

Abstract
Zinc plays a crucial role in the biology of p53 in that p53 binds to DNA through a structurally complex domain stabilized by zinc atom. The p53 negative regulator MDM2 protein also carries a C-terminal RING domain that coordinates two zinc atoms which are responsible for p53 nuclear export and proteasomal degradation. In this clinically translatable study, we explored the critical role of zinc on p53 re-activation by MDM2-inhibitor MI-219 in colon and breast cancer cells. ZnCl2 enhanced MI-219 activity (MTT, apoptosis and colony formation), and chelation of zinc not only blocked the activity of MI-219, it also suppressed re-activation of the p53 and its downstream effector molecules p21WAF1 and Bax. TPEN, a specific zinc chelator but not Bapta-AM, a calcium chelator, blocked MI-219-induced apoptosis. Nuclear localization is a pre-requisite for proper functioning of p53 and our results confirm that TPEN and not Bapta-AM could abrogate p53 nuclear localization and interfered with p53 transcriptional activation. Addition of zinc suppressed the known p53 feedback MDM2 activation which could be restored by TPEN. Co-immunoprecipitation studies verified that MI-219-mediated MDM2-p53 disruption could be suppressed by TPEN and restored by zinc. As such, single agent therapies that target MDM2 inhibition, without supplemental zinc, may not be optimal in certain patients due to the less recognized mild zinc deficiency among the “at risk population” as in the elderly which are more prone to cancers. Therefore, use of supplemental zinc with MI-219 will benefit the overall efficacy of MDM2 inhibitors and this potent combination warrants further investigation.
Keywords: MDM2 and p53, Small molecule inhibitors, Zinc, Apoptosis
Introduction
The tumor-suppressor p53 is the most frequently mutated gene in human cancers. However, approximately 50% of all human tumors retain normal or wild type p53 (wt-p53) (Lane and Fischer 2004). Direct activation of p53 in these tumors could in principle be used as a means to eradicate tumor cells (Brown et al. 2009). p53 is activated in response to a variety of stresses, such as DNA damage, nutrient deprivation or oncogenic activation, resulting in the transcriptional activation of target genes involved in growth arrest and apoptosis (Feng et al. 2008). To protect healthy cells from the deleterious effects of uncontrolled p53 activation, p53 is subject to a negative feedback loop activated by the protein product of one of its target genes, MDM2 (Marine and Lozano 2010). The protein MDM2 binds to p53, inhibits its transcriptional activation, causes nuclear export and acts as an E3 ligase to target p53 for proteasomal degradation (Kubbutat et al. 1997). Thus, there is a fine balance between MDM2, p53 and the need for p53 activation to promote cell survival or apoptosis following DNA damage or other cellular stresses. Unfortunately, in many cancers, the MDM2 protein is over expressed and suppresses the activation of even the functional wt-p53, thereby disrupting the finely-tuned balance of cell survival versus cell death. The end result is a loss of control of the normal apoptotic processes and contributes to drug resistance. One potential approach for re-activating p53 in tumor cells is to disrupt the interaction between MDM2 and p53 with the MDM2-targeting small molecule MI-219 or related inhibitors (Shangary and Wang 2009; Shangary et al. 2008; Verma et al. 2010; Vassilev 2007). MI-219 binds to MDM2, thereby preventing the interaction with p53 and causing p53 to be stabilized. We and others have shown that MI-219 can induce growth inhibition and apoptosis in multiple cancer cell lines and also induce growth arrest in corresponding tumor xenografts (Yu et al. 2009; Canner et al. 2009; Mohammad et al. 2009; Shangary and Wang 2009; Shangary et al. 2008).
Wt-p53 is one of the best recognized “zinc-finger” transcription factors and binds DNA through a sequence-specific DNA-binding domain (p53DBD) extending from amino acid residues 96–308 (Bargonetti et al. 1993). The p53DBD incurs an unusually high number of mutations that consequently results in a failure to bind DNA and prevention of p53-induced transcription (Levine et al. 1995; Levine 1997). This fact strongly suggests that sequence-specific DNA binding and transactivation are the key activities that control the biological functions of p53 (Meek 1998). The crystal structure of p53DBD reveals that the p53 core domain structure consists of a beta sandwich that serves as a scaffold for two large loops (L2 and L3) and a loop–sheet–helix motif (L1) (Pavletich et al. 1993). Zn2+ is coordinated to C176 and H179 of the L2 loop and C238 and C242 of the L3 loop (Pavletich et al. 1993; Cho et al. 1994). Zinc coordination has been demonstrated to be necessary for the proper folding of the p53 core domain in vitro and disruption of this interaction greatly reduces or abrogates p53 DNA binding and transactivation of target genes (Meplan et al. 2000). NMR spectra reveal that the DNA-binding surface is altered by removing zinc ion and fluorescence anisotropy studies show that zinc ion removal abolishes site-specific DNA-binding activity (Butler and Loh 2007; Butler and Loh 2003). Using a cell-permeable metal chelator, previous investigators were able to show that depletion of intracellular zinc could induce a change in p53 protein conformation, with loss of DNA-binding capacity, that was reversible upon removal of the chelator from the culture medium or the addition of zinc to the media (Verhaegh et al. 1998). The amount of supporting information certainly highlights the crucial role of zinc in the biology of p53 protein and its importance for DNA binding as well as stability of this important tumor suppressor.
The p53 negative regulator MDM2 protein contains a C-terminal RING domain (aa 430-480) which coordinates two molecules of Zinc (Wawrzynow et al. 2009a; Wallace et al. 2006). This unique RING finger is responsible for shuttling p53 out of the nucleus for proteasomal degradation and is also responsible for MDM2 auto-ubiquitination (Itahana et al. 2007; Lindstrom et al. 2007; Alshatwi et al. 2006). The intrinsic E3 activity of MDM2 is dependent on its zinc-coordinated RING finger domain. The capacity to mediate MDM2's own ubiquitination requires no eukaryotic protein other than E1 and E2. Therefore, Zinc acts as a “double-edged sword” promoting p53 activity while simultaneously causing MDM2 auto-ubiquitination.
Mild zinc deficiency is a common occurrence in the elderly, a population that is well known to be more prone to cancers (Prasad 2004; Prasad 2001; Prasad et al. 1993). The observed deficiency of zinc in the elderly has been attributed to a combination of factors that includes poor zinc absorption as well as consumption of low-zinc diets (Hambidge et al. 1998). Low intracellular zinc has been found in different tumors and has been correlated to induction of oxidative DNA damage, disruption of p53, AP1 DNA binding and affects DNA repair (Ho and Ames 2002; Ho 2004; Ho and Song 2009). Therefore, therapies that are based on using small molecule inhibitors to target MDM2 may not be fully successful in the clinical setting or in patients due to unrecognized mild to moderate zinc deficiency. The studies presented in this paper demonstrate that zinc is crucial for the activity of p53 re-activated by MI-219 and provides evidence for using combination therapies with MI-219 that include supplemental zinc for the treatment of wt-p53 tumors. Prior to MDM2 inhibitors making their way into the clinic for patients, we believe that the results of our findings will have high impact towards the design of novel treatment strategies (i.e combination with zinc) for achieving better survival outcome.
