

Abstract
Integrins are heterodimeric membrane-spanning adhesion receptors that are essential for a wide range of biological functions. Control of integrin conformational states is required for bidirectional signalling across the membrane. Key components of this control mechanism are the transmembrane and cytoplasmic domains of the α and β subunits. These domains are believed to interact, holding the integrin in the inactive state, while inside-out integrin activation is accompanied by domain separation. Although there are strong indications for domain interactions, the majority of evidence is insufficient to precisely define the interaction interface. The current best model of the complex, derived from computational calculations with experimental restraints, suggests that integrin activation by the cytoplasmic protein talin is accomplished by steric disruption of the α/β interface. Better atomic-level resolution structures of the α/β transmembrane/cytoplasmic domain complex are still required for the resting state integrin to corroborate this. Integrin activation is also controlled by competitive interactions involving the cytoplasmic domains, particularly the β-tails. The concept of the β integrin tail as a focal adhesion interaction ‘hub’ for interactions and regulation is discussed. Current efforts to define the structure and affinity of the various complexes formed by integrin tails, and how these interactions are controlled, e.g. by phosphorylation and localisation, are described.
Keywords: Integrin cytoplasmic tails, transmembrane helices, talin, protein-protein interactions, integrin activation
Introduction
Integrins are cell surface receptors that connect the interior of the cell to the extracellular environment, forming both a structural connection and a bi-directional signalling pathway across the cell membrane. These heterodimeric proteins are comprised of α and β subunits, each containing a large extracellular domain, of approximately 80–150 kDa, a single transmembraneα-helix and a short, largely unstructured cytoplasmic domain of 10–70 residues. In mammals, one of 18 possible α subunits heterodimerises with one of 8 β subunits to form 24 distinct integrin proteins, each with specific but overlapping functions [1].
Integrins are central components of focal adhesion complexes, in which the extracellular domains bind to ligands such as fibronectin and collagen, while the flexible internal tails interact with the actin cytoskeleton via additional cytoplasmic adaptor proteins, including talin and filamin. These interactions are finely controlled so that focal adhesions are formed and degraded appropriately to allow cell migration and adhesion. Integrins are also key components of a variety of signal transduction pathways and have roles in processes such as apoptosis, gene expression and cell differentiation [2]. Intriguingly, integrins can transmit information in either direction across the cell membrane. Thus binding of activating proteins, such as talin, to the β-cytoplasmic domain causes conformational changes in the integrin ectodomain that lead to increased ligand affinity (inside-out signalling), while engagement of extracellular matrix ligands can result in changes to the cytoplasmic regions and further downstream signalling (outside in signalling). Following activation and ligand binding, clustering of the integrins occurs within the resulting focal adhesions [3].
The crystal structure of the extracellular domains of αvβ3 integrin published in 2001 [4] revealed that the αv and β3 subunits can be considered as a ‘head domain’, formed by the αv subunit β-propeller domain, the β3 subunit βA domain and an immunoglobulin (Ig)-like ‘hybrid’ domain, and two ‘legs’. The αv ‘leg’ is comprised of the Ig-like ‘thigh’ domain and two β-sandwich ‘calf’ domains. The β leg consists of a PSI domain [5] four EGF domains and a β-tail domain (β-TD) (see Figure 1).
Figure 1.

Schematic view of integrin activation. The integrin α/β heterodimer is believed to convert between the bent structure of the resting state and the extended arrangement of the activated state. Binding of talin to the membrane proximal and NPLY regions of the β3 cytoplasmic tail shifts the equilibrium to the right, activating the integrin. Binding of other PTB domains, such as dok1, can occur in the resting state. Phosphorylation of the β3-integrin tail can switch the affinity preference from talin to dok1 and may play a role in regulating activation [56]. A) View of intact integrins. B) Zoomed in view of the membrane and cytoplasmic region. The figures incorporate structures 1JV2, 2h7d and 2v76 from the PDB, and the Gottschalk model of the α/β TM complex [16]. In the online version talin is coloured yellow, the αIIb subunit is cyan and the β3 integrin subunit is red. The nomenclature for the different integrin regions is indicated.
In the crystal structure, the legs are folded at the knee or ‘genu’ between the αv thigh and calf-1 domain and between the EGF domains 1 and 2, such that the head domain is in proximity with the lower leg [4]; there is evidence to suggest that this represents the resting-state (Figure 1). Upon activation, the integrin is believed to move to a more extended form, accompanied by conformational changes within the head domain, including a significant swing in the hybrid domain, that facilitates ligand binding – the so-called ‘switch-blade’ mechanism. However, some of the details of this mechanism remain controversial (see [6–8] for reviews). Here we concentrate on integrin regions that were absent from the celebrated crystal structure, namely the relatively diminutive transmembrane (TM) and cytoplasmic domains. We discuss the evidence for interactions between the α and β subunits in these regions, as well as how these interactions may be affected by the process of inside-out activation, for example by the protein talin. The importance of the β cytoplasmic domains for protein-protein interactions is also examined. The sequences of these domains are given in Figure 2.
Figure 2.

Sequences of the transmembrane (TM) and cytoplasmic tail domains of the αIIb and β3 integrin subunits. The residues believed to reside within the bilayer are boxed. The borders were determined by sequence comparison to other α and β subunits whose membrane bordering residues have been determined experimentally [36]. The TM domain residue pairs identified as being at the heterodimeric interface by cysteine-scanning experiments [11], are indicated by lines, while the salt-bridge between αIIb-R995 and β3-D723, inferred from mutational experiments on intact integrins [25], is indicated by +/− symbols. These pairs were used as structural restraints for modelling the α/β complex interface [16]. The binding regions for talin [49], filamin [54], dok1 [56] and kindlin3 [59] are indicated on the β3-tail. Dok1 binding is much more significant in phosphorylated β3; its binding region is distinguished by a dashed line. The two β NPxY-type sequences and the αIIb double proline motif are highlighted in bold.
Transmembrane domain interactions
Interaction between the TM domains has been visualised directly using cryomicroscopy and single particle reconstruction of detergent-solubilised intact αIIb/β3 integrins [9]. The final image revealed regions of electron density consistent with a right-handed coiled-coil arrangement of the TM helices, with the membrane proximal α and β cytoplasmic tails in close spatial proximity. Interestingly, the ectodomain in this image appears to be in an unusual conformation, neither ‘jack-knifed’, as in the crystal structure, nor fully extended, as in other electron microscopy (EM) images. Echistatin was shown to bind to the integrin causing a conformational change, consistent with activation; this was taken as evidence that the original images corresponded with the low-affinity, resting state. A limitation of cryo-EM is that, at the resolution available, different conformations of the integrin cannot be distinguished from different orientations of a single conformation [10]. However, fitting the TM model to the electron density suggested single rather than multiple conformations of this region.
