

Abstract
Phosphodiesterase-6 (PDE6) is the key effector enzyme of the phototransduction cascade in rods and cones. The catalytic core of rod PDE6 is a unique heterodimer of PDE6A and PDE6B catalytic subunits. The functional significance of rod PDE6 heterodimerization and conserved differences between PDE6AB and cone PDE6C and the individual properties of PDE6A and PDE6B are unknown. To address these outstanding questions, we expressed chimeric homodimeric enzymes, enhanced GFP (EGFP)-PDE6C-A and EGFP-PDE6C-B, containing the PDE6A and PDE6B catalytic domains, respectively, in transgenic Xenopus laevis. Similar to EGFP-PDE6C, EGFP-PDE6C-A and EGFP-PDE6C-B were targeted to the rod outer segments and concentrated at the disc rims. PDE6C, PDE6C-A, and PDE6C-B were isolated following selective immunoprecipitation of the EGFP fusion proteins. All three enzymes, PDE6C, PDE6C-A, and PDE6C-B, hydrolyzed cGMP with similar Km (20–23 μm) and kcat (4200–5100 s−1) values. Likewise, the Ki values for PDE6C, PDE6C-A, and PDE6C-B inhibition by the cone- and rod-specific PDE6 γ-subunits (Pγ) were comparable. Recombinant cone transducin-α (Gαt2) and native rod Gαt1 fully and potently activated PDE6C, PDE6C-A, and PDE6C-B. In contrast, the half-maximal activation of bovine rod PDE6 required markedly higher concentrations of Gαt2 or Gαt1. Our results suggest that PDE6A and PDE6B are enzymatically equivalent. Furthermore, PDE6A and PDE6B are similar to PDE6C with respect to catalytic properties and the interaction with Pγ but differ in the interaction with transducin. This study significantly limits the range of mechanisms by which conserved differences between PDE6A, PDE6B, and PDE6C may contribute to remarkable differences in rod and cone physiology.
Keywords: G Proteins, Phosphodiesterases, Photoreceptors, Phototransduction, Transgenic
Introduction
Vertebrates rely on two types of photoreceptor cells, rods and cones, for vision. The phototransduction cascades in rods and cones are principally similar. The central components of the rod and cone signaling pathways, visual pigments, transducins (Gt), and retinal cGMP-phosphodiesterases (PDE6)2 are distinct but highly homologous proteins (1–3). In contrast, the physiology of rods and cones is strikingly different. Rods are exceptionally sensitive to light and provide for nighttime (scotopic) vision, whereas cones are markedly less sensitive and signal during daytime (photopic receptors). Cone electrical responses to light are smaller in amplitude and much faster than rod responses. Furthermore, cones adapt to a much broader range of illumination conditions than rods and can function in intensely bright light (1–3). The molecular origin(s) of the differences in physiology of rods and cones is one of the key unresolved questions of vertebrate phototransduction (3). The physiological differences may be due to sequence and concentration differences between signaling proteins in rods and cones, as well as to characteristic photoreceptor morphologies of rods and cones (3, 4).
Sequence differences in rod and cone transduction components are limited, but well conserved, among vertebrate species. Thus, they may lead to differences in protein structure and biochemical properties that underlie the distinct physiology of the two types of photoreceptors. Supporting this notion, a much lower efficiency of transducin activation by visual pigment was reported in carp cones in vitro compared with transducin activation in rods (5). The resulting low signal amplification may explain low sensitivity of cone photoreceptors. Current evidence suggests that the signaling properties of rod and cone visual pigments are nearly identical. Human rhodopsin and red cone pigment expressed in Xenopus cones and rods, respectively, produced responses identical to native responses of Xenopus photoreceptors (6). The input of different transducin-α subunits (Gαt) into characteristic responses of rods and cones is controversial. Rod and cone Gαt subunits were able to functionally substitute for each other when expressed exogenously in the opposite photoreceptor cell type in mutant mice lacking one or both Gαt subunits (7). However, a more recent analysis of transgenic mice with rods expressing cone Gαt2 instead of rod Gαt1 showed the hallmarks of cone phototransduction such as decreased rod sensitivity, reduced rate of activation, and more rapid recovery (8). PDE6 is the key remaining molecule whose contribution (or lack thereof) to the rod/cone differences is unknown. An original characterization of bovine cone PDE6 unexpectedly revealed that the cone enzyme is remarkably more sensitive to activation by Gαt1 than the rod enzyme (9). In contrast to this finding, PDE6 activation by transducin in carp cones appears to be less effective than in rods (5).
