kHz) ABR thresholds were assessed at P17. We found none of the Cdh23v-2J/v-2J mutants that received Z-VAD-FMK had detectable ABR at 100 dB but the Cdh23v-2J/+ mice had normal ABR thresholds as expected at P17 (data not shown). Figure S2 shows the results of H&E staining of the basal turn of cochleae from mice at P17 (a–c), with enlarged views of the organ of Corti (OC) region in panels (d–f). Outer hair cells (OHC) were lost in the DMSO sham-treated Cdh23v-2J/v-2J mice (a, d), and only support cells remained (arrowhead in d). However, as with the DMSO-treated Cdh23v-2J/+ mice (c, f), the Z-VAD-FMK (in DMSO) treated Cdh23v-2J/v-2J mice showed intact OHCs (4 arrows in e) in the organ of Corti in multiple serial sections. The results indicate that Z-VAD-FMK treatment can protect OHCs in Cdh23v-2J/v-2J mice, but fails to improve the hearing of the mice. SV, scala vestibuli; SM, scala media; RM, Reissner’s membrane; StV, Stria vascularis; SL, spiral ligament; SG, spiral ganglion. Scale bars = 50 μm.
Acknowledgments
This work was supported by NIH R01DC007392 and R01DC009246 to QYZ. The mutagenesis program at the Jackson Laboratory was funded by NIH (U01NS041215). We wish to thank Shengli Li and Baiya Li for technical assistance, Louise Dionne for initial colony management and Lucy Rowe for her genotyping Service. Hui E Chen is supported by CWRU Summer Program in Undergraduate Research (SPUR).
Footnotes
Conflict of interest
Conflict of interest statement: QYZ, F.H. and H. Y. have filed a patent covering the general approach of Z-VAD-FMK as a therapeutic approach for genetic hearing loss and using the erl mouse model for evaluating otoprotective drugs.
References
- 1.Di Palma F, Holme RH, Bryda EC, Belyantseva IA, Pellegrino R, Kachar B, et al. Mutations in Cdh23, encoding a new type of cadherin, cause stereocilia disorganization in waltzer, the mouse model for Usher syndrome type 1D. Nat Genet. 2001;27(1):103–107. doi: 10.1038/83660. [DOI] [PubMed] [Google Scholar]
- 2.Di Palma F, Pellegrino R, Noben-Trauth K. Genomic structure, alternative splice forms and normal and mutant alleles of cadherin 23 (Cdh23) Gene. 2001;281(1–2):31–41. doi: 10.1016/s0378-1119(01)00761-2. [DOI] [PubMed] [Google Scholar]
- 3.Siemens J, Lillo C, Dumont RA, Reynolds A, Williams DS, Gillespie PG, et al. Cadherin 23 is a component of the tip link in hair-cell stereocilia. Nature. 2004;428(6986):950–955. doi: 10.1038/nature02483. [DOI] [PubMed] [Google Scholar]
- 4.Noben-Trauth K, Zheng QY, Johnson KR. Association of cadherin 23 with polygenic inheritance and genetic modification of sensorineural hearing loss. Nat Genet. 2003;35(1):21–23. doi: 10.1038/ng1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bolz H, von Brederlow B, Ramirez A, Bryda EC, Kutsche K, Nothwang HG, et al. Mutation of CDH23, encoding a new member of the cadherin gene family, causes Usher syndrome type 1D. Nat Genet. 2001;27(1):108–112. doi: 10.1038/83667. [DOI] [PubMed] [Google Scholar]
- 6.Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. American journal of human genetics. 2001;68(1):26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Astuto LM, Bork JM, Weston MD, Askew JW, Fields RR, Orten DJ, et al. CDH23 mutation and phenotype heterogeneity: a profile of 107 diverse families with Usher syndrome and nonsyndromic deafness. Am J Hum Genet. 2002;71(2):262–275. doi: 10.1086/341558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng QY, Johnson KR, Erway LC. Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses. Hear Res. 1999;130(1–2):94–107. doi: 10.1016/s0378-5955(99)00003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson KR, Erway LC, Cook SA, Willott JF, Zheng QY. A major gene affecting age-related hearing loss in C57BL/6J mice. Hear Res. 1997;114(1–2):83–92. doi: 10.1016/s0378-5955(97)00155-x. [DOI] [PubMed] [Google Scholar]