Additional evidence for an inter-subunit interaction of the TM domains has come from cysteine-scanning experiments using intact αIIb/β3 integrin [11]. Single-point cysteine mutations were created in the N-terminal region of the α and β TM domains and formation of disulphide bonds in the intact integrin receptors was taken to indicate interaction between TM helices (Figure 2). The pattern of disulphide bonds resulting from this array of mutations indicated a preferred α/β interface and a specific interaction between the αIIb and β3 TM helices. Since neither disulphide-bonded integrins, nor single-cysteine mutant constructs could bind soluble fibrinogen, the observed TM interactions were taken to arise from the structural properties of the inactive integrin state. The same experiments, performed on integrins with constitutively activating mutations (i.e. Δ991GFFKR995 or 992FF993/AA αIIb [12]), revealed a pattern of disulphide bond formation, that suggested a non-specific interaction. Consistent with a model in which the TM domains are separated in the active state, disulphide bonded constructs were shown to be inactive, despite the presence of activating mutations, while non-disulphide bonded constructs bound soluble ligand [11].
Interestingly, disulphide bonded constructs in both wild-type and ‘activated’ integrin receptors could be activated in an ‘outside-in’ manner by addition of Mn2+ or antibodies. Thus ‘outside-in’ activation does not appear to require the separation of the TM domains, while ‘inside-out’ activation does.
Later studies on intact integrins showed that mutation of residues throughout the TM and membrane proximal regions of the β3 subunit can lead to integrin activation [13]. Mutations predicted to disrupt the β-TM helix resulted in constitutively activated integrins, consistent with a shift in equilibrium towards helix separation. A subset of activating mutations not anticipated to affect the structure of the TM helix were suggested to be directly involved in a coiled-coil interface. These results were used to drive a Monte Carlo simulation that predicted a right-handed coiled-coil interface between the α and β subunits [13].
Gottschalk and co-workers also used computer simulations to predict the nature of the α/β TM interface [14–16]. In their most recent study, TM and membrane proximal α and β subunits were restrained at the N-terminus using data obtained from the cysteine-scanning experiments of Luo et al. [11], and at the C-terminus by including a restraint for the putative MP salt bridge (Figure 2 and see below). With these experimental restraints, the simulation suggested that the helices form a right-handed coiled-coil arrangement. Both the α subunit and the β-subunit have sequence motifs (972GxxxG976 and 692SxxxA696, respectively) that are homologous to motifs from glycophorin A (GpA) – a protein known to form a membrane spanning coiled-coil interface [17]. The α GpA-like motif was at the interface of the computed integrin dimer, consistent with studies showing that mutation of these residues results in increased ligand binding [18]. The β tail GpA-like motif was not at the interface, however, suggesting that the integrin interaction is not the same as the one observed in the GpA dimer.
Li et al. [19] performed experiments on isolated α and β TM-cytoplasmic domains dissolved in dodecylphosphocholine (DPC) micelles, using a variety of techniques including nuclear magnetic resonance (NMR). This approach failed to detect any interaction between the subunits; instead it was found that the subunits formed homo-oligomers (trimers in the case of β3 and dimers for αIIβ), in equilibrium with the monomeric form. The absence of observed heterodimeric interactions could be due to low affinity of the inter-subunit interaction (relatively weak interactions are expected in a system that has to switch between resting and ligand-binding states) and to limitations in the detergent micelle system. The absence of inter-subunit connections provided by the extracellular domains would also be expected to decrease heterodimeric interactions in the isolated TM/cytoplasmic tail constructs. The possibility that the observed homo-oligomers [19] are relevant to integrin clustering is also supported by mutation studies [20].
The capacity of a range of different α and β TM fragments, including αIIb and β3, to homo- or hetero-dimerise within the membrane of E. coli, was investigated using the GALLEX assay [21]. This assay involves TM domains fused to LexA DNA binding domains. Interaction of the TM domains within the bacterial cell membrane allows dimerisation of the LexA domain, which can then repress the activity of the lacz gene. The GALLEX assay has the advantage that it can be modified to assay for heterodimerisation – even in a background of homodimerisation. This contrasts with the experimental setup of Li et al. [19] where the absence of extracellular domains is likely to promote formation of homodimers.
The results indicated that all TM domains tested homodimerised weakly, apart from β7 which homodimerised as strongly as GpA protein [21]. α/β pairs were tested for heterodimerisation using a modified version of the assay. All hetero- combinations dimerised to some degree, although in all cases more weakly than the GpA homodimer. Mutating the glycine residues in the β7 GxxxG GpA-like motif decreased both homo-and hetero-association [21] although the predicted interface would produce a different pattern of disulphide bonding to that observed in the cysteine-scanning experiments [11]. These apparently incompatible results could be due to the use of truncated peptide constructs in the GALLEX assay, in particular the absence of the 989GFFKR995 motif from the αIIβ tail that is critically important for maintaining the inactive state. Extending the TM sequences used in the GALLEX assays, to include the membrane proximal cytoplasmic regions, may improve the reliability of the results. In general these experiments, together with cysteine scanning [11], suggest that homodimerisation is unlikely to be an initiator of integrin clustering although it may occur at a later stage in the clustering process.
α-β Cytoplasmic tail interface
Fluorescence resonance energy transfer (FRET) studies on intact αLβ2 integrins suggested that cytoplasmic domain separation occurs during activation [22]. Cells were transfected with αL and β2 integrin subunits fused at their C-termini to cyan fluorescent protein (CFP) and yellow fluorescent protein (YFP) respectively. Fluorescence transfer from CFP to YFP was observed for the αLβ2 integrin in the resting state, indicating that the cytoplasmic tails of the α and β subunits were close together.
‘Inside-out’ activation of the integrin receptors by various methods, such as binding the talin head domain, mutation of the αL GFFKR motif, or by chemokine/G-protein coupled receptor interactions, all led to significant reductions in FRET [22]. This suggests that, in addition to separation of the TM domains, inside-out activation results in separation of the α and β cytoplasmic tails by more than 100. On the other hand, outside-in activation by Mn2+ resulted in no change in the basal FRET signal, indicating that tail separation is not required for this process, although subsequent extracellular ligand binding did reduce FRET [22]. This is consistent with cysteine scanning experiments [11], which showed that separation of the TM regions of α and β integrin subunits was not induced by Mn2+.
Conservation of sequences in both α and β cytoplasmic domains at the membrane interfacial region led to speculation that they may be important in activation, perhaps forming an interaction interface. Studies on intact integrins confirmed that these regions are important for normal integrin function [23–25]. For example, truncation of the αIIb subunit following residue N996 had no impact on integrin activation, while truncation after G991 produced a constitutively active integrin, revealing the importance of the intervening residues 992FFKR995 [23]. Similarly, deletion of residues 717LLITIHD723 from the β3 integrin tail results in integrin activation [24].