The most obvious distinction between the rod and cone effector enzymes is the heterodimerization of rod PDE6 catalytic subunits. Rod PDE6 is unique among all 11 families of cyclic nucleotide phosphodiesterases that are typically represented by homodimeric enzymes (10). In various species, except chicken, rod holo-PDE6 is composed of two large homologous catalytic α- and β-subunits (PDE6A and PDE6B, respectively) and two copies of an inhibitory γ-subunit (Pγ) (11). No PDE6A subunit is found in chicken (12). Cone PDE6 is composed of two identical α′-subunits (PDE6C), each associated with a cone-specific inhibitory Pγ subunit (11, 13). The obligatory heterodimerization of PDE6A and PDE6B raises a number of outstanding questions. Because the PDE6AB dimer is functionally inseparable, and heterologous expression of the PDE6 catalytic subunits has not been achieved, the catalytic properties of PDE6A and PDE6B and their individual interactions with Pγ are still uncharacterized. The possibility exists that one subunit, perhaps PDE6A, is catalytically deficient. Consistent with this possibility, two binding sites for Pγ on rod PDE6 had been reported, with only one of the two sites mediating PDE6 inhibition (14). In addition, several studies have shown that just one Gαt molecule can maximally activate rod PDE6 (15, 16). This finding may indicate that PDE6A-Pγ and PDE6B-Pγ have significantly different affinities for Gαt-GTP and that the binding of Gαt to the lower affinity site does not lead to PDE6 activation. Other studies have demonstrated that one Gαt molecule effectively relieves Pγ inhibition at one PDE6 site and that this leads to one-half of the maximal PDE6 activity (17, 18). The heterogeneity of transducin-binding sites on rod PDE6 could originate from potential differences in PDE6A-Pγ and PDE6B-Pγ interactions, resulting in different mechanisms of PDE6 activation in rods and cones. Here, we utilized transgenic Xenopus laevis for expression of chimeric homodimeric PDE6 enzymes containing the PDE6A or PDE6B catalytic domain. This approach allowed direct analysis of essential properties of PDE6A and PDE6B.
EXPERIMENTAL PROCEDURES
Generation of Transgenic X. laevis
The constructs for PDE6 chimeras containing the N-terminal regulatory GAF domains of human cone PDE6C and the C-terminal catalytic domain of PDE6A or PDE6B were generated using the previously described pXOP(−508/+41)-EGFP-PDE6C vector (19). First, a thrombin cleavage site was created in the linker between enhanced GFP (EGFP) and PDE6C sequences by PCR-directed mutagenesis. Subsequently, a silent EcoRV site was introduced into the PDE6C cDNA sequence at Asp450-Ile451 with PCR-based mutagenesis. The C-terminal sequences of human PDE6A (amino acids 449–860) and PDE6B (amino acids 447–853) were PCR-amplified from a human retinal cDNA library and inserted into the pXOP(−508/+41)-EGFP-PDE6C vector using the EcoRV and XmaI restriction sites. All sequences were verified by automated DNA sequencing. Transgenic X. laevis frogs expressing EGFP-PDE6C-A and EGFP-PDE6C-B in rods were produced using the method of restriction enzyme-mediated integration (20) as described previously (19). Adult transgenic frogs were mated to produce transgenic tadpoles for biochemical characterization of PDE6C, PDE6C-A, and PDE6C-B.
PDE6C, PDE6C-A, and PDE6C-B were extracted and immunoprecipitated with sheep anti-GFP antibodies according to a published protocol (19). Immunoprecipitated PDE6C, PDE6C-A, and PDE6C-B were eluted by incubating beads (15 μl) with 20 μl of 20 mm Tris-HCl buffer (pH 7.5) containing 500 mm NaCl, 1 mm MgSO4, 1 mm β-mercaptoethanol, and 5 μg of trypsin (15 min, +4 °C) or 2 units of thrombin (restriction-grade; Novagen) for 2 h at 25 °C. All samples were analyzed immediately or stored at −20 °C in the presence of 40% glycerol.
Cloning, Expression, and Purification of Human Cone Gαt2
The Gαt2 cDNA was amplified from a human retinal cDNA library and subcloned into the pET15b vector using the XhoI and SpeI sites. Overnight expression of Gαt2 in BL21-CodonPlus Escherichia coli cells at 13 °C was induced with the addition of 15 μm isopropyl β-d-thiogalactopyranoside. His-tagged Gαt2 was purified on nickel-nitrilotriacetic acid resin (Novagen) as described previously (21). Gαt2 was incubated with 200 μm GTPγS in 20 mm Tris-HCl buffer (pH 8.0) containing 4 mm MgSO4 and 2 mm β-mercaptoethanol (buffer A) for 12 h at 4 °C and purified using a Mono Q HR 5/5 column or a UNO Q1 column with a 0–500 mm gradient of NaCl in buffer A. Bovine Gαt1-GTPγS was isolated as described previously (22).
Immunoblotting
Samples were subjected to SDS-PAGE on 7.5% gels, electrotransferred to nitrocellulose membranes, and probed with anti-GFP monoclonal antibody B-2 (1:2000 dilution; Santa Cruz Biotechnology), anti-bovine rod holo-PDE6 antibody MOE (1:2000; Cytosignal), and anti-bovine PDE6B-(397–417) antibody 63F (1:6000; gift of Dr. R. Cote, University of New Hampshire). Sequence alignment of bovine PDE6B-(397–417) with the corresponding sequences of human PDE6C and frog PDE6A and PDE6B indicated that antibody 63F recognizes PDE6C, PDE6C-A, and PDE6C-B and frog PDE6A and PDE6B equally well (supplemental Fig. 1). For detection of Pγ, electrophoresis was performed on 10% gels using the Tris/Tricine buffer system (23), and the blots were probed with anti-Pγ-(63–87) antibody (1:3000; gift of Dr. R. Cote). The antibody-antigen complexes were detected using anti-rabbit or anti-mouse antibodies conjugated to horseradish peroxidase (Santa Cruz Biotechnology) and ECL reagent (Amersham Biosciences.).