- 10.Shnerson A, Devigne C, Pujol R. Age-related changes in the C57BL/6J mouse cochlea. II. Ultrastructural findings. Brain research. 1981;254(1):77–88. doi: 10.1016/0165-3806(81)90060-2. [DOI] [PubMed] [Google Scholar]
- 11.Hequembourg S, Liberman MC. Spiral ligament pathology: a major aspect of age-related cochlear degeneration in C57BL/6 mice. Journal of the Association for Research in Otolaryngology. 2001;2(2):118–129. doi: 10.1007/s101620010075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bryda EC, Ling H, Flaherty L. A high-resolution genetic map around waltzer on mouse chromosome 10 and identification of a new allele of waltzer. Mamm Genome. 1997;8(1):1–4. doi: 10.1007/s003359900336. [DOI] [PubMed] [Google Scholar]
- 13.Wada T, Wakabayashi Y, Takahashi S, Ushiki T, Kikkawa Y, Yonekawa H, et al. A point mutation in a cadherin gene, Cdh23, causes deafness in a novel mutant, Waltzer mouse niigata. Biochem Biophys Res Commun. 2001;283(1):113–117. doi: 10.1006/bbrc.2001.4724. [DOI] [PubMed] [Google Scholar]
- 14.Lefevre G, Michel V, Weil D, Lepelletier L, Bizard E, Wolfrum U, et al. A core cochlear phenotype in USH1 mouse mutants implicates fibrous links of the hair bundle in its cohesion, orientation and differential growth. Development. 2008;135(8):1427–1437. doi: 10.1242/dev.012922. [DOI] [PubMed] [Google Scholar]
- 15.Schwander M, Xiong W, Tokita J, Lelli A, Elledge HM, Kazmierczak P, et al. A mouse model for nonsyndromic deafness (DFNB12) links hearing loss to defects in tip links of mechanosensory hair cells. Proc Natl Acad Sci U S A. 2009;106(13):5252–5257. doi: 10.1073/pnas.0900691106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deol M. A gene for uncomplicated deafness in the mouse. J Embryol Exp Morphol. 1956;4(2):190–195. [Google Scholar]
- 17.Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, et al. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12(23):3075–3086. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor BA, Navin A, Phillips SJ. PCR-amplification of simple sequence repeat variants from pooled DNA samples for rapidly mapping new mutations of the mouse. Genomics. 1994;21(3):626–632. doi: 10.1006/geno.1994.1323. [DOI] [PubMed] [Google Scholar]
- 19.Polak M, Eshraghi AA, Nehme O, Ahsan S, Guzman J, Delgado RE, et al. Evaluation of hearing and auditory nerve function by combining ABR, DPOAE and eABR tests into a single recording session. J Neurosci Methods. 2004;134(2):141–149. doi: 10.1016/j.jneumeth.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 20.Zheng QY, Ding D, Yu H, Salvi RJ, Johnson KR. A locus on distal chromosome 10 (ahl4) affecting age-related hearing loss in A/J mice. Neurobiol Aging. 2009;30(10):1693–1705. doi: 10.1016/j.neurobiolaging.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mizutari K, Matsunaga T, Kamiya K, Fujinami Y, Fujii M, Ogawa K. Caspase inhibitor facilitates recovery of hearing by protecting the cochlear lateral wall from acute cochlear mitochondrial dysfunction. J Neurosci Res. 2008;86(1):215–222. doi: 10.1002/jnr.21470. [DOI] [PubMed] [Google Scholar]
- 22.Johnson KR, Zheng QY, Erway LC. A major gene affecting age-related hearing loss is common to at least ten inbred strains of mice. Genomics. 2000;70(2):171–180. doi: 10.1006/geno.2000.6377. [DOI] [PubMed] [Google Scholar]