Alanine scanning of the 991GFFKR995 motif in αIIb showed that the two phenylalanine residues, as well as the positively charged arginine residue are important for maintaining the integrin resting state, possibly through inter-subunit interactions [25]. D723A substitution in the β-subunit was also found to activate, and the fact that R995 in the α-tail is expected to have a similar position in relation to the membrane led to the proposal that R995 and D723 form an inter-subunit salt-bridge, helping to maintain the integrin in the resting state (Figure 2). Consistent with this proposal, the double charge-reversal mutant, R995D/D723R, had low activity [25].
Attempts to observe interactions between the α and β cytoplasmic domains directly have produced conflicting results. Early studies used fluorescence quenching, mass spectrometry and circular dichroism detected an interaction under low ionic strength conditions [26]. Surface plasmon resonance (SPR) experiments carried out at physiological pH and salt concentrations, later showed that free αIIb cytoplasmic domain bound to a GST-fusion β3 tail construct in a concentration dependent manner [27]. This interaction was inhibited by deletion of the conserved KVGFFKR region or an R995A mutation.
Later, NMR was used to examine the structure of the α/β complex. Ulmer et al. [28] looked for α/β tail interactions using coiled-coil fusion constructs, where each tail was fused to one half of a heterodimeric coiled-coil via a triple-glycine linker. The idea behind this was that by covalently linking the two subunits, the equilibrium would shift toward complex formation, without overly restricting the orientational flexibility of the peptide tails. In these constructs, the tails appeared to adopt random coil type conformations. However, β3 residues R724-A735 did show a propensity for α-helical structure, while the residues of the 744NPLY747 motif had a preference for forming a reverse turn [28].
Spectra from a coiled-coil construct attached to both αIIb and β3 tails were compared to spectra from the same construct with a truncatedα tail (αIIbΔ996-β3). Very small spectral perturbations were detected within the N-terminal region of the βtail and along the length of the α-tail but the same experiments, comparing β3-β3 orαIIb-αIIb constructs, revealed a similar pattern of shift changes. This was taken to indicate that any interaction that was present must be very weak and non-specific. 15N-1H heteronuclear NOEs also showed that the tail regions were significantly more flexible than the coiled coil region – again suggesting that a tight α/β complex was not formed in solution.
In contrast, α/β complexes were observed by Weljie and co-workers [29] using peptides corresponding to truncated αIIb (M987-R997) and β3 (K716-D740) tails. Evidence was found for two different structural conformers of the complex, and these conformers were suggested to represent the inactive and active integrin states since only one of the two conformers was compatible with an intermolecular salt-bridge. The accuracy of these designations is debatable, however, and the existence of two entirely different complexes in a mixture of flexible peptides is hard to understand.
An alternative view of the α/β cytoplasmic complex was presented by Vinogradova et al. [30]. Data were obtained from mixtures of full-length αIIb and β3 cytoplasmic domain peptides in water, revealing very small chemical shift changes from unbound values. Residues along the length of theαIIb tail were perturbed, while only the N-terminal end of the β3 tail was affected. The perturbations were of similar magnitude and distribution to those found by Ulmer et al. (see supplementary data in [28]). Ulmer and co-workers believed these shifts were non-specific effects, since similar patterns and shift sizes were observed for homo-dimeric subunit pairs. Vinogradova et al [30], however, also observed a number of intermolecular transferred-NOEs between an MBP-β3 fusion construct and free αIIb cytoplasmic tail. The role of the MBP protein was to increase both the solubility of the β-integrin tail and its apparent molecular weight (to ~50kDa) thus enabling the detection of the transferred NOEs (these can be observed indirectly because of rapid exchange between free α peptide and the α/β complex).
The structure obtained using these 11 intermolecular NOEs as restraints, revealed an interface between the α and β cytoplasmic peptides that was stabilised by both hydrophobic and electrostatic interactions. The structure was compatible with a salt-bridge between residues αIIb-R995 and β3-D723, but additional electrostatic interactions were also observed. Mutation of residues in the αIIb tail that are critically involved in the interface (R995, F992) abolished observed interactions between the α and β tails and resulted in constitutively active integrins [25]. Similarly, addition of talin head domain, a potent ‘inside-out’ activator of integrins [31], eliminated the interaction detected by α/β transferred NOEs.
The structure of the αIIb tail in theα/β complex is remarkably similar to that determined for a myristoylated αIIb tail in DPC micelles [32]. Here the C-terminal portion of the α-tail forms an extended structure that folds back on the α-helical N-terminus, with the bend occurring at the site of two consecutive proline residues (Figure 2). The relevance of this N/C-terminal interaction is inferred by the finding that P998A/P999A substitution in the intact receptor produces a constitutively active integrin [32].
The β3 peptide, on the other hand, formed anα-helix over residues K716-K738, with the remaining residues unstructured. This is in contrast to two independent NMR studies on the β3 subunit on its own that showed that the region between the two NPxY-like motifs (Y747-T755) forms a second α-helix in the presence of DPC micelles [33, 34]. This C-terminal helix was shown to be anchored to the micelle by the 744NPLY747 residues, and it was postulated that this may play a role in restricting the lateral motion of the integrin within the membrane, and that binding of talin to the integrin tail could relieve this restriction [33]. Formation of this second α-helix may require a membrane environment, however, which could explain why it is not seen in the α/β complex structure [30].
The small size of the secondary chemical shifts observed in theα/β tail complex suggests that the interaction between the αIIb and β3 peptides is extremely weak. SPR experiments, which tend to indicate stronger interactions than ITC or NMR, determined a Kd for the interaction of 7μM [27]. However, the close proximity of the tails in the intact integrin receptor and the likely interaction of the TM domains are expected to increase the probability of interaction.
The structure determined by Vinogradova et al. [30] is strikingly different from the conformers found by Weljie and co-workers [29], with the α/β interface located on the opposite face of the αIIb tail. In the Weljie structures, the side-chains of residues R995 and F992 are pointing into solution, rather than being located at the interface as they are in the Vinogradova structure. The importance of these residues in integrin activation suggests that the Vinogradova structure is the more physiologically relevant. The different structures obtained with the truncated β3 tails could be due to the absence of the C-terminal half of the αIIb tail, which seems to play a role in the structure of the complex. Limitations of the Vinogradova structure may in turn come from the absence of the TM domains and/or a membrane bilayer environment. The complex interface is small and relatively poorly defined, so the details of the interface may be different in the intact integrin.
Gottschalk et al. [15] performed calculations to investigate the compatibility of the Vinogradova structure [30] with α-helices extending into the TM region. Incorporation of the 11 intermolecular experimental restraints failed to give convergence, and the majority of calculated α/β heterodimers were found to align in a head-to-tail fashion i.e. with N- and C-termini together. It was also found that by taking the Vinogradova structure directly, and extending the cytoplasmic α-helices into the TM region, steric clashes between the α and β peptides result. These findings indicate that the NMR structure is underdetermined by the NOE restraints. The structures could be led to convergence, and head-to-head dimer formation, by incorporation of the proposed salt-bridge between the α and β subunits; however the orientation of the cytoplasmic domains was different to that obtained by Vinogradova et al. [30]. The most recent calculated structure for the α/β interface, which was calculated using full TM constructs and the membrane proximal region (up to residueαIIb R997 and β3 F727), is also incompatible with the Vinogradova structure [16]. Thus there is a continuing need to try to define the full structure of the interface between the α and β TM and cytoplasmic domains.