Results
Metal chelation in general and zinc-specific depletion suppresses MI-219-induced growth inhibition and apoptosis
In order to verify the role of metal ions on the activity of MI-219 we performed growth inhibition and apoptosis assays in media chelated to remove trace and non-trace metals (chelexed media). Assessment of chelation using atomic absorption spectroscopy confirmed chelation of zinc (and minimal chelation of copper) (Supplementary Table 1). Our laboratory has previously standardized the procedure of zinc chelation that minimally alters the status of other trace metal ions such as iron, copper and magnesium (Bao et al. 2006; Prasad et al. 2002; Prasad et al. 2001). HCT-116 and MCF-7 cells (wt-p53) were passaged in chelexed media and then treated with MI-219 (0-10 μM for 72 hrs) followed by analysis of growth inhibition by MTT assay. In normal media the IC50 of MI-219 in HCT-116 and MCF-7 cells is 4μM and 3.5 μM respectively. However, in chelexed media depleted of trace metals, the IC50 increased to >10 μM (Fig 1A). Similar to the results obtained from MI-219-induced growth inhibition (0-10 μM for 72 hrs), MI-219 was also less effective in inducing apoptosis in chelexed media (Fig 1B). We did not observe any appreciable toxicity represented as change in growth patterns in cells grown in chelexed media vs those grown in normal media (Fig 1C). Based on our preliminary studies to define the optimal conditions, we found that specifically chelating Ca (using Bapta-AM) or copper (using Bath) had no effect on the events studied here and these specific chelators were used as negative controls in studies designed to determine the specific effect of zinc in MI-219-induced p53 re-activation. To delineate the crucial role of zinc on the efficacy of MI-219 we first evaluated the effect of ZnCl2 (0-32 μM) on both HCT-116 and MCF-7 cells. As can be seen from results of Fig 2A. ZnCl2 alone did not induce any appreciable growth inhibition in both cell lines. We then performed dose kinetics experiment to verify the effect of ZnCl2 on MI-219 mediated cell growth inhibition. As can be seen from our novel results of Fig 2B ZnCl2 at increasing doses progressively enhanced cell growth inhibition by MI-219. We also tested the effect of this combination on colony formation capability in HCT-116 cells. Fig 2C shows colony formation in control and zinc alone treated cells. MI-219 alone as expected suppressed colony formation. However the most important and clinically relevant results are observed in the combination treatment where no colonies were visible even after 4 weeks. Taken together these results indicate that MI-219 requires trace metal ions specifically zinc for its proper activity. Since Chelex is non specific metal chelator, in the next experiments we used TPEN (a well recognized membrane permeable zinc-specific chelator) to verify the exclusive role of zinc in mediating MI-219-induced apoptotic effects. Histone DNA ELISA results shown in Fig 3A, confirm that addition of TPEN (1 μM) significantly blocks MI-219-mediated apoptosis and this could be restored by the addition of 16 μM ZnCl2 to both HCT-116 and MCF-7 cells. We also performed Annexin V FITC apoptosis analysis to verify that MI-219 mediated apoptosis could only be blocked by TPEN and not other metal chelators such as the calcium specific Bapta-AM or copper chelator bathocuproine (Fig 3B). Taken together these results highlight a significant role of zinc in the activity of MI-219 on wt-p53 cells. We then tested the role of zinc on the re-activation of p53 pathway in both HCT-116 and MCF-7 cells.
Figure 1. Chelation suppresses MI-219-mediated growth inhibition and apoptosis.
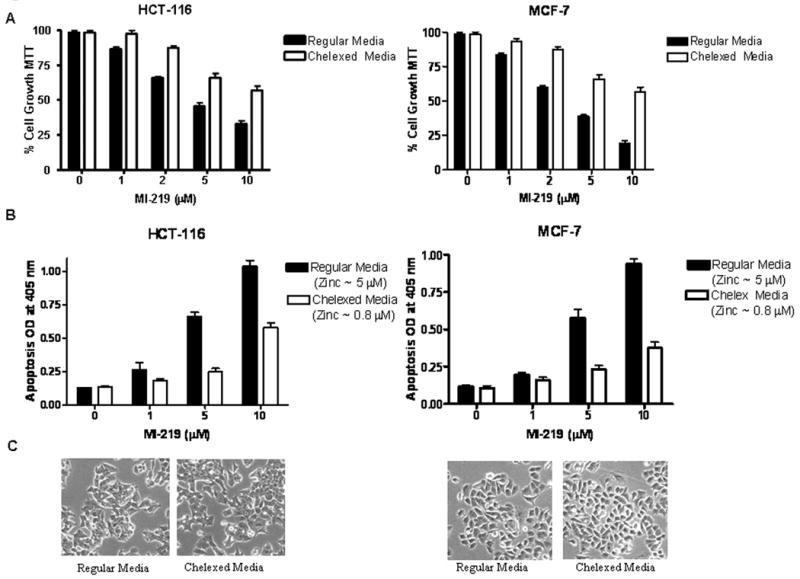
[A] Wt-p53 HCT-116 and MCF-7 cells were treated with MI-219 (0-10 μM for 72 hrs) in either regular or chelexed media. Note: Growth inhibition (MTT assay) by MI-219 is diminished in chelexed media. [B] Chelation reduced media zinc levels from 5 μM to 0.8 μM (AAS analysis Supplementary Table 1) and this was associated with reduced MI-219-induced apoptosis (detected by Histone/DNA ELISA assay) in both cell lines (right and left panels). [C] Microphotographs of HCT-116 and MCF-7 cells showing no apparent toxicity reflected as changes in growth pattern of cells grown in either regular or chelexed media. All experiments were performed in triplicates, data presented represents mean of three independent experiments.
Figure 2. Zinc enhances MI-219 mediated inhibition of cell growth and colony formation.