- 23.Johnson KR, Zheng QY, Noben-Trauth K. Strain background effects and genetic modifiers of hearing in mice. Brain Res. 2006;1091(1):79–88. doi: 10.1016/j.brainres.2006.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng QY, Johnson KR. Hearing loss associated with the modifier of deaf waddler (mdfw) locus corresponds with age-related hearing loss in 12 inbred strains of mice. Hear Res. 2001;154(1–2):45–53. doi: 10.1016/s0378-5955(01)00215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zheng QY, Yan D, Ouyang XM, Du LL, Yu H, Chang B, et al. Digenic inheritance of deafness caused by mutations in genes encoding cadherin 23 and protocadherin 15 in mice and humans. Hum Mol Genet. 2005;14(1):103–111. doi: 10.1093/hmg/ddi010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996;266:540–553. doi: 10.1016/s0076-6879(96)66034-0. [DOI] [PubMed] [Google Scholar]
- 27.Yin XM. Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways. Cell Res. 2000;10(3):161–167. doi: 10.1038/sj.cr.7290045. [DOI] [PubMed] [Google Scholar]
- 28.Creagh EM, Conroy H, Martin SJ. Caspase-activation pathways in apoptosis and immunity. Immunological reviews. 2003;193:10–21. doi: 10.1034/j.1600-065x.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 29.George SJ, Beeching CA. Cadherin:catenin complex: a novel regulator of vascular smooth muscle cell behaviour. Atherosclerosis. 2006;188(1):1–11. doi: 10.1016/j.atherosclerosis.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Zhu W, Leber B, Andrews DW. Cytoplasmic O-glycosylation prevents cell surface transport of E-cadherin during apoptosis. Embo J. 2001;20(21):5999–6007. doi: 10.1093/emboj/20.21.5999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, et al. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14(3):347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- 32.Kazmierczak P, Sakaguchi H, Tokita J, Wilson-Kubalek EM, Milligan RA, Muller U, et al. Cadherin 23 and protocadherin 15 interact to form tip-link filaments in sensory hair cells. Nature. 2007;449(7158):87–91. doi: 10.1038/nature06091. [DOI] [PubMed] [Google Scholar]
- 33.Yap AS, Brieher WM, Gumbiner BM. Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol. 1997;13:119–146. doi: 10.1146/annurev.cellbio.13.1.119. [DOI] [PubMed] [Google Scholar]
- 34.Howard J, Hudspeth AJ. Compliance of the hair bundle associated with gating of mechanoelectrical transduction channels in the bullfrog’s saccular hair cell. Neuron. 1988;1(3):189–199. doi: 10.1016/0896-6273(88)90139-0. [DOI] [PubMed] [Google Scholar]
- 35.Jia S, Dallos P, He DZ. Mechanoelectric transduction of adult inner hair cells. J Neurosci. 2007;27(5):1006–1014. doi: 10.1523/JNEUROSCI.5452-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan DK, Lieberman DM, Musatov S, Goldfein JA, Selesnick SH, Kaplitt MG. Protection against cisplatin-induced ototoxicity by adeno-associated virus-mediated delivery of the X-linked inhibitor of apoptosis protein is not dependent on caspase inhibition. Otol Neurotol. 2007;28(3):417–425. doi: 10.1097/01.mao.0000247826.28893.7a. [DOI] [PubMed] [Google Scholar]
- 37.de Brouwer AP, Pennings RJ, Roeters M, Van Hauwe P, Astuto LM, Hoefsloot LH, et al. Mutations in the calcium-binding motifs of CDH23 and the 35delG mutation in GJB2 cause hearing loss in one family. Hum Genet. 2003;112(2):156–163. doi: 10.1007/s00439-002-0833-0. [DOI] [PubMed] [Google Scholar]
- 38.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol Cell Biol. 2001;21(6):2213–2220. doi: 10.1128/MCB.21.6.2213-2220.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Erez N, Zamir E, Gour BJ, Blaschuk OW, Geiger B. Induction of apoptosis in cultured endothelial cells by a cadherin antagonist peptide: involvement of fibroblast growth factor receptor-mediated signalling. Exp Cell Res. 2004;294(2):366–378. doi: 10.1016/j.yexcr.2003.11.033. [DOI] [PubMed] [Google Scholar]