In summary, there is evidence that the isolated cytoplasmic tails interact, albeit very weakly [29, 30]. As a result of this weak affinity, as well as the small interaction interface, the reliability of the structures so far determined is relatively low, as demonstrated by the variety of complex structures obtained. Extending the constructs to include the TM domains is the next logical step to obtaining a more reliable view of the α/β complex. Attempts to detect interactions between such constructs in micelles have, however, so far failed [19]. This may be due to limitations of micelles as a membrane mimic and similar experiments in bicelles may prove more successful. The compelling evidence from the cysteine-scanning [11], mutational [13] and cryo-EM experiments [9], suggests that the interaction is present, at least in intact integrins. The latest computational determined model of the α/β TM and membrane proximal interaction is based on the cysteine-scanning experiments, as well as the presence of the putative salt-bridge [16]. Currently the evidence for the salt-bridge comes only from indirect mutational experiments [25]. Many of the structural studies place great emphasis on this interaction so it is highly desirable that direct evidence is obtained to confirm its existence. A future challenge is therefore to answer the many remaining questions about the TM and cytoplasmic domain interactions in integrins.
Positioning of the α and β subunits within the membrane
Various models have been proposed to explain how conformational changes in the cytoplasmic tails influence the conformation of the ectodomain. These generally involve changes in the position of the TM domains within the bilayer; this, in turn, causes the connecting extracellular domains to move to the active state. For example, in the ‘scissor’ model the TM domains interact with a small crossing angle in the resting state, and scissor to a larger crossing angle in the activated state [14]. In the ‘piston’ model, on the other hand, the TM domains to move vertically in the membrane during activation, acting as pistons [35]. Evidence has also been put forward for a TM domain separation model (see above). Locating the α and β subunits within the lipid bilayer is therefore important for differentiating between these models.
Glycosylation mapping experiments have been used to identify the residues that lie at the lipid/membrane interface of microsomal membranes [36]. These showed that 26 residues of the β1 integrin are located within the membrane, finishing at residue I721. Thus the positively charged lysine residue K716 is situated within the membrane, allowing the subsequent hydrophobic sequence 717LLITI721 to reside in the bilayer. Similarly, the α2 and α5 integrins also incorporate a lysine residue within the membrane at a similar position. By sequence alignment, the αIIb and β3 integrins are expected to have TM domains of 27 and 26 residues, respectively as shown in Figure 2, with αIIb-F993 and β3-I721 the final lipid buried residues. This is consistent with NMR spectroscopy experiments in DPC micelles that showed αIIb K989-F993 and β3 K716-I721 were in close proximity to the micellar lipids [33].
The structure and orientation of the β3 TM domain has also been investigated in bicelles [37]. Here, the TM helix was found to extend from residue D692 to H722, with H722 located at the lipid/water interface, again consistent with the glycosylation mapping experiments [36]. The length of the β3 TM helix was determined to be approximately 43Å. To accommodate a helix of this length within a lipid bilayer it would be necessary for the helix to adopt a tilt angle of 20–30° [37]. Such an angle would allow the side-chain of K716 to ‘snorkel’ to the interface where it could interact with the negatively charged phosphate groups of the lipid head-groups, as well as allow the indole group of W715 to lie near the membrane/water interface, a location strongly preferred by tryptophan residues [38]. In support of this, the χ1 angle (dihedral angle defined by N-Cα-Cβ-Cγ) of W715 was found to be well-defined for the β3 TM domain in bicelles, but rotationally averaged in the presence of micelles [37]. Given the comparable length of the αIIb TM domain, and the presence of a similarly positioned 988WK989 motif (Figure 2), it seems likely that the TM domain of the αIIb subunit will also adopt an oblique orientation within the membrane.
Interestingly, β3 residues L717-I721 were not forced into the aqueous medium when in micelles, where tilting of the TM-helix cannot occur. Instead, the full TM helix was accommodated within the micelle by deformation of the helix, and distortion of the spherical micelle structure. Shortening the lipid chain-length from 16/18 carbons to 14/14 carbons had no effect on the partitioning of these residues in bicelles, suggesting it is energetically less costly to tilt the helix further, than to repartition the C-terminal residues into the aqueous phase [37]. Thus, Lau et al. [37] suggest that the role of these residues in integrin function may be to fix the tilt angle of the TM helix. However, glycosylation mapping studies showed that addition of a second lysine residue in the β1 TM domain can induce repartitioning of the subsequent residues into the cytoplasm, without changing the residues at the extracellular side [36]. These results suggest that it is unlikely that the TM helices reposition themselves vertically within the bilayer as in the ‘piston’ model of integrin activation. However, it may be possible that the TM domains change their angle of insertion in the membrane from their complexed resting state, to the activated TM domain-separated state. This would probably occur during the process of domain separation, rather than from a scissor-like motion, since the cysteine-scanning experiments showed no evidence for maintenance of a specific interface in the activated state [11].
It should be noted that all of the experiments on the positioning of the TM domains within bilayers have examined the α and β subunits in isolation. Thus, the results obtained are likely to represent the activated integrin state with separatedα and β subunits. It will be of considerable interest to determine the orientation and membrane insertion properties of a TM domain complex representative of the inactive state. However, as we have seen, this is difficult to do experimentally and ‘course-grained’ computational simulations, in combination with mutational experiments may give further insight into the nature of the integrin resting state [39].
Inside-out activation by talin
The inside-out activation process requires cytoplasmic ligands to interact with the integrin tails, to transmit the information to the extracellular ligand-binding domains, via the TM and stalk regions. One such ligand identified is the cytoplasmic protein talin [40]. Talin consists of a ~50kDa head region that contains a 300-residue FERM domain, and a ~200kDa rod segment, the structure of which is being revealed as a series of α-helical bundles [41]. Talin has been shown to bind to integrin cytoplasmic domains, vinculin and actin filaments suggesting that it forms a link between the cytoskeleton and the extracellular matrix. The intact talin protein is relatively ineffectual at activating integrins, and needs to be stimulated in some way to trigger activation, implying that the integrin binding site/s are masked in the intact molecule. Cleavage of talin between the head and rod domains by the protease calpain, for example, results in 16-fold increase in binding to β3 tails [42]. Similarly, binding of PIP2 to talin induces a conformational change that increases β-tail binding [43].
Integrin activation is mediated via the talin head domain [31], which can be broken down into four regions, known as the F0, F1, F2 and F3 domains, the latter 3 making up a FERM domain [44]. The phosphotyrosine binding domain (PTB)-like F3 domain is capable of activating the αIIb/β3 integrin on its own [45]. The canonical ligands of PTB domains are peptides containing an NPxY type sequence (either phosphorylated or unphosphorylated), and the majority of β-integrin tails contain at least one of these motifs. Mutation of Y747A within the first NPxY-like motif in the β3 tail inhibits integrin activation by talin, indicating an interaction at this position [45].