PDE Activity Assay and Data Analysis
PDE activity was measured using 5 μm [3H]cGMP and 1 pm PDE6 in Pγ inhibition assays or 50 μm [3H]cGMP and 100 pm PDE6 in Gαt-GTPγS activation assays (24, 25). To determine Km values for cGMP, PDE activity was measured using 5–500 μm cGMP, and the data were fit to the following equation: Y = Vmax*X/(Km + X). The kcat values for cGMP hydrolysis were calculated as Vmax/[PDE]. [PDE] were determined by densitometric analysis of immunoblots of PDE6C, PDE6C-A, and PDE6C-B samples with anti-PDE6 antibody 63F using ImageJ and purified recombinant PDE6C-His6 as the standard. The human cone and bovine rod Pγ subunits were subcloned into the pET15b vector, expressed in E. coli, and purified using His·Bind resin and reverse-phase HPLC as described previously (19, 26). The Ki values for PDE6 inhibition by Pγ were calculated by fitting data to the following equation: Y (%) = 100/(1 + 10^(X − log Ki)), where X is the logarithm of the total Pγ concentration. The K½ values for PDE6 activation by Gαt-GTPγS were calculated by fitting data to the following equation: Y (%) = B + (T − B)/(1 + 10^(log Ki − X)), where B is PDE6 activity in the absence of Gαt, T is the maximal Gαt-stimulated PDE6 activity expressed as a percent of the trypsin-activated PDE6 activity, and X is the logarithm of the total concentration Gαt-GTPγS. Fitting the experimental data to equations was performed with nonlinear least-squares criteria using GraphPad Prism 4 software. Experimental results are shown as the mean ± S.E.
RESULTS
Expression and Compartmentalization of Chimeric EGFP-PDE6 in Transgenic Rods
The N-terminal regulatory GAF domains of PDE6 contain major structural determinants for the selectivity of dimerization of PDE6 catalytic subunits (27). Thus, the PDE6C-A and PDE6C-B chimeras containing the GAF domains of PDE6C and the C-terminal catalytic domains of PDE6A or PDE6B were designed to produce homodimeric PDE6 enzymes in the rods of transgenic X. laevis (Fig. 1A). Previously described transgenic X. laevis tadpoles expressing EGFP-PDE6C in rods were used to obtain PDE6C (19). In transgenic PDE6C-A and PDE6C-B tadpoles, EGFP fluorescence was confined to the rod outer segment in the frog retina, indicating correct targeting of the chimeric proteins (Fig. 1B). The striated peripheral pattern of EGFP fluorescence in transgenic EGFP-PDE6C-A and EGFP-PDE6C-B rods was indistinguishable from the distribution of EGFP-PDE6C observed previously (Fig. 1B and supplemental Fig. 2) (19). This pattern suggests that similar to PDE6C, PDE6C-A and PDE6C-B concentrate at the rim region and incisures of membrane discs. Bands of the predicted size (∼125 kDa) for EGFP fusion proteins of PDE6C, PDE6C-A, and PDE6C-B were recognized in immunoblots by anti-GFP antibodies (Fig. 2A). Although expression of the EGFP fusion proteins varied between transgenic tadpoles, the average levels of PDE6C, PDE6C-A, and PDE6C-B were comparable and below the level of endogenous Xenopus PDE6 (Fig. 2B).
FIGURE 1.

Expression of EGFP-PDE6C-A and EGFP-PDE6C-B in transgenic rods. A, map of the transgene. The XhoI site was used to linearize the pXOP(508/+41)-EGFP-PDE6C plasmid for production of transgenic X. laevis embryos. T, thrombin cleavage site. B, EGFP fluorescence in living photoreceptor cells expressing EGFP-PDE6C-A and EGFP-PDE6C-B. Scale bars = 20 μm.
FIGURE 2.