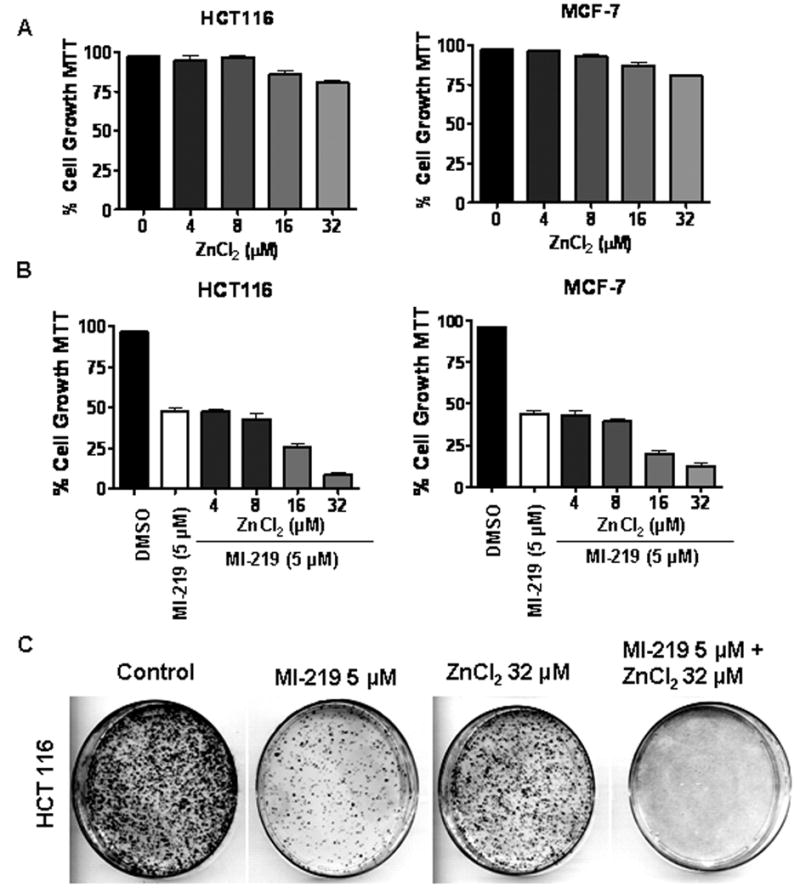
[A] HCT-116 and MCF-7 cells were exposed to increasing concentrations of ZnCl2 (0-32 μM) for 72 hrs and cell growth inhibition was detected using MTT assay. [B] Cells were exposed to either MI-219 5 μM in combination with ZnCl2 (0-32 μM) for 72 hrs and cell growth inhibition was detected by MTT assay. [C] Microphotographs of cell survival of HCT-116 cells at indicated treatments and evaluated by the clonogenic assay. Note significant reduction in the colony formation in the combination compared to cells treated with either MI-219 (10 μM) or ZnCl2 (32 μM) alone. All experiments were performed in triplicates, data presented represents mean of three independent experiments.
Figure 3. TPEN a zinc specific chelator blocks apoptosis by MI-219.
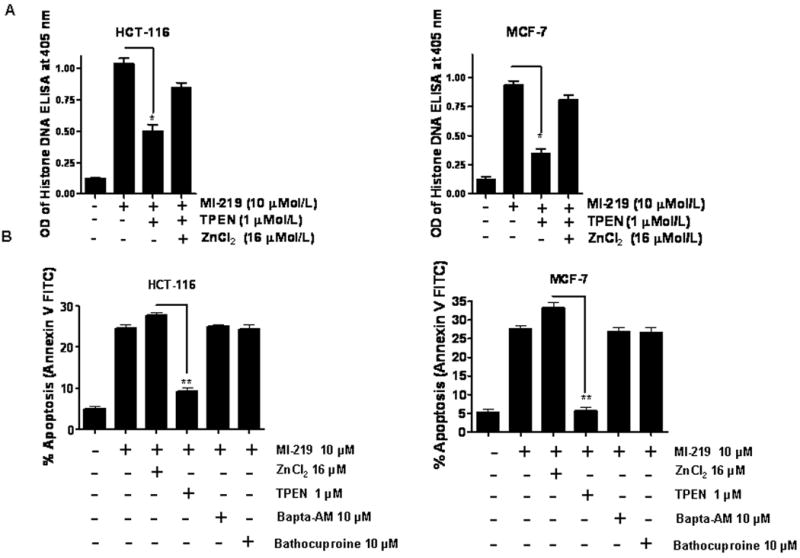
[A] Cells were treated with MI-219 at indicated concentration for 72 hrs. TPEN blocked MI 219-induced apoptosis (Histone/DNA ELISA) which could be restored by addition of physiological levels (16 μM) of ZnCl2. [B] Zinc augments MI-219-induced apoptosis (Annexin V FITC assay) which can be abrogated by TPEN. Depletion of calcium using the calcium specific chelator, Bapta-AM or copper, using a specific copper chelator, bathocuproine, had no effect on MI-219-induced apoptosis in either cell line. All experiments were performed in triplicate, data presented represents mean of three independent experiments. * represents p<0.05 and ** p<0.01.
Zinc depletion blocks MI-219 mediated p53 re-activation
It is well known that for its transcriptional activity, p53 requires a specific DNA binding sequence that is coordinated by a complex helix domain motif stabilized by a zinc atom. It is therefore expected, that by modifying zinc levels, one would affect the DNA binding capacities of p53 which would result in altering the re-activation of p53 induced by MI-219. We performed western blot analysis to determine the specific role of zinc on p53 reactivation. As can be seen from results of Fig 4A & B, in regular media MI-219 treatment (0-10 μM for 24 hrs) in both HCT-116 and MCF-7 cells resulted in sequential up-regulation of p53 along with downstream effecter molecules such as the cell cycle regulator p21 and the pro-apoptotic Bax. However, using chelexed media, we found negligible to modest expression of p53. Furthermore, the downstream effector's (p21 and Bax) were also not up-regulated. We then used TPEN to evaluate the exclusive role of zinc on p53 re-activation on cell growth in normal media. In line with our cell growth inhibition and apoptosis studies using chelexed media, TPEN (1 μM) significantly blocked p53 pathway re-activation in both HCT-116 and MCF-7 cells which could be partially restored by addition of 16 μM ZnCl2 (Fig 4C). These results confirm our hypothesis that zinc is required for the proper reactivation of p53 by MI-219.
Figure 4. Zinc chelation blocks p53 re-activation by MI-219.