Ulmer et al. [46] looked at the interaction between talin head fragments and the β3-integrin tail fused to a coiled-coil construct using NMR and found that while binding of the talin F3 domain affects residues in the NPxY site, it also influenced residues along the majority of the β3-integrin sequence. Solubility limitations of the cytoplasmic tail constructs, as well as the low affinity of the interaction meant that determination of the structure of the integrin/talin complex was not feasible. Crystallography studies using the whole β3 cytoplasmic domain and an F2F3 fragment of talin were also unsuccessful, presumably because of the same limitations [47]. However, the F2F3 talin fragment was successfully crystallised on its own, and the particular arrangement of molecules within the crystal suggested that a chimeric talin-integrin molecule could be designed so as to encourage complex formation; N-terminal residues in the F2 domain were replaced with theβ-integrin sequence W739-A750, thus allowing crystallisation of a talin-integrin complex [47]. The structure revealed that β3 residue W739 inserts into a well-defined pocket on the talin surface, with the subsequent residues forming a β-strand that extends the sheet formed by strands 5, 6 and 7 of the talin F3 domain. The 744NPLY747 sequence forms a reverse turn, as expected, but the number of residues between W739 and the NPLY motif is such that a bulge occurs around residues A742 and N743. This effect is different from the canonical PTB interactions and may explain the lower than expected affinity for the talin-integrin complex.
This work provided information about the structure of the talin F2F3 domain, and its interaction with the integrin NPxY motif, but it remained unclear how talin activates integrins, while other PTB domains, although capable of binding [48] do not activate. Shift-mapping and intensity changes in NMR spectra had indicated additional interactions between the membrane proximal β-integrin residues and talin [30, 46], but these initially eluded high-resolution scrutiny. This led to another chimeric trick being employed to determine the structure; this time the membrane proximal β-tail residues K716-W739 were combined with residues V743-R752 from the C-terminal tail of PIPKI(γ), which had previously been shown to bind tightly to the canonical NPxY binding region of talin [49]. This resulted in a high-affinity talin ligand that allowed structure determination of the complex, thus revealing that the membrane proximal βtail residues form an α-helix that lies across the talin PTB domain, interacting with β-strands 1 and 2 and the intervening loop. Mutation of interfacial integrin residues, particularly F727 or F730, or talin residues L325 or Q381, resulted in loss of this membrane proximal interaction, as observed by NMR, and decreases in integrin activation, as observed by whole cell FACS analysis, confirmed the importance of the interface in the activation process [49].
The current model is that talin binding to the membrane proximal region initiates integrin activation by interfering with the interface between the α and β cytoplasmic domains. Overlaying the talin F3/chimeric peptide structure on the Gottschalk model of the αIIb/β3 TM domains [16] leads to steric clashes between talin and the αIIb tail in the vicinity of the membrane/water interface, suggesting that talin binds to the β3 tail and sterically interrupts the α/β interactions [49] (Figure 3). The talin/β3 tail interaction also puts a positively charged talin loop (between S1 and S2) close to the membrane, and interactions between membrane-facing lysine residues and the negatively charged membrane lipids could enhance talin binding to the integrin in the cellular context. In support of this, mutation of a membrane-facing lysine (K322D) prevented integrin activation, while mutation of a lysine that faced away from the membrane (K320D) had no effect [49].
Figure 3.
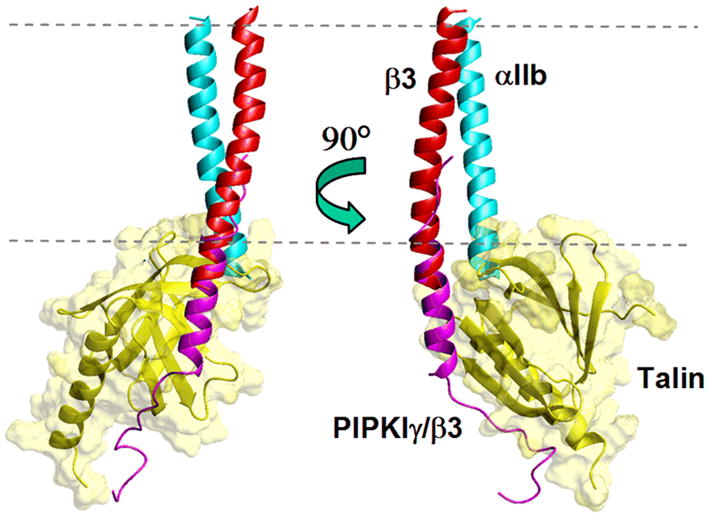
Overlay of the talin/PIPKIγβ3 complex [49], with the calculated model of the integrin αIIb/β3 transmembrane and membrane proximal complex (co-ordinates kindly provided by K. Gottschalk [16]), over β3 residues 722–727. The talin domain overlaps with the αIIb structure. In the online version the talin PTB domain is coloured yellow, the chimeric PIPKIγ/β3 peptide is magenta, the αIIb subunit is cyan and the β3 integrin subunit is red. The dashed lines represent the approximate location of the membrane. Figure prepared using Molmol [62].
Similar structural overlays between the talin/integrin complex and the experimentally determined α/β cytodomain complex [30] showed few direct steric clashes between talin and the αIIb tail, although the two entities are very close Experimental determination of the full α/β TM and cytoplasmic domain interfaces in a membrane bilayer environment should give a better description of how talin is involved, as well as validate the current computational and experimental models of integrin interactions.
Another possibility is that the entire talin head domain is required for activation. Recently it has been shown that β3 activation is enhanced in the presence of the whole head domain, over that induced by F3 alone [44]. There also appear to be differences in activation mechanisms between different β-subunits, as the F3 domain alone is not able to activate α5β1 integrin, something that is only achieved by the full head domain [44]. How the additional domains are involved in the activation mechanism is unclear at this time.
β-Tails as a protein interaction hub
In addition to talin, which seems to play a special role by binding to the membrane proximal region of the β-integrin tails, both β and α cytoplasmic regions are known to bind to many other proteins [50]. These interactions have roles both in forming mechanical links to the cytoskeleton and in downstream signalling. The formation of focal adhesions, a large assembly of cytoskeletal and signalling proteins, is critically dependent on these tails. Interactions with the α tails are relatively rare, one of the best characterised being the interaction of α4 with paxillin [51]. In contrast, the β-tails have a large number of well documented interactions with partners that include α-actinin, ICAP1, filamin, Src, CD98, and skelemin [50, 52].