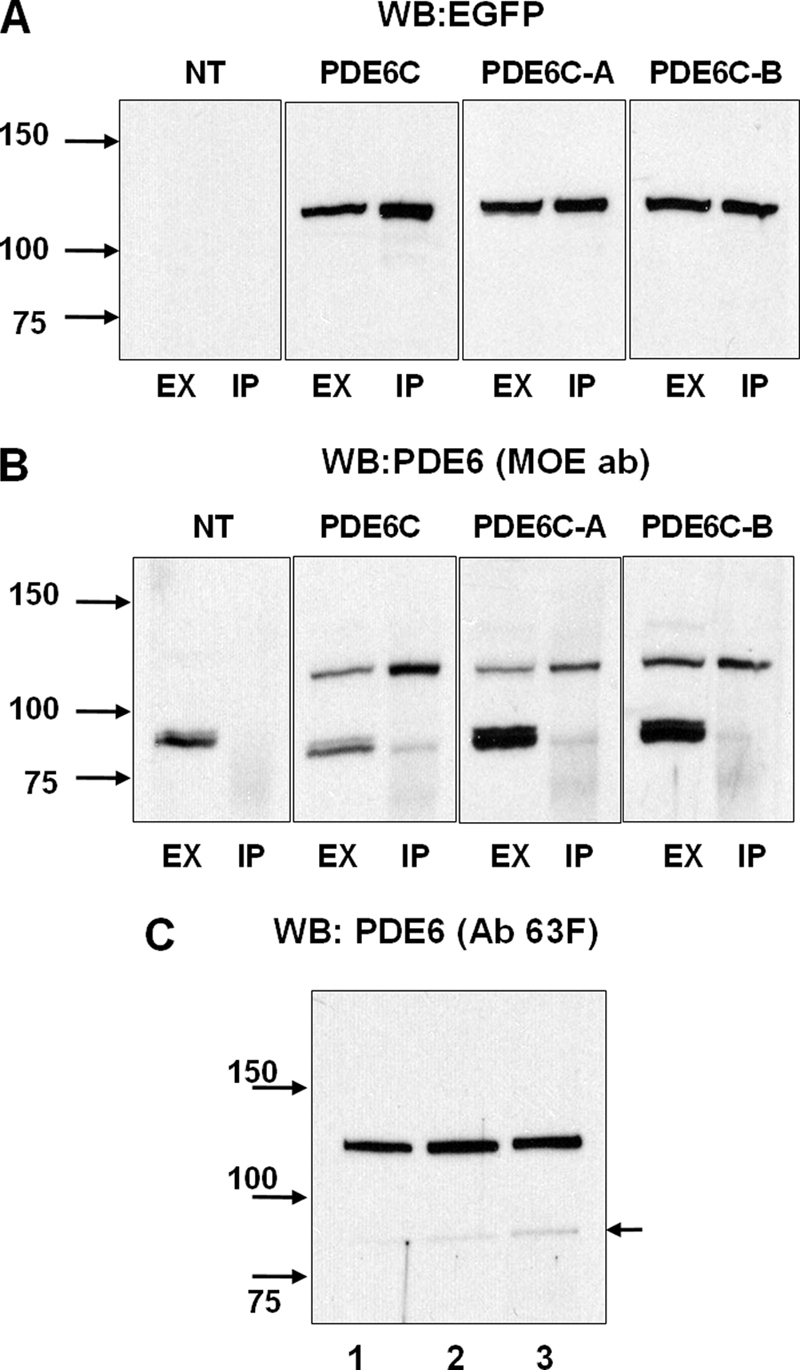
Immunoprecipitation of EGFP-PDE6C, EGFP-PDE6C-A, and EGFP-PDE6C-B. Extracts (EX) and immunoprecipitates with sheep anti-GFP antibodies (IP) from retinas of non-transgenic (NT) and transgenic X. laevis were immunoblotted with anti-GFP monoclonal antibody B-2 (A), stripped, and reprobed with anti-PDE6 antibody (ab) MOE (B). C, immunoprecipitates of PDE6C (lane 1), PDE6C-A (lane 2), and PDE6C-B (lane 3) with sheep anti-GFP antibodies were immunoblotted with anti-PDE6 antibody 63F. The level of coprecipitation (co-dimerization) with frog PDE6AB (indicated by the arrow) was <3%. WB, Western blot.
Properties of PDE6C-A and PDE6C-B
PDE6C, PDE6C-A, and PDE6C-B were immunoprecipitated from retinal extracts with anti-GFP antibodies (Fig. 2, A and B). Anti-PDE6 antibody MOE readily recognized endogenous Xenopus PDE6 in the retinal extracts, but only minute amounts of frog PDE6 in comparison with the EGFP-fused PDE6 proteins were detectable in the immunoprecipitated samples (IPs) (Fig. 2B). No frog PDE6AB was seen in control IPs using retinal extracts from non-transgenic tadpoles. Thus, the presence of trace amounts of frog PDE6AB in the IPs from transgenic animals was due to very weak heterodimerization of PDE6A or PDE6B with PDE6C, PDE6C-A, and PDE6C-B. Anti-PDE6 antibody MOE was raised against bovine rod holo-PDE6 and recognizes rod PDE6 better than cone PDE6. Consequently, contaminations of the PDE6C, PDE6C-A, and PDE6C-B proteins by frog PDE6AB are even smaller than appears from the immunoblotting with the MOE antibody. To quantify the presence of frog PDE6AB, we utilized anti-PDE6 antibody 63F, which recognizes PDE6C, PDE6C-A, PDE6C-B, and frog PDE6A and PDE6B equally well (supplemental Fig. 1). IPs from PDE6C, PDE6C-A, and PDE6C-B retinal extracts were analyzed by Western blotting with antibody 63F (Fig. 2C), which quantitatively showed a level of coprecipitation of frog PDE6AB of <3% (data not shown).