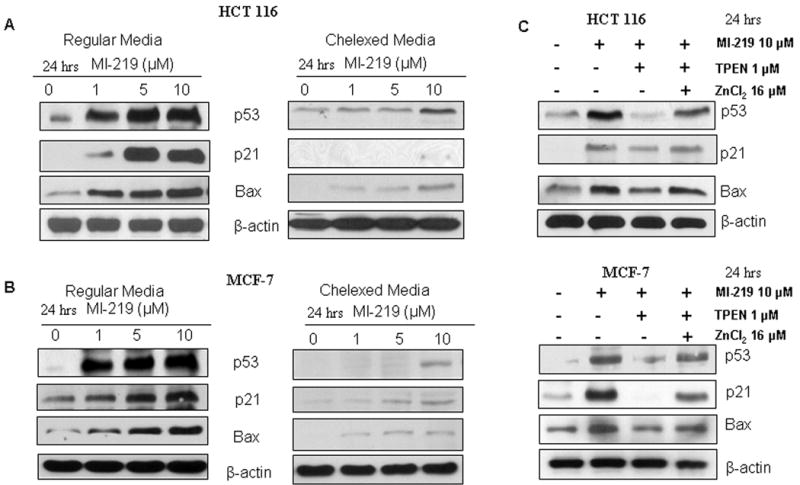
HCT-116 and MCF-7 cells were grown in either regular or chelexed media and treated with increasing doses of MI-219 (0-10 μM for 24 hrs). Protein lysates were analyzed using western blot analysis for activation of the p53 pathway. [A & B] Cells grown in chelexed media exhibited insignificant amounts of p53 or its downstream p21 and Bax compared to cells grown in regular media. [C] MI-219 inhibition of MDM2 allowed for increased p53 and downstream Bax and p21 expression which could be abrogated by TPEN (a zinc specific chelator) in normal (non-chelexed) media and then reversed by the addition of physiological levels of ZnCl2. HCT-116 and MCF-7 cell were exposed to either DMSO; MI-219 (10μM); MI-219 (10 μM) + TPEN (1μM) or MI-219 (10 μM) + TPEN (1μM) + ZnCl2 (16 μM) (24 hrs) and western blot analysis was performed on lysates isolated from treated cells. Blots are representative of three independent experiments. β-actin was used as loading control.
Zinc chelation blocks p53 nuclear localization
Nuclear localization of p53 is a necessary pre-requisite for its proper function (O'Brate and Giannakakou 2003). Therefore, we tested whether zinc chelation could alter the nuclear localization of p53. To achieve this, we used a fluorescent microscopy p53-Cell-Based Activation/Translocation assay kit coupled with a highly specific p53 primary monoclonal antibody and DyLight™ conjugated secondary antibody. As can be seen from results shown in Fig 5A, in both HCT-116 (upper panel) and MCF-7 (lower panel) cells, MI-219 (5 μM) could induce p53 nuclear localization in normal media but not in chelexed media. Chelation of zinc with TPEN could suppress p53 localization that again, could be restored by addition of ZnCl2 (16 μM). To further confirm that MI-219 activity is specifically directed towards wt-p53, we used siRNA against p53 and as can be seen from results of Fig 5B, in both HCT-116 and MCF-7 cells, p53 knockdown shows neither p53 activation nor translocation induced by MI-219. To reaffirm siRNA silencing of p53, we also performed western blot analysis to demonstrate negative expression of p53 in p53 siRNA treated and not control siRNA treated cells (Fig 5C). These results for the first time, confirm that zinc is crucial for the proper re-activation of p53 as well as its nuclear localization induced by the MDM2 inhibitor, MI-219.
Figure 5. TPEN suppresses p53 nuclear localization.
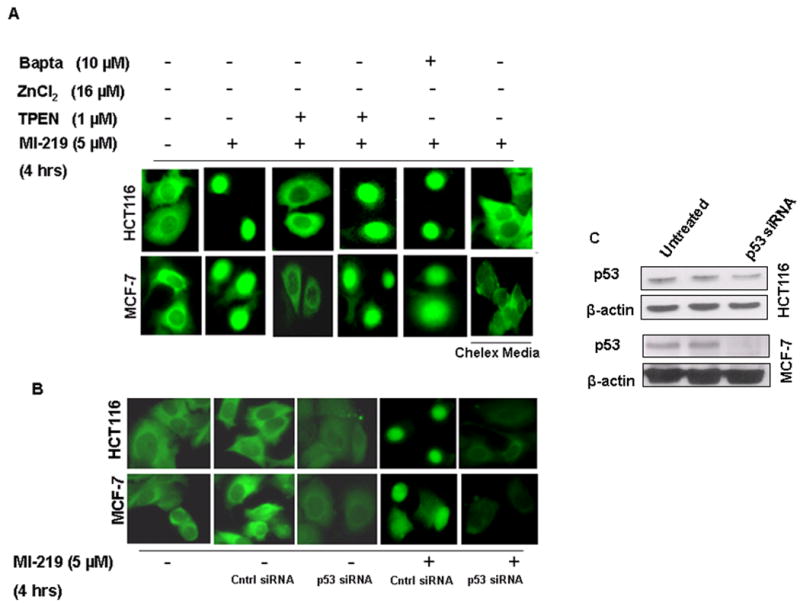
[A] MCF-7 and HCT-116 cells (1000) were grown in triplicate in 96 well plates and treated with either vehicle (DMSO); MI-219 (5 μM); MI-219 (5 μM) + TPEN (1 μM); MI-219 (5 μM) + TPEN (1 μM) + ZnCl2 (16 μM) or MI-219 (5 μM) + Bapta-AM (10 μM) for 4 hrs. p53 activation/localization assay was performed using p53 localization kit according to manufacturer's protocol (Cayman Chemicals Ann Arbor, MI, USA). Note: MI-219-induced p53 localization in the nucleus is blocked by TPEN. Zinc treatment restores p53 in the nucleus even in the presence of TPEN. Bapta-AM, a calcium chelator, had no effect on p53 nuclear localization. Cell grown in chelex media are also resistant to MI-219 mediated p53 nuclear localization. [B] p53 siRNA suppresses p53 nuclear localization/activation. HCT-116 and MCF-7 cells were grown in 96 well plates and treated with either control siRNA or p53 siRNA for 5 hrs in the absence or presence of MI-219 5 μM for 4 hrs and p53 localization was detected. siRNA treated MCF-7 and HCT-116 cells show reduced p53 in the nucleus even in the presence of MI-219. [C] Western blot analysis showing siRNA mediated suppression of p53 in both HCT-116 and MCF-7 cells.
Zinc augments p53 transcription in HCT-116 and MCF-7 cells
We tested the p53 transcription activity using a sensitive non-radioactive method for detecting specific transcription factor DNA binding activity in nuclear extracts. In this assay, a specific double-stranded DNA (dsDNA) sequence containing the p53 response element is immobilized onto the wells of a 96-well plate. p53 present in a nuclear extract, binds specifically to the p53 response element and is detected by addition of a specific primary antibody directed against p53. A secondary antibody conjugated to HRP is added to provide a sensitive colorimetric readout at 450 nm. As can be seen from results of Fig 6A MI-219 (10 μM) induces p53 transcription that is significantly augmented by addition of ZnCl2 (16μM). In line with our growth inhibition, apoptosis, western blot and p53 nuclear localization results, TPEN suppressed MI-219-mediated p53 transcription that could be restored by addition of zinc. Furthermore, the MI-219 inactive analog, MI-10, did not induce p53 transcriptional activity. These results are in further support of our hypothesis that zinc plays an important role in the biology of p53 and its use in conjunction of such a targeted therapy.