It has been noted that intrinsically disordered regions, such as the β-tail, generally appear to be well suited for forming ‘hubs’ for a large numbers of protein interactions [53]. Reversible modification of side-chains, for example phosphorylation-dephosphorylation, can also affect the number and type of interactions available and therefore greatly increase the versatility of a disordered peptide as a binding partner. The majority of β-tails are comprised of 40–60 residues (β4 is a notable exception with around 1000 amino-acids). Although short, they can extend over a significant distance due to their disorder (e.g. the β-tail extends over 8nm in structure 1M80, deposited in the PDB) and they can provide different binding sequences along their length. For example, a number of PTB domain-containing proteins have been shown to bind one or other of the two NPxY-type sequences usually found within the β-tails [48], while filamin binds to residues between these two motifs [54] and, as discussed above, the talin PTB domain binds to the membrane proximal β3 residues as well as to the central NPLY sequence [49] (Figure 2).
Phosphotyrosine binding (PTB) domains generally function as adaptors in the formation of signalling complexes in wide-ranging physiological processes (52). They bind both peptides and head-groups of phosphatidylinositides on different sites in a domain with a conserved core structure. PTB domains mainly bind peptides with an NPxY motif but have differing requirements for phosphorylation of the tyrosine within this recognition sequence. The talin PTB domain is specifically suited to bind the membrane proximal β-tail residues due to the length and flexibility of the loop betweenβ-strands 1 and 2. In most PTB domains an α-helix is inserted between these strands [55], preventing interaction with the β3 helix. In addition, talin contains a hydrophobic residue (L325) that is appropriately placed to interact with the F727 and F730 from the tail, shielding them from the aqueous environment. In a survey of PTB domains, including those that form part of FERM domain complexes, hydrophobic residues equivalent to those in talin were not found [49].
The Dok1 PTB domain was shown to bind weakly, only to the central NPxY motif in the β3 cytoplasmic domain [56]. The affinity of the interaction was increased from the millimolar range to micromolar by phosphorylation of Y747. Binding of talin, on the other hand, was inhibited by phosphorylation, suggesting that phosphorylation at this position can act as a switch by reversing the preference for the β3-tail from talin binding to Dok1 (Figure 1) [56]. Thus, side-chain modification can increase the number of ligands that can interact with the β-tails, and provide a means for controlling which ligands are binding. This kind of phosphorylation switch is expected to apply to other PTB interactions. For example it was recently shown that integrin cytoplasmic domain associated protein 1 (ICAP-1) slows down FA assembly by affecting the high affinity state favoured by talin [57].
Another group of FERM domain containing proteins, called kindlins, has recently been shown to be important for integrin activation [58, 59]. Mutagenesis results have indicated the kindlin-3 F3 sub-domain interacts directly with the β3 cytoplasmic domain. However, intriguingly this interaction appears to be mediated at the second NPxY-like domain 756NITY759 rather than the first, and further studies will be required to dissect kindlins’ role in activation [59]. Mutation of residues from the 756NITY759 motif have previously been shown to abrogate cell spreading [60].
A demonstration of the versatility of the β-integrin cytoplasmic domains comes from its interaction with filamin. The crystal structure of filamin domain 21 in complex with a β7-tail fragment shows that residues 745PLY747 from the first NPxY motif are arranged in a linear manner as part of the β-strand that complements one of the β-sheets in the filamin Ig-like domain [54]. However, when this motif binds to the talin PTB domain, it becomes part of a reverse turn structure [47]. This means that the same β-tail residues can form different structural arrangements in order to adapt to different binding partners. The residues following the first NPLY motif are also in a β-strand arrangement in the filamin complex. As mentioned previously, this region of the β3 integrin tail has been shown to be α-helical in DPC micelles [33, 34], further showing that the β-tail is capable of forming disparate conformations depending on the environment. The biologically relevant S752P mutation lies within this region of the β3 tail, suggesting that it may exert its effects by disrupting secondary structure [61].
Conclusions
Subtle control of integrin activation state is required for many cell functions. Integrins are intact receptors operating in a regulated environment but this review has concentrated on activation control by the membrane spanning and cytoplasmic regions of the integrin heterodimer. These regions are demonstrably essential to the activation process since interference with their structure can either lock the system in an inactive state (e.g. S-S bridges) or cause constitutive activation (e.g. tail truncation). Considerable progress has been made in recent years both in our understanding of the various interactions between these regions and of how specific binding to the β-tail (e.g. by talin) can lead to activation. Our understanding of the role of the tails in forming links to the cytoskeleton (e.g. via filamin) has also advanced. This is, however, a difficult system to study by structural biology. Ideally one would have a complete structure of active and inactive states in the membrane and a view of the various competing interactions for the tails. Since this ideal has not been realised at present we must try to extrapolate from imperfect data. Future investigations will require ingenuity to circumvent the experimental difficulties to provide us with a more complete view of integrin activation.
Acknowledgments
IDC acknowledges the Wellcome Trust and KLW thanks the NIH Cell migration consortium (Grant U54 GM64346 from the National Institute of General Medical Sciences), for funding.
Contributor Information
KATE L. WEGENER, Email: kate.wegener@bioch.ox.ac.uk.
IAIN D. CAMPBELL, Email: iain.campbell@bioch.ox.ac.uk.