Beads with IPs were treated with trypsin or thrombin to remove the GFP tag and to release the enzymes into solution. The trypsin treatment of PDE6C, PDE6C-A, and PDE6C-B IPs released soluble PDE6 enzymes of the same size (∼88 kDa) (Fig. 3A) as the treatment of PDE6C-A and PDE6C-B with thrombin (data not shown), indicating the proximity of the cleavage sites. The trypsin treatment of PDE6C, PDE6C-A, and PDE6C-B IPs was also accompanied by robust PDE6 activation similar to that described previously (19). Thus, PDE6C, PDE6C-A, and PDE6C-B immunoprecipitated in complex with the endogenous frog Pγ subunit, which was cleaved by trypsin during solubilization (supplemental Fig. 3).
FIGURE 3.

Solubilization and quantification of PDE6C, PDE6C-A, and PDE6C-B. A, the beads with bound EGFP-PDE6C (lane 1), EGFP-PDE6C-A (lane 2), and EGFP-PDE6C-B (lane 3) were treated with trypsin, and proteins released into the soluble fraction were analyzed by immunoblotting with anti-PDE6 antibody (Ab) 63F. Purified recombinant PDE6C-His6 was used as the standard. WB, Western blot. B, PDE6C, PDE6C-A, and PDE6C-B were quantified by measuring the integrated densities of the corresponding bands and PDE6C-His6 bands corrected for the background using ImageJ.
Soluble PDE6C, PDE6C-A, and PDE6C-B proteins released with trypsin were quantified by immunoblotting with antibody 63F and purified bacterially expressed PDE6C-His6 as the standard (Fig. 3). Such PDE6 samples were used in the comparative analysis of the catalytic properties of PDE6C, PDE6C-A, and PDE6C-B. PDE6C, PDE6C-A, and PDE6C-B hydrolyzed cGMP with similar Km values of 23 ± 1, 20 ± 2, and 22 ± 2 μm, respectively (Fig. 4). For comparison, a Km value of 21 ± 3 cGMP μm was determined for purified trypsin-activated bovine rod PDE6 under the same experimental conditions. The kcat values for PDE6C, PDE6C-A, and PDE6C-B were 5100 ± 100, 4200 ± 150, and 4300 ± 150 s−1, respectively (Fig. 4). The catalytic constant for trypsin-activated bovine rod PDE was comparable at 4500 ± 200 s−1 (data not shown).
FIGURE 4.

Catalytic properties of PDE6C, PDE6C-A, and PDE6C-B. The rates of cGMP hydrolysis by PDE6C, PDE6C-A, and PDE6C-B are plotted as a function of cGMP concentration. The Km and kcat values are as follows, respectively: PDE6C, 23 ± 1 μm and 5100 ± 100 s−1; PDE6C-A, 20 ± 2 μm and 4200 ± 150 s−1; and PDE6C-B, 22 ± 2 μm and 4300 ± 150 s−1. Results from one of three similar experiments are shown.
The full catalytic competence of PDE6C-A and PDE6C-B allowed examination of the inhibition of the chimeric enzymes by rod and cone Pγ subunits. Both PDE6C-A and PDE6C-B were potently and similarly inhibited by both Pγ subunits, with Ki values ranging from 33 to 46 pm (Fig. 5). The inhibition analysis revealed no significant differences between PDE6C-A and PDE6C-B or between the chimeras and PDE6C (Fig. 5). Trypsin-activated native bovine rod PDE6 was inhibited by rod and cone Pγ subunits, with Ki values of ∼80 and 90 pm, respectively (data not shown).
FIGURE 5.

Inhibition of PDE6C, PDE6C-A, and PDE6C-B by the cone and rod Pγ subunits. Various concentrations of rod Pγ (A) and cone Pγ (B) were added to trypsin-released PDE6C, PDE6C-A, and PDE6C-B (1 pm each). The Ki values for inhibition with rod Pγ are as follows: PDE6C, 35 ± 5 pm; PDE6C-A, 38 ± 4 pm; and PDE6C-B, 46 ± 6 pm. The Ki values for inhibition with cone Pγ are as follows: PDE6C, 33 ± 4 pm; PDE6C-A, 40 ± 5 pm; and PDE6C-B, 45 ± 4 pm. Results from one of three similar experiments are shown.
Activation of PDE6C, PDE6C-A, and PDE6C-B by Cone and Rod Transducins
To examine the interactions of PDE6C, PDE6C-A, and PDE6C-B with Gαt, soluble PDE6 enzymes were released from beads with IPs using thrombin. Although the PDE6C construct did not contain the signature thrombin cleavage site LVPRGS (Fig. 1A) (19), thrombin cleaved off EGFP and produced soluble PDE6C. The molecular masses of thrombin- and trypsin-released PDE6C, PDE6C-A, and PDE6C-B were practically indistinguishable by Western blotting, indicating that the trypsin cleavage site(s) were in close proximity to the PDE6C N terminus and the thrombin site (data not shown). Thrombin-released PDE6C, PDE6C-A, and PDE6C-B were partially activated (∼20–25% of the trypsin-activated level), apparently due to fractional loss of Pγ during the prolonged thrombin treatment procedure (supplemental Fig. 3).