Figure 6. Zinc enhances MI-219 mediated p53 transcription that is blocked by TPEN.
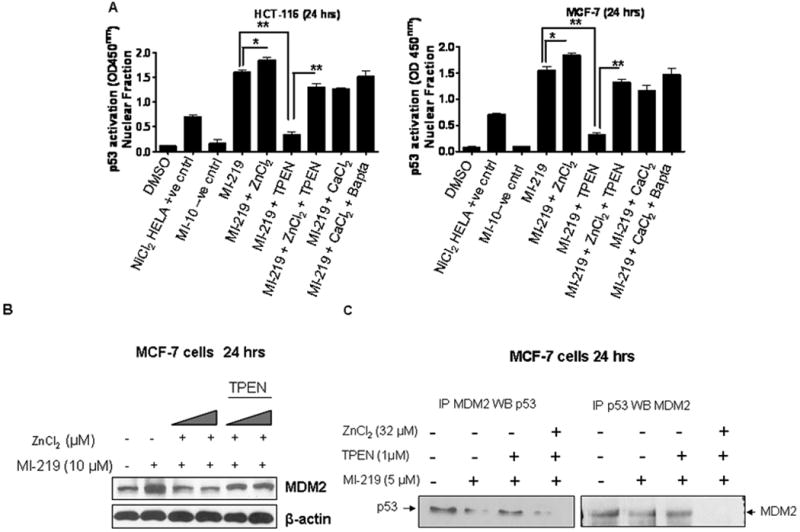
[A & B] Semi-confluent HCT-116 and MCF-7 cells grown in triplicate were exposed to either vehicle (DMSO); MI-219 (10 μM); MI-219 (10 μM) + ZnCl2 (16 μM); MI-219 (10 μM) + ZnCl2 (16 μM) + TPEN (1 μM); MI-219 (10 μM) + CaCl2 (15 μM) or MI-219 (10 μM) + CaCl2 (15 μM) + Bapta-AM (10 μM) for 24 hrs in 6 well plates. Nuclear lysates were isolated and p53 transcription assay was performed in 96 well plates according to manufacturer's protocol (Cayman Chemicals Ann Arbor, MI, USA). ZnCl2 (16 μM) enhances MI-219 (10 μM) mediated p53 transcription that is blocked by TPEN. Calcium does not induce p53 transcription and the calcium chelator, Bapta-AM has no affect. NiCl2 treated HELA cell lysates were used as positive control (provided in kit) while MI-10 the inactive analogue of MI-219 was used as negative control. * represents p<0.05 and ** p< 0.01. [C] ZnCl2 suppresses MDM2 protein expression and enhances MI-219-induced MDM2-p53 disruption. MCF-7 cells were exposed to either vehicle (DMSO); MI-219 (10 μM); MI-219 (10 μM) + ZnCl2 (16 μM); MI-219 (10 μM) + ZnCl2 (32 μM); MI-219 (10 μM) + ZnCl2 (16 and 32 μM) in the presence of TPEN (1 μM) for 24 hrs and protein lysates were subjected to western blot analysis followed by probing with MDM2 antibody. ZnCl2 (16 and 32 μM) suppressed MDM2 expression [D] Co-immunoprecipitation studies of MCF-7 cells demonstrate reduced disruption of MDM2-p53 interaction in the presence of TPEN. Again ZnCl2 abrogates the effects of TPEN. Blots are representative of three independent experiments.
Zinc suppresses p53-MDM2 regulatory feedback mechanism
MDM2 contains an auto-regulatory RING finger domain that coordinates zinc atoms. Therefore, we sought to assess the role of zinc addition and chelation on the activity of MDM2. As can be seen from results shown in Fig 6B, MI-219 alone at 10 μM induces MDM2 expression which was expected since activated p53 positively regulates MDM2 (Levine 1997). However, in the presence of zinc (ZnCl2 16 & 32 μM), the induction of MDM2 was suppressed but could be restored by addition of TPEN. These results provide evidence that zinc is required for proper positive regulation of MDM2 by p53. We further performed Co-immunoprecipitation experiments to verify the affect of zinc chelation/addition on disruption of MDM2-p53 complex. As can be seen from the results shown in Fig 6C, MI-219 disrupts the MDM2-p53 interaction (with less protein precipitated in the treated samples) and this could be reversed by the addition of TPEN. The addition of supplemental ZnCl2 (32 μM) could negate the effect of zinc chelator TPEN, thereby restoring the MI-219-induced disassociation between MDM2 and p53.
Discussion
This clinically translatable study, for the first time, provides insight into the mechanisms by which zinc affects the activity of the MDM2 inhibitor MI-219 in mediating proper reactivation of p53 leading to apoptosis in wild type p53 cancer cells. MI-219 is entering phase I clinical trials. Prior to MDM2 inhibitors making their way for treatment of cancer patients, we believe that our findings will positively impact the design of MDM2 inhibitor therapy.
According to the International Zinc Nutrition Consultative Group (IZNCG) 20% of the world's population lack sufficient zinc in their diet, while one-third live in countries considered at high risk of zinc deficiency (Brown et al. 2004). Zinc is required by at least 200 different proteins and transcription factors (Christianson 1991). Zinc binding by p53 was confirmed when the partial crystal structure of the protein was published (Collins et al. 1997). The zinc atom has an essential structural role in stabilizing the architecture of the DNA-binding domain of p53 and it has clearly been shown that zinc is an important co-factor for p53 DNA-binding activity in vitro. Most of this evidence relies on the fact that metal chelators can remove zinc from p53, turning the protein to a ‘mutant-like’ form with loss of such sequence-specific DNA-binding activity (Meplan et al. 1999). Likewise the p53 regulator MDM2 carries a RING domain that co-ordinates two zinc atoms (Wawrzynow et al. 2009b). The major functions of this RING domain in to mediate nuclear export of p53 for proteasomal degradation and also induce MDM2 auto-ubiquitination. Therefore, we believe that zinc acts as a double edged sword on one hand promoting p53 re-activation and on the other causing MDM2 degradation. Most of intracellular zinc is immobilized in proteins, however, cells also contain a pool of labile zinc, which is in dynamic equilibrium with the extracellular medium (Zalewski et al. 1993; Zalewski et al. 1994). This labile zinc can undergo fluctuations (3-5 fold) depending on the availability of zinc. Such changes in zinc levels (1-10 μM range) might have regulatory effects on specific, intracellular metalloproteins including the all important p53.