- 40.Melki SJ, Heddon CM, Frankel JK, Levitt AH, Momin SR, O’Brien RG, et al. Pharmacological Protection of Hearing Loss in the Mouse Model of Endolymphatic Hydrops. The Laryngoscope. 2010 doi: 10.1002/lary.21018. In press. [DOI] [PubMed] [Google Scholar]
- 41.Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, et al. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6(7):797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- 42.Zender L, Hutker S, Liedtke C, Tillmann HL, Zender S, Mundt B, et al. Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc Natl Acad Sci U S A. 2003;100(13):7797–7802. doi: 10.1073/pnas.1330920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehret G. Masked auditory thresholds, critical ratios, and scales of the basilar membrane of the housemouse (Mus musculus) J Comp Physiol. 1975;130:329–341. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table. S1. Mapping of the mutation in erlong mice. (A) DNA from affected mice and unaffected mice was analyzed for MIT markers (D10Mit166, D10Mit194 and D10Mit42) on chromosome 10, which are listed in the top row and indicate the base pair position (NCBI Build 37). The mouse identification numbers (ped#) (column K) and phenotypes (ABR thresholds, db SPL, column E-H at five month of age column B) are indicated in the 2 top rows. The genotype for each marker is given as: b, homozygous C57BL/6J genotype; c, homozygous C3H/HeJ; and h, heterozygous. The chromosomal intervals that segregate with the mutant phenotype are highlighted with boxes.
Four Cdh23erl/erl and 4 Cdh23ahl/ahl mice at the ages of 2 weeks and 2 months, respectively, were used. The primers for GAPDH and Cdh23 are described in the methods and the other primers are listed in Table 2. The relative mRNA levels of Cdh23, TNF-α, caspase-12, m-Calpain and μ-Calpain were averaged for each genotype and plotted, with error bars indicating standard error from the mean. The results indicated that there was no significant difference for the relative mRNA levels of Cdh23 between the Cdh23ahlahl and Cdh23erl/erl mouse groups (P > 0.05). However, the relative mRNA levels of TNF-α, caspase-12, m-Calpain and μ-Calpain were significantly up-regulated in the Cdh23erl/erl mice at both time points of 2 weeks (a, c) and 2 months (b, d), indicating that multiple apoptosis pathways are involved in the hearing loss of the mutant mice. *P < 0.05; **P < 0.01
Six littermate Cdh23v-2J/v-2J mice and 3 Cdh23v-2J/+ mice at the age of P8 were used. Three Cdh23v-2J/v-2J mice received 4 doses of Z-VAD-FMK (1.5mg/kg mouse weight, IP injection in DMSO solvent) at P8, P9, P10 and P13. Three Cdh23v-2J/v-2J and three Cdh23v-2J/+ mice were simultaneously injected with an equivalent dose of DMSO without Z-VAD-FMK. Click and pure tone (8 kHz, 16 kHz, and 32 kHz) ABR thresholds were assessed at P17. We found none of the Cdh23v-2J/v-2J mutants that received Z-VAD-FMK had detectable ABR at 100 dB but the Cdh23v-2J/+ mice had normal ABR thresholds as expected at P17 (data not shown). Figure S2 shows the results of H&E staining of the basal turn of cochleae from mice at P17 (a–c), with enlarged views of the organ of Corti (OC) region in panels (d–f). Outer hair cells (OHC) were lost in the DMSO sham-treated Cdh23v-2J/v-2J mice (a, d), and only support cells remained (arrowhead in d). However, as with the DMSO-treated Cdh23v-2J/+ mice (c, f), the Z-VAD-FMK (in DMSO) treated Cdh23v-2J/v-2J mice showed intact OHCs (4 arrows in e) in the organ of Corti in multiple serial sections. The results indicate that Z-VAD-FMK treatment can protect OHCs in Cdh23v-2J/v-2J mice, but fails to improve the hearing of the mice. SV, scala vestibuli; SM, scala media; RM, Reissner’s membrane; StV, Stria vascularis; SL, spiral ligament; SG, spiral ganglion. Scale bars = 50 μm.