To activate PDE6, we utilized purified bovine rod Gαt1 and recombinant human cone Gαt2. Although expression of active rod Gαt1 in bacteria had not been reported, a functional chimeric Gαt1-Gαi protein containing only 16 Gαi residues can be produced in E. coli (21, 28). We screened for conditions slowing protein synthesis and aggregation and determined that expression of His-tagged human Gαt1 and cone Gαt2 at 13 °C dramatically increased the solubility of the recombinant proteins. After isolation of recombinant proteins using nickel-nitrilotriacetic acid resin, Gαt1 was inactive, whereas a significant fraction of Gαt2 (∼20%) was functional on the basis of GTPγS binding assay (data not shown). Active Gαt2 was separated from the nonfunctional protein by chromatography on a Mono Q HR 5/5 column. The resulting preparation of Gαt2 was ∼70% pure (supplemental Fig. 4A). The trypsin protection test demonstrated the ability of purified Gαt2 to adopt an active conformation and confirmed proper folding of the protein (supplemental Fig. 4B).
Recombinant Gαt2 and native Gαt1 at low nanomolar concentrations activated PDE6C, PDE6C-A, and PDE6C-B in solution to the maximal level obtained with trypsin treatment (Fig. 6). The activation potencies of Gαt2 and Gαt1 were comparable. The K½ values for activation of PDE6C and PDE6C-A were similar and 2–4 fold lower than that for PDE6C-B (Fig. 6). In comparison with PDE6C, bovine rod PDE6 was activated by Gαt2 and native Gαt1 much less effectively. The K½ values for rod PDE6 activation by Gαt2 and Gαt1 were ∼170–200-fold greater than the respective K½ values for PDE6C (Fig. 6). The maximal Gαt2- or Gαt1-stimulated activity of rod PDE6 did not exceed 60% of the trypsin-activated PDE6 activity. To test whether the difference in transducin activation of PDE6C and rod PDE6 resulted from the thrombin treatment of PDE6C, a similar treatment was applied to rod PDE6. This treatment elevated the basal activity of rod PDE6 (∼10% of the trypsin-activated level) but did not significantly alter the dose dependence or the maximal activation of the enzyme (supplemental Fig. 5).
FIGURE 6.

Activation of PDE6C, PDE6C-A, PDE6C-B, and rod PDE6 by Gαt2 and Gαt1. Various concentrations of GTPγS-bound recombinant Gαt2 (A) and native bovine Gαt1 (B) were added to thrombin-released PDE6C, PDE6C-A, PDE6C-B, or rod PDE6 isolated from bovine rod outer segments. PDE activities are expressed as a percent of the maximal level obtained with trypsin treatment. The K½ values for activation with cone Gαt2 are as follows: PDE6C, 6.5 ± 0.8 nm; PDE6C-A, 5.5 ± 1.1 nm; PDE6C-B, 22 ± 2 nm; and rod PDE6, 1100 ± 150 nm. The K½ values for activation with rod Gαt1 are as follows: PDE6C, 4.0 ± 0.4 nm; PDE6C-A, 5.2 ± 1.0 nm; PDE6C-B, 10.5 ± 1.5 nm; and rod PDE6, 780 ± 90 nm. Results from one of three similar experiments are shown.
DISCUSSION
PDE6 may contribute to the differences in signaling in rods and cones by various means. The differences may arise from distinct catalytic efficiencies of the PDE6A, PDE6B, and PDE6C active sites. PDE6A and PDE6B may bind Pγ with different affinities, which in turn may differ from the avidity of the PDE6C-Pγ interaction (24). Variations in the PDE6-Pγ subunit interactions may lead to distinct efficiencies of rod and cone PDE6 activation by transducins and thereby underlie the observed heterogeneity of transducin-dependent activation of rod PDE6 (18). In this study, we have demonstrated that the catalytic PDE6A and PDE6B subunits are enzymatically equivalent. Chimeric PDE6C-A and PDE6C-B catalyze hydrolysis of cGMP with equivalent Km and kcat values. Furthermore, the enzymatic properties of PDE6C-A and PDE6C-B are similar to those of PDE6C and native rod and cone PDE6 (9). Thus, the catalytic efficiencies (kcat/Km) of the rod and cone enzymes are essentially analogous. Also, PDE6C-A, PDE6C-B, and PDE6C are similarly inhibited by rod and cone Pγ subunits. Because the three recombinant PDE6 proteins share the same PDE6C GAFa and GAFb domains, we infer that the catalytic domains of PDE6A, PDE6B, and PDE6C bind Pγ similarly. The Pγ inhibition of PDE6C is somewhat more potent than the inhibition of native cone PDE6 reported previously (25), possibly due to differences in the isolation procedures for PDE6 and the Pγ subunits.