Strategies that utilize MDM2 inhibition to re-activate p53 pathway may not be fully successful in zinc deficient environments/patients. Based on this assumption, we investigated in wt-p53 cancer cells (HCT-116 and MCF-7) and demonstrated that, zinc is necessary to the activity of MDM2 inhibitor in mediating enhanced reactivation of p53 leading to apoptosis. Our findings confirmed that in the absence zinc, the efficacy of MI-219 is diminished. Following that, we verified the effect of zinc chelation on MI-219-induced p53 re-activation. As expected, p53 pathway including downstream affector p21 and Bax, re-activation was markedly suppressed by zinc chelation and most significantly, this activity was restored by the addition of zinc. These results, provide irrevocable evidence for the crucial requirement of zinc on the activity of MI-219 in restoring superior p53 functioning.
Cytoplasmic p53 is rapidly degraded by MDM2 mediated ubiquitination; a reason for inhibiting MDM2 activity using MI-219. However, under stress, p53 shuttles to nucleus where it trans-activates numerous genes regulating cell cycle, apoptosis and senescence (Chen et al. 2006). It is logical to hypothesize that by blocking MDM2-p53 binding, p53 would be allowed to localize into the nucleus and initiate transcription of target genes. To demonstrate that this is a zinc dependent phenomenon, we found that in the presence of zinc chelator TPEN, p53 nuclear localization was significantly diminished and that the addition of zinc could restore this function (Figure 5). Using siRNA against p53 to silence the p53 expression reaffirmed the nuclear localization data observed above which further re-instates that zinc is a crucial component of the p53 localization signaling pathways. On a cautionary note, we believe that it is too early to fully explain how zinc chelation blocks p53 nuclear localization. One possibility is that zinc deficiency may induce p53 conformational change that would hinder proper alignment of p53 to the nuclear localization signal sequence (NLS) and thus block its nuclear translocation. Another explanation for the observed effects comes from an earlier study done by Giannakakou and group, where it was shown that in low zinc environment the microtubule assembly required to position p53 on NLS is disturbed and results in reduced nuclear localization (Giannakakou et al. 2002). However, it is understood that more work is needed in this direction before concluding to the exact mechanism behind such reduced p53 nuclear localization. Pavletich et al., have reported that for proper transcription, p53 requires specific DNA binding sequence that is regulated by zinc atoms (Pavletich et al. 1993). In this study we also found that by lowering zinc levels p53 transcription activity was suppressed (Figure 6). Fortified with the above strong evidence, we correctly hypothesized that zinc was required for the efficacy of MI-219 on the re-activation of p53.
It is well recognized that MDM2 expression is under a positive feedback loop of p53. However, over-expression of MDM2 as may occur in cancer cells inhibits the p53 re-activation. This would make the cancerous/tumor cells to multiply continuously or become resistant to chemotherapy. To this end we explored whether zinc could affect the MI-219-induced inhibition of MDM2 and causes p53 release and thus, p53 re-activation. Our results show that the addition of zinc can significantly enhance MI-219-induced suppression of MDM2-p53 interaction. That this suppression could be reversed by a specific zinc chelator, TPEN, provides clear proof for the role of zinc in this process. We believe that zinc chelation may change the p53 binding pocket in MDM2, it is possible that zinc binding to the sites on MDM2 may alter its conformation and thus its p53 binding affinity (summarized in Figure 7). Such binding studies using x-ray crystallography are currently under study in our laboratory. In conclusion therapies which use target p53 reactivation using MDM2 inhibitor approach may still not be fully successful in zinc deficient environment that is commonly found in a sizable patient population (Prasad et al. 1993; Prasad 2001; Prasad 2004). Therefore, a logical step forward is to use supplemental zinc to enhance the efficacy of MI-219 for treatment of cancers.
Figure 7. Zinc plays a crucial role in the biology of p53 (a mechanistic summary).
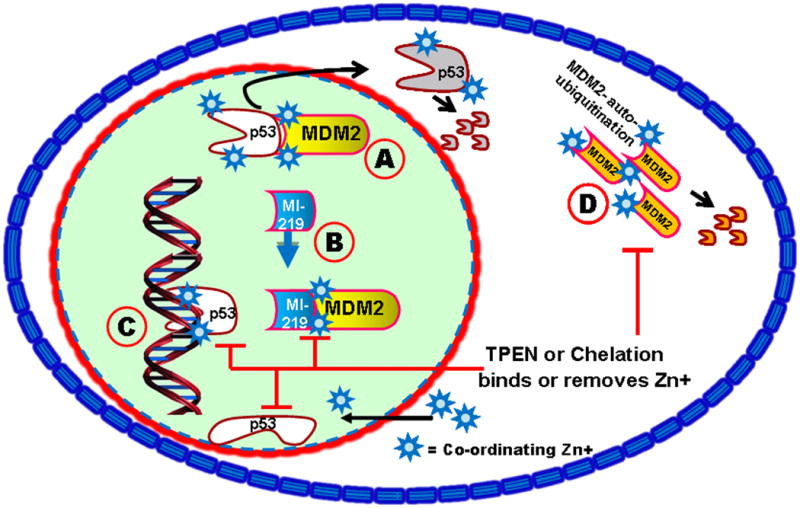
[A]. MDM2 binds to p53 and targets p53 for nuclear export and proteasomal degradation. [B] MI-219's ability to block this interaction is enhanced by the presence of zinc, releasing p53 to bind to DNA. [C] p53 sequence specific DNA binding requires crucial zinc atoms. If present, p53-initiated transcription blocks p53-MDM2 feedback loop and promotes MDM2 autoubiquitination. Chelation or TPEN, a membrane permeable metal chelator, abrogates zinc mediated effects by removing zinc from p53 and causing a conformational change to prevent DNA binding. Changes in binding affinity induced by zinc (or its removal) may also apply to MI-219-MDM2 and MDM2-p53 interaction as well. [D]. TPEN also blocks zinc-mediated MDM2 auto-ubiquitination.
Materials and Methods
Cell culture, experimental reagents and chemicals
HCT-116 (wt-p53) was obtained from Dr. Bert Vogelstein's lab. MCF-7 wt-p53 breast cancer cell line was purchased from ATCC. The cell lines have been tested and authenticated in our core facility, Applied Genomics Technology Center at Wayne State University, as late as March 13, 2009. The method used for testing was short tandem repeat (STR) profiling using the PowerPlex® 16 System from Promega (Madison, WI). Primary antibodies for p53, Bax, MDM2 and p21 were purchased from Cell Signaling (Beverly MA). All secondary antibodies were obtained from Sigma (St. Louis, MO). MI-219 was synthesized by using our previously published methods (Ding et al. 2005; Ding et al. 2006). Chelex 100 resin, the zinc chelator N,N,N′N′-tetrakis(-)[2-pyridylmethyl]-ethylenediamine (TPEN), the calcium chelator 1,2-bis-(o-Aminophenoxy)- ethane-N,N,N′,N′-tetraacetic acid (BAPTA-AM), the copper chelator, bathocuprione disulfonate (Bath) and ZnCl2 were purchased from Sigma.