References
- 1.Hynes RO. Integrins: Bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 2.van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285–298. doi: 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- 3.Cluzel C, Saltel F, Lussi J, Paulhe F, Imhof BA, Wehrle-Haller B. The mechanisms and dynamics of {alpha}v{beta}3 integrin clustering in living cells. J Cell Biol. 2005;171:383–392. doi: 10.1083/jcb.200503017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong JP, Stehle T, Diefenbach B, Zhang R, Dunker R, Scott DL, Joachimiak A, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin alpha vbeta3. Science. 2001;294:339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi M, Sundramurthy K, Liu B, Tan SM, Law SK, Lescar J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/i-egf1 segment from the human integrin beta2 subunit at 1.8-a resolution. J Biol Chem. 2005;280:30586–30593. doi: 10.1074/jbc.M502525200. [DOI] [PubMed] [Google Scholar]
- 6.Luo B-H, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arnaout MA, Goodman SL, Xiong J-P. Structure and mechanics of integrin-based cell adhesion. Curr Opin Cell Biol. 2007;19:495–507. doi: 10.1016/j.ceb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mould AP, Humphries MJ. Regulation of integrin function through conformational complexity: Not simply a knee-jerk reaction? Curr Opin Cell Biol. 2004;16:544–551. doi: 10.1016/j.ceb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Adair BD, Yeager M. Three-dimensional model of the human platelet integrin alpha iibbeta 3 based on electron cryomicroscopy and x-ray crystallography. Proc Natl Acad Sci U S A. 2002;99:14059–14064. doi: 10.1073/pnas.212498199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luo B-H, Springer TA. Integrin structures and conformational signaling. Curr Opin in Cell Biol. 2006;18:579–586. doi: 10.1016/j.ceb.2006.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo B-H, Springer TA, Takagi J. A specific interface between integrin transmembrane helices and affinity for ligand. PLoS Biology. 2004;2:153. doi: 10.1371/journal.pbio.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu CF, Springer TA. The alpha subunit cytoplasmic domain regulates the assembly and adhesiveness of integrin lymphocyte function-associated antigen-1. J Immunol. 1997;159:268–278. [PubMed] [Google Scholar]
- 13.Partridge AW, Liu S, Kim S, Bowie JU, Ginsberg MH. Transmembrane domain helix packing stabilizes integrin alphaiibbeta3 in the low affinity state. J Biol Chem. 2005;280:7294–7300. doi: 10.1074/jbc.M412701200. [DOI] [PubMed] [Google Scholar]
- 14.Gottschalk KE, Adams PD, Brunger AT, Kessler H. Transmembrane signal transduction of the {alpha}iib{beta}3 integrin. Protein Sci. 2002;11:1800–1812. doi: 10.1110/ps.4120102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gottschalk KE, Kessler H. Evidence for hetero-association of transmembrane helices of integrins. FEBS Lett. 2004;557:253–258. doi: 10.1016/s0014-5793(03)01443-1. [DOI] [PubMed] [Google Scholar]
- 16.Gottschalk KE. A coiled-coil structure of the alphaiibbeta3 integrin transmembrane and cytoplasmic domains in its resting state. Structure. 2005;13:703–712. doi: 10.1016/j.str.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 17.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: Structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 18.Luo BH, Carman CV, Takagi J, Springer TA. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci U S A. 2005;102:3679–3684. doi: 10.1073/pnas.0409440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li R, Babu CR, Lear JD, Wand AJ, Bennett JS, DeGrado WF. Oligomerization of the integrin alphaiibbeta3: Roles of the transmembrane and cytoplasmic domains. Proc Natl Acad Sci U S A. 2001;98:12462–12467. doi: 10.1073/pnas.221463098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Metcalf DG, Gorelik R, Li R, Mitra N, Nanda V, Law PB, Lear JD, Degrado WF, Bennett JS. A push-pull mechanism for regulating integrin function. Proc Natl Acad Sci U S A. 2005;102:1424–1429. doi: 10.1073/pnas.0409334102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider D, Engelman DM. Involvement of transmembrane domain interactions in signal transduction by {alpha}/{beta} integrins. J Biol Chem. 2004;279:9840–9846. doi: 10.1074/jbc.M312749200. [DOI] [PubMed] [Google Scholar]
- 22.Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi: 10.1126/science.1084174. [DOI] [PubMed] [Google Scholar]
- 23.O’Toole TE, Katagiri Y, Faull RJ, Peter K, Tamura R, Quaranta V, Loftus JC, Shattil SJ, Ginsberg MH. Integrin cytoplasmic domains mediate inside-out signal transduction. J Cell Biol. 1994;124:1047–1059. doi: 10.1083/jcb.124.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hughes PE, O’Toole TE, Ylanne J, Shattil SJ, Ginsberg MH. The conserved membrane-proximal region of an integrin cytoplasmic domain specifies ligand binding affinity. J Biol Chem. 1995;270:12411–12417. doi: 10.1074/jbc.270.21.12411. [DOI] [PubMed] [Google Scholar]
- 25.Hughes PE, Diaz-Gonzalez F, Leong L, Wu C, McDonald JA, Shattil SJ, Ginsberg MH. Breaking the integrin hinge. A defined structural constraint regulates integrin signaling. J Biol Chem. 1996;271:6571–6574. doi: 10.1074/jbc.271.12.6571. [DOI] [PubMed] [Google Scholar]
- 26.Haas TA, Plow EF. The cytoplasmic domain of alphaiib/beta(3) J Biol Chem. 1996;271:6017–6026. doi: 10.1074/jbc.271.11.6017. [DOI] [PubMed] [Google Scholar]
- 27.Vallar L, Melchior C, Plancon S, Drobecq H, Lippens G, Regnault V, Kieffer N. Divalent cations differentially regulate integrin alphaiib cytoplasmic tail binding to beta3 and to calcium- and integrin-binding protein. J Biol Chem. 1999;274:17257–17266. doi: 10.1074/jbc.274.24.17257. [DOI] [PubMed] [Google Scholar]
- 28.Ulmer TS, Yaspan B, Ginsberg MH, Campbell ID. Nmr analysis of structure and dynamics of the cytosolic tails of integrin alpha iib beta 3 in aqueous solution. Biochemistry. 2001;40:7498–7508. doi: 10.1021/bi010338l. [DOI] [PubMed] [Google Scholar]
- 29.Weljie AM, Hwang PM, Vogel HJ. Solution structures of the cytoplasmic tail complex from platelet integrin alpha iib- and beta 3-subunits. Proc Natl Acad Sci U S A. 2002;99:5878–5883. doi: 10.1073/pnas.092515799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vinogradova O, Velyvis A, Velyviene A, Hu B, Haas T, Plow E, Qin J. A structural mechanism of integrin alpha(iib)beta(3) “Inside-out” Activation as regulated by its cytoplasmic face. Cell. 2002;110:587–597. doi: 10.1016/s0092-8674(02)00906-6. [DOI] [PubMed] [Google Scholar]
- 31.Calderwood DA, Zent R, Grant R, Rees DJ, Hynes RO, Ginsberg MH. The talin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrin activation. J Biol Chem. 1999;274:28071–28074. doi: 10.1074/jbc.274.40.28071. [DOI] [PubMed] [Google Scholar]