The catalytic domains of PDE6 bind the C terminus of Pγ, allowing Pγ to block PDE6 active sites (29–31). The second main binding site between PDE6 and Pγ involves the GAFa domains and the central Pro-rich polycationic region of Pγ (32–34). The finding that PDE6C, PDE6C-A, and PDE6C-B are inhibited by the Pγ subunits comparably to the inhibition of native rod PDE6 therefore also suggests that the GAF domains of rod and cone PDE6 bind Pγ similarly as well. The possibility that the PDE6C regulatory region differentially alters the enzymatic and inhibitory properties of the PDE6A and PDE6B catalytic domains and that the properties of native PDE6A and PDE6B are significantly different seems to be very unlikely.
PDE6C, PDE6C-A, and PDE6C-B complexed with endogenous frog Pγ were potently and fully activated (100% of the trypsin-activated level) by Gαt1 or Gαt2 in solution. In comparison, activation of bovine rod holo-PDE6 by Gαt1 or Gαt2 was 50–200 fold less potent and only to ∼60% of the trypsin-activated level. It is unlikely that the observed difference was due to the recombinant nature or the isolation procedure of PDE6C because our results parallel well the previous findings with native bovine cone PDE6 (9). The transducin activation analysis indicates that the N-terminal GAF domains of cone PDE6 increase the enzyme sensitivity to transducin activation in solution. This effect might be linked to non-catalytic cGMP binding by the PDE6 GAFa domains. Rod PDE6 binds non-catalytic cGMP tighter than cone PDE6 (9, 35). Dissociation of non-catalytic cGMP upon holo-PDE6C interaction with Gαt may decrease PDE6C affinity for Pγ and facilitate the enzyme activation (32). The relative ease of PDE6C activation by transducin in solution is puzzling and seemingly inconsistent with the low sensitivity of cones. Efficient activation of rod PDE6 by transducin requires membranes (36), and the relative potencies of transducin activation of rod and cone PDE6 in vivo remain unknown. The apparent enzymatic equivalence of the PDE6A and PDE6B catalytic subunits supports the idea that two Gαt molecules are necessary to elicit maximal activation of PDE6 regardless of whether it is actually achieved in vivo (18, 37). PDE6C-A was activated by transducin somewhat more potently than PDE6C-B. Thus, the two Gαt-GTP-binding sites on rod holoenzyme are possibly not equivalent due to the two distinct catalytic subunits leading to a biphasic activation of rod PDE6 (18). PDE6C activation by transducin in solution does not appear to be biphasic.
What is the functional significance of conserved sequence differences between PDE6AB and PDE6C besides noticeable dissimilarities in the non-catalytic cGMP binding and interactions with transducin? These conserved differences apparently include determinants for homodimerization of PDE6C and heterodimerization of PDE6AB. Heterodimerization of rod PDE6 is a potential mechanism to control enzyme expression, folding, and assembly via a rate-limiting translation of one of the catalytic subunits (38). In addition, rod and cone PDE6 may be specialized for selective interactions with regulatory proteins. GARP2 (glutamic acid-rich protein-2), a splice variant of the rod cGMP-gated channel β-subunit, is expressed exclusively in rods, where it is a major binding partner of PDE6 (39–43). GARP2 suppresses basal PDE6 activity and thereby may regulate rod sensitivity (43). This study considerably narrows the potential pathways for PDE6 contribution to the physiological differences of rods and cones.
Supplementary Material
Acknowledgment
We thank Dr. R. Cote for the gift of anti-PDE6 antibody 63F and anti-Pγ-(63–87) antibody.
This work was supported, in whole or in part, by National Institutes of Health Grants EY-10843 and EY-12682 (to N. O. A.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–5.
- PDE6
- photoreceptor phosphodiesterase-6
- EGFP
- enhanced GFP
- GTPγS
- guanosine 5′-O-(thiotriphosphate)
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- IPs
- immunoprecipitated samples.