Metal deficient media
Chelation of media and other solution with chelex 100 resin is a non-specific method of removing unbound or excess trace and to some extent non-trace metals. The advantage of its use is that the resin bound to the metals can be physically removed from the media. Metal ions were removed from the culture media, DMEM or McCoy 5 A by incubating regular media (DMEM supplemented with FBS penicillin and streptomycin and 10% glutamine or McCoys 5A media supplemented with FBS penicillin and streptomycin, 10% glutamine and 10% HEPES) with chelex 100 resin (1 gram per 50 ml media) for 1 hr at 37°C. After incubation, the media was vacuum filtered to remove the metal bound resin. The zinc and copper content was determined using atomic absorption spectroscopy (AAS) using our previously established methods (Beck et al. 2004) and other electrolytes plus iron determined by the clinical laboratories at the Detroit medical center. Chelation of particular metal ions was accomplished by adding metal-specific chelators to normal media and include TPEN for zinc, BAPTA-AM for Calcium, Bath for copper and desferroxamine for Iron.
Cell growth inhibition studies by MTT and clonogenic assay
HCT-116 and MCF-7 cells (3 × 103) were seeded in a 96-well culture plate either in regular or chelexed media and treated with MI-219 (0 or 10 μM) for 72 hrs and MTT assay was performed as described earlier (Azmi et al. 2008). The results were plotted as means ± SD of three separate experiments using six determinations per experiment for each experimental condition. Clonogenic assay for cell survival on HCT-116 cells was performed according to previously described methods (Azmi et al. 2010).
Quantification of apoptosis by annexin V FITC flow cytometry and ELISA
Apoptosis in HCT-116 and MCF-7 cells was determined using Annexin V FITC apoptosis kit (Biovision Research Products) and ELISA detection kit (Roche, Palo Alto, CA) according to manufacturer's protocol.
siRNA and transfections
To study the effect of MI-219 on re-activating p53 in the presence of silenced wt-p53, we utilized siRNA techniques in both HCT-116 and MCF-7 cells. The p53 siRNA and control siRNA were obtained from Cell Signaling. Cells were transfected with respective siRNAs for 5 hrs using LipofectAMINE 2000 as described by the manufacturer's protocol (Cell Signaling).
Western blot analysis
HCT-116 or MCF-7 were exposed to different treatments for 24 hrs followed by extraction of protein for western blot analysis. Procedure for cells lyses, protein concentration determination and SDS-PAGE analysis has been described in our previous publication (Azmi et al. 2009).
P53 activation and translocation
Activation and translocation of p53 post MI-219, chelation or siRNA treatments was detected using “Cayman's p53 Cell-Based Activation/Translocation Assay Kit” (Ann Arbor. MI) according to the manufacturer's protocol.
p53 transcript DNA binding assay
Specific transcription factor DNA binding in nuclear extract post treatments was detected using the sensitive non-radioactive “Cayman's p53 Transcription Factor Assay kit” (Ann Arbor, MI). The procedure for nuclear extract preparation and transcription activity was done according to the manufacturer's protocol.
Supplementary Material
Acknowledgments
National Cancer Institute, NIH grant R01CA109389 (R.M. Mohammad) and NIH grant 5R01CA101870 (F.H. Sarkar) are acknowledged. We sincerely acknowledge the Guido foundation for their support.
Footnotes
Conflict of Interest: The University of Michigan has patented MI-219, which has been licensed by Ascenta Therapeutics, Inc. The University of Michigan and S. Wang and Dajun Yang own equity in Ascenta.
Reference List
- Alshatwi AA, Han CT, Schoene NW, Lei KY. Exp Biol Med (Maywood) 2006;231:611–618. doi: 10.1177/153537020623100516. [DOI] [PubMed] [Google Scholar]
- Azmi AS, Aboukameel A, Banerjee S, Wang Z, Mohammad M, Wu J, Wang S, Yang D, Philip PA, Sarkar FH, Mohammad RM. Eur J Cancer. 2010;46:1122–1131. doi: 10.1016/j.ejca.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azmi AS, Wang Z, Burikhanov R, Rangnekar VM, Wang G, Chen J, Wang S, Sarkar FH, Mohammad RM. Mol Cancer Ther. 2008;7:2884–2893. doi: 10.1158/1535-7163.MCT-08-0438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao B, Prasad A, Beck FW, Suneja A, Sarkar F. Toxicol Lett. 2006;166:222–228. doi: 10.1016/j.toxlet.2006.07.306. [DOI] [PubMed] [Google Scholar]
- Bargonetti J, Manfredi JJ, Chen X, Marshak DR, Prives C. Genes Dev. 1993;7:2565–2574. doi: 10.1101/gad.7.12b.2565. [DOI] [PubMed] [Google Scholar]
- Beck FW, Prasad AS, Butler CE, Sakr WA, Kucuk O, Sarkar FH. Prostate. 2004;58:374–381. doi: 10.1002/pros.10344. [DOI] [PubMed] [Google Scholar]
- Brown CJ, Lain S, Verma CS, Fersht AR, Lane DP. Nat Rev Cancer. 2009;9:862–873. doi: 10.1038/nrc2763. [DOI] [PubMed] [Google Scholar]
- Brown KH, Rivera JA, Bhutta Z, Gibson RS, King JC, Lonnerdal B, Ruel MT, Sandtrom B, Wasantwisut E, Hotz C. Food Nutr Bull. 2004;25:S99–203. [PubMed] [Google Scholar]
- Butler JS, Loh SN. Biochemistry. 2003;42:2396–2403. doi: 10.1021/bi026635n. [DOI] [PubMed] [Google Scholar]
- Butler JS, Loh SN. Biochemistry. 2007;46:2630–2639. doi: 10.1021/bi062106y. [DOI] [PubMed] [Google Scholar]
- Canner JA, Sobo M, Ball S, Hutzen B, DeAngelis S, Willis W, Studebaker AW, Ding K, Wang S, Yang D, Lin J. Br J Cancer. 2009;101:774–781. doi: 10.1038/sj.bjc.6605199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GD, Chou CM, Hwang SP, Wang FF, Chen YC, Hung CC, Chen JY, Huang CJ. Biochem Biophys Res Commun. 2006;344:272–282. doi: 10.1016/j.bbrc.2006.03.136. [DOI] [PubMed] [Google Scholar]
- Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Christianson DW. Adv Protein Chem. 1991;42:281–355. doi: 10.1016/s0065-3233(08)60538-0. [DOI] [PubMed] [Google Scholar]
- Collins K, Jacks T, Pavletich NP. Proc Natl Acad Sci U S A. 1997;94:2776–2778. doi: 10.1073/pnas.94.7.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S. J Am Chem Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]
- Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qiu S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J Med Chem. 2006;49:3432–3435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Rajagopal G, Levine AJ. Cell Cycle. 2008;7:842–847. doi: 10.4161/cc.7.7.5657. [DOI] [PubMed] [Google Scholar]
- Giannakakou P, Nakano M, Nicolaou KC, O'Brate A, Yu J, Blagosklonny MV, Greber UF, Fojo T. Proc Natl Acad Sci U S A. 2002;99:10855–10860. doi: 10.1073/pnas.132275599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambidge KM, Krebs NF, Miller L. Am J Clin Nutr. 1998;68:410S–413S. doi: 10.1093/ajcn/68.2.410S. [DOI] [PubMed] [Google Scholar]
- Ho E. J Nutr Biochem. 2004;15:572–578. doi: 10.1016/j.jnutbio.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Ho E, Ames BN. Proc Natl Acad Sci U S A. 2002;99:16770–16775. doi: 10.1073/pnas.222679399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho E, Song Y. Curr Opin Clin Nutr Metab Care. 2009;12:640–645. doi: 10.1097/MCO.0b013e32833106ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itahana K, Mao H, Jin A, Itahana Y, Clegg HV, Lindstrom MS, Bhat KP, Godfrey VL, Evan GI, Zhang Y. Cancer Cell. 2007;12:355–366. doi: 10.1016/j.ccr.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Kubbutat MH, Jones SN, Vousden KH. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Lane DP, Fischer PM. Nature. 2004;427:789–790. doi: 10.1038/427789a. [DOI] [PubMed] [Google Scholar]
- Levine AJ. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Levine AJ, Wu MC, Chang A, Silver A, Attiyeh EF, Lin J, Epstein CB. Ann N Y Acad Sci. 1995;768:111–128. doi: 10.1111/j.1749-6632.1995.tb12115.x. [DOI] [PubMed] [Google Scholar]
- Lindstrom MS, Jin A, Deisenroth C, White WG, Zhang Y. Mol Cell Biol. 2007;27:1056–1068. doi: 10.1128/MCB.01307-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine JC, Lozano G. Cell Death Differ. 2010;17:93–102. doi: 10.1038/cdd.2009.68. [DOI] [PubMed] [Google Scholar]
- Meek DW. Cell Signal. 1998;10:159–166. doi: 10.1016/s0898-6568(97)00119-8. [DOI] [PubMed] [Google Scholar]
- Meplan C, Richard MJ, Hainaut P. Oncogene. 2000;19:5227–5236. doi: 10.1038/sj.onc.1203907. [DOI] [PubMed] [Google Scholar]
- Meplan C, Verhaegh G, Richard MJ, Hainaut P. Proc Nutr Soc. 1999;58:565–571. doi: 10.1017/s0029665199000749. [DOI] [PubMed] [Google Scholar]
- Mohammad RM, Wu J, Azmi AS, Aboukameel A, Sosin A, Wu S, Yang D, Wang S, Al-Katib AM. Mol Cancer. 2009;8:115. doi: 10.1186/1476-4598-8-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brate A, Giannakakou P. Drug Resist Updat. 2003;6:313–322. doi: 10.1016/j.drup.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Pavletich NP, Chambers KA, Pabo CO. Genes Dev. 1993;7:2556–2564. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- Prasad AS. Met Ions Biol Syst. 2004;41:103–137. [PubMed] [Google Scholar]
- Prasad AS. Nutrition. 2001;17:685–687. doi: 10.1016/s0899-9007(01)00598-6. [DOI] [PubMed] [Google Scholar]
- Prasad AS, Bao B, Beck FW, Sarkar FH. J Lab Clin Med. 2001;138:250–256. doi: 10.1067/mlc.2001.118108. [DOI] [PubMed] [Google Scholar]
- Prasad AS, Bao B, Beck FW, Sarkar FH. J Lab Clin Med. 2002;140:272–289. doi: 10.1067/mlc.2002.127908. [DOI] [PubMed] [Google Scholar]
- Prasad AS, Fitzgerald JT, Hess JW, Kaplan J, Pelen F, Dardenne M. Nutrition. 1993;9:218–224. [PubMed] [Google Scholar]
- Shangary S, Qin D, McEachern D, Liu M, Miller RS, Qiu S, Nikolovska-Coleska Z, Ding K, Wang G, Chen J, Bernard D, Zhang J, Lu Y, Gu Q, Shah RB, Pienta KJ, Ling X, Kang S, Guo M, Sun Y, Yang D, Wang S. Proc Natl Acad Sci U S A. 2008;105:3933–3938. doi: 10.1073/pnas.0708917105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shangary S, Wang S. Annu Rev Pharmacol Toxicol. 2009;49:223–241. doi: 10.1146/annurev.pharmtox.48.113006.094723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT. Trends Mol Med. 2007;13:23–31. doi: 10.1016/j.molmed.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Verhaegh GW, Parat MO, Richard MJ, Hainaut P. Mol Carcinog. 1998;21:205–214. doi: 10.1002/(sici)1098-2744(199803)21:3<205::aid-mc8>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Verma R, Rigatti MJ, Belinsky GS, Godman CA, Giardina C. Biochem Pharmacol. 2010;79:565–574. doi: 10.1016/j.bcp.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace M, Worrall E, Pettersson S, Hupp TR, Ball KL. Mol Cell. 2006;23:251–263. doi: 10.1016/j.molcel.2006.05.029. [DOI] [PubMed] [Google Scholar]
- Wawrzynow B, Pettersson S, Zylicz A, Bramham J, Worrall E, Hupp TR, Ball KL. J Biol Chem. 2009b;284:11517–11530. doi: 10.1074/jbc.M809294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wawrzynow B, Pettersson S, Zylicz A, Bramham J, Worrall E, Hupp TR, Ball KL. J Biol Chem. 2009a;284:11517–11530. doi: 10.1074/jbc.M809294200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu S, Qin D, Shangary S, Chen J, Wang G, Ding K, McEachern D, Qiu S, Nikolovska-Coleska Z, Miller R, Kang S, Yang D, Wang S. J Med Chem. 2009;52:7970–7973. doi: 10.1021/jm901400z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalewski PD, Forbes IJ, Betts WH. Biochem J. 1993;296(Pt 2):403–408. doi: 10.1042/bj2960403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalewski PD, Forbes IJ, Seamark RF, Borlinghaus R, Betts WH, Lincoln SF, Ward AD. Chem Biol. 1994;1:153–161. doi: 10.1016/1074-5521(94)90005-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.