- 32.Vinogradova O, Haas T, Plow EF, Qin J. A structural basis for integrin activation by the cytoplasmic tail of the alpha iib-subunit. Proc Natl Acad Sci U S A. 2000;97:1450–1455. doi: 10.1073/pnas.040548197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vinogradova O, Vaynberg J, Kong X, Haas TA, Plow EF, Qin J. Membrane-mediated structural transitions at the cytoplasmic face during integrin activation. Proc Natl Acad Sci U S A. 2004;101:4094–4099. doi: 10.1073/pnas.0400742101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li R, Babu CR, Valentine K, Lear JD, Wand AJ, Bennett JS, DeGrado WF. Characterization of the monomeric form of the transmembrane and cytoplasmic domains of the integrin beta 3 subunit by nmr spectroscopy. Biochemistry. 2002;41:15618–15624. doi: 10.1021/bi026822l. [DOI] [PubMed] [Google Scholar]
- 35.Williams MJ, Hughes PE, O’Toole TE, Ginsberg MH. The inner world of cell adhesion: Integrin cytoplasmic domains. Trends Cell Biol. 1994;4:109–112. doi: 10.1016/0962-8924(94)90059-0. [DOI] [PubMed] [Google Scholar]
- 36.Stefansson A, Armulik A, Nilsson I, von Heijne G, Johansson S. Determination of n- and c-terminal borders of the transmembrane domain of integrin subunits. J Biol Chem. 2004;279:21200–21205. doi: 10.1074/jbc.M400771200. [DOI] [PubMed] [Google Scholar]
- 37.Lau T-L, Partridge AW, Ginsberg MH, Ulmer TS. Structure of the integrin alpha-iib/beta-3 transmembrane segment in phospholipid bicelles and detergent micelles. Biochemistry. 2008 doi: 10.1021/bi800107a. [DOI] [PubMed] [Google Scholar]
- 38.Yau WM, Wimley WC, Gawrisch K, White SH. The preference of tryptophan for membrane interfaces. Biochemistry. 1998;37:14713–14718. doi: 10.1021/bi980809c. [DOI] [PubMed] [Google Scholar]
- 39.Sansom MS, Scott KA, Bond PJ. Coarse-grained simulation: A high-throughput computational approach to membrane proteins. Biochem Soc Trans. 2008;36:27–32. doi: 10.1042/BST0360027. [DOI] [PubMed] [Google Scholar]
- 40.Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, Ginsberg MH, Calderwood DA. Talin binding to integrin beta tails: A final common step in integrin activation. Science. 2003;302:103–106. doi: 10.1126/science.1086652. [DOI] [PubMed] [Google Scholar]
- 41.Critchley DR, Gingras AR. Talin at a glance. J Cell Sci. 2008;121:1345–1347. doi: 10.1242/jcs.018085. [DOI] [PubMed] [Google Scholar]
- 42.Yan B, Calderwood DA, Yaspan B, Ginsberg MH. Calpain cleavage promotes talin binding to the beta 3 integrin cytoplasmic domain. J Biol Chem. 2001;276:28164–28170. doi: 10.1074/jbc.M104161200. [DOI] [PubMed] [Google Scholar]
- 43.Martel V, Racaud-Sultan C, Dupe S, Marie C, Paulhe F, Galmiche A, Block MR, Albiges-Rizo C. Conformation, localization, and integrin binding of talin depend on its interaction with phosphoinositides. J Biol Chem. 2001;276:21217–21227. doi: 10.1074/jbc.M102373200. [DOI] [PubMed] [Google Scholar]
- 44.Bouaouina M, Lad Y, Calderwood DA. The n-terminal domains of talin cooperate with the phosphotyrosine binding-like domain to activate {beta}1 and {beta}3 integrins. J Biol Chem. 2008;283:6118–6125. doi: 10.1074/jbc.M709527200. [DOI] [PubMed] [Google Scholar]
- 45.Calderwood DA, Yan B, de Pereda JM, Alvarez BG, Fujioka Y, Liddington RC, Ginsberg MH. The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem. 2002;277:21749–21758. doi: 10.1074/jbc.M111996200. [DOI] [PubMed] [Google Scholar]
- 46.Ulmer TS, Calderwood DA, Ginsberg MH, Campbell ID. Domain-specific interactions of talin with the membrane-proximal region of the integrin beta3 subunit. Biochemistry. 2003;42:8307–8312. doi: 10.1021/bi034384s. [DOI] [PubMed] [Google Scholar]
- 47.Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, Ginsberg MH, Liddington RC. Structural determinants of integrin recognition by talin. Mol Cell. 2003;11:49–58. doi: 10.1016/s1097-2765(02)00823-7. [DOI] [PubMed] [Google Scholar]
- 48.Calderwood DA, Fujioka Y, de Pereda JM, Garcia-Alvarez B, Nakamoto T, Margolis B, McGlade CJ, Liddington RC, Ginsberg MH. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: A structural prototype for diversity in integrin signaling. Proc Natl Acad Sci U S A. 2003;100:2272–2277. doi: 10.1073/pnas.262791999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wegener KL, Partridge AW, Han J, Pickford AR, Liddington RC, Ginsberg MH, Campbell ID. Structural basis of integrin activation by talin. Cell. 2007;128:171–182. doi: 10.1016/j.cell.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 50.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113 (Pt 20):3563–3571. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 51.Rose DM, Liu S, Woodside DG, Han J, Schlaepfer DD, Ginsberg MH. Paxillin binding to the {alpha}4 integrin subunit stimulates lfa-1 (integrin {alpha}l{beta}2)-dependent t cell migration by augmenting the activation of focal adhesion kinase/proline-rich tyrosine kinase-2. J Immunol. 2003;170:5912–5918. doi: 10.4049/jimmunol.170.12.5912. [DOI] [PubMed] [Google Scholar]
- 52.Lad Y, Harburger DS, Calderwood DA. Integrin cytoskeletal interactions. Methods in Enzymol. 2007;426:69–84. doi: 10.1016/S0076-6879(07)26004-5. [DOI] [PubMed] [Google Scholar]
- 53.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. FEBS Journal. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 54.Kiema T, Lad Y, Jiang P, Oxley CL, Baldassarre M, Wegener KL, Campbell ID, Ylanne J, Calderwood DA. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell. 2006;21:337–347. doi: 10.1016/j.molcel.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 55.Uhlik MT, Temple B, Bencharit S, Kimple AJ, Siderovski DP, Johnson GL. Structural and evolutionary division of phosphotyrosine binding (ptb) domains. J Mol Biol. 2005;345:1–20. doi: 10.1016/j.jmb.2004.10.038. [DOI] [PubMed] [Google Scholar]
- 56.Oxley CL, Anthis NJ, Lowe ED, Vakonakis I, Campbell ID, Wegener KL. An integrin phosphorylation switch: The effect of beta3 integrin tail phosphorylation on dok1 and talin binding. J Biol Chem. 2008;283:5420–5426. doi: 10.1074/jbc.M709435200. [DOI] [PubMed] [Google Scholar]
- 57.Millon-Fremillon A, Bouvard D, Grichine A, Manet-Dupe S, Block MR, Albiges-Rizo C. Cell adaptive response to extracellular matrix density is controlled by icap-1-dependent {beta}1-integrin affinity. J Cell Biol. 2008;180:427–441. doi: 10.1083/jcb.200707142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi X, Ma YQ, Tu Y, Chen K, Wu S, Fukuda K, Qin J, Plow EF, Wu C. The mig-2/integrin interaction strengthens cell-matrix adhesion and modulates cell motility. J Biol Chem. 2007;282:20455–20466. doi: 10.1074/jbc.M611680200. [DOI] [PubMed] [Google Scholar]
- 59.Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14:325–330. doi: 10.1038/nm1722. [DOI] [PubMed] [Google Scholar]
- 60.Ylänne J, Huuskonen J, O’Toole TE, Ginsberg MH, Virtanen I, Gahmberg CG. Mutation of the cytoplasmic domain of the integrin beta3 subunit. J Biol Chem. 1995;270:9550–9557. doi: 10.1074/jbc.270.16.9550. [DOI] [PubMed] [Google Scholar]
- 61.Chen YP, O’Toole TE, Ylanne J, Rosa JP, Ginsberg MH. A point mutation in the integrin beta 3 cytoplasmic domain (s752-->p) impairs bidirectional signaling through alpha iib beta 3 (platelet glycoprotein iib-iiia) Blood. 1994;84:1857–1865. [PubMed] [Google Scholar]
- 62.Koradi R, Billeter M, Wuthrich K. Molmol: A program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]