REFERENCES
- 1.Burns M. E., Arshavsky V. Y. (2005) Neuron 48, 387–401 [DOI] [PubMed] [Google Scholar]
- 2.Lamb T. D., Pugh E. N., Jr. (2006) Invest. Ophthalmol. Vis. Sci. 47, 5137–5152 [DOI] [PubMed] [Google Scholar]
- 3.Fu Y., Yau K. W. (2007) Pflugers Arch. 454, 805–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X., Wensel T. G., Kraft T. W. (2003) J. Neurosci. 23, 1287–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tachibanaki S., Tsushima S., Kawamura S. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 14044–14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kefalov V., Fu Y., Marsh-Armstrong N., Yau K. W. (2003) Nature 425, 526–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deng W. T., Sakurai K., Liu J., Dinculescu A., Li J., Pang J., Min S. H., Chiodo V. A., Boye S. L., Chang B., Kefalov V. J., Hauswirth W. W. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 17681–17686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C. K., Woodruff M. L., Chen F. S., Shim H., Cilluffo M. C., Fain G. (2010) J. Physiol. 588, 3231–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gillespie P. G., Beavo J. A. (1988) J. Biol. Chem. 263, 8133–8141 [PubMed] [Google Scholar]
- 10.Conti M., Beavo J. (2007) Annu. Rev. Biochem. 76, 481–511 [DOI] [PubMed] [Google Scholar]
- 11.Zhang X., Cote R. H. (2005) Front. Biosci. 10, 1191–1204 [DOI] [PubMed] [Google Scholar]
- 12.Huang D., Hinds T. R., Martinez S. E., Doneanu C., Beavo J. A. (2004) J. Biol. Chem. 279, 48143–48151 [DOI] [PubMed] [Google Scholar]
- 13.Hamilton S. E., Hurley J. B. (1990) J. Biol. Chem. 265, 11259–11264 [PubMed] [Google Scholar]
- 14.Berger A. L., Cerione R. A., Erickson J. W. (1999) Biochemistry 38, 1293–1299 [DOI] [PubMed] [Google Scholar]
- 15.Bruckert F., Catty P., Deterre P., Pfister C. (1994) Biochemistry 33, 12625–12634 [DOI] [PubMed] [Google Scholar]
- 16.Melia T. J., Malinski J. A., He F., Wensel T. G. (2000) J. Biol. Chem. 275, 3535–3542 [DOI] [PubMed] [Google Scholar]
- 17.Norton A. W., D'Amours M. R., Grazio H. J., Hebert T. L., Cote R. H. (2000) J. Biol. Chem. 275, 38611–38619 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y. T., Matte S. L., Corbin J. D., Francis S. H., Cote R. H. (2009) J. Biol. Chem. 284, 31541–31547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muradov H., Boyd K. K., Haeri M., Kerov V., Knox B. E., Artemyev N. O. (2009) J. Biol. Chem. 284, 32662–32669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kroll K. L., Amaya E. (1996) Development 122, 3173–3183 [DOI] [PubMed] [Google Scholar]
- 21.Skiba N. P., Bae H., Hamm H. E. (1996) J. Biol. Chem. 271, 413–424 [DOI] [PubMed] [Google Scholar]
- 22.Kleuss C., Pallast M., Brendel S., Rosenthal W., Schultz G. (1987) J. Chromatogr. 407, 281–289 [DOI] [PubMed] [Google Scholar]
- 23.Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 24.Muradov H., Boyd K. K., Artemyev N. O. (2006) Vision Res. 46, 860–868 [DOI] [PubMed] [Google Scholar]
- 25.Hamilton S. E., Prusti R. K., Bentley J. K., Beavo J. A., Hurley J. B. (1993) FEBS Lett. 318, 157–161 [DOI] [PubMed] [Google Scholar]
- 26.Muradov H., Boyd K. K., Kerov V., Artemyev N. O. (2007) Biochemistry 46, 9992–10000 [DOI] [PubMed] [Google Scholar]
- 27.Muradov K. G., Boyd K. K., Martinez S. E., Beavo J. A., Artemyev N. O. (2003) J. Biol. Chem. 278, 10594–10601 [DOI] [PubMed] [Google Scholar]
- 28.Natochin M., Granovsky A. E., Muradov K. G., Artemyev N. O. (1999) J. Biol. Chem. 274, 7865–7869 [DOI] [PubMed] [Google Scholar]
- 29.Artemyev N. O., Natochin M., Busman M., Schey K. L., Hamm H. E. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 5407–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Granovsky A. E., Natochin M., Artemyev N. O. (1997) J. Biol. Chem. 272, 11686–11689 [DOI] [PubMed] [Google Scholar]
- 31.Barren B., Gakhar L., Muradov H., Boyd K. K., Ramaswamy S., Artemyev N. O. (2009) EMBO J. 28, 3613–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mou H., Cote R. H. (2001) J. Biol. Chem. 276, 27527–27534 [DOI] [PubMed] [Google Scholar]
- 33.Muradov K. G., Granovsky A. E., Schey K. L., Artemyev N. O. (2002) Biochemistry 41, 3884–3890 [DOI] [PubMed] [Google Scholar]
- 34.Guo L. W., Muradov H., Hajipour A. R., Sievert M. K., Artemyev N. O., Ruoho A. E. (2006) J. Biol. Chem. 281, 15412–15422 [DOI] [PubMed] [Google Scholar]
- 35.Gillespie P. G., Beavo J. A. (1989) Proc. Natl. Acad. Sci. U.S.A. 86, 4311–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Malinski J. A., Wensel T. G. (1992) Biochemistry 31, 9502–9512 [DOI] [PubMed] [Google Scholar]
- 37.Leskov I. B., Klenchin V. A., Handy J. W., Whitlock G. G., Govardovskii V. I., Bownds M. D., Lamb T. D., Pugh E. N., Jr., Arshavsky V. Y. (2000) Neuron 27, 525–537 [DOI] [PubMed] [Google Scholar]
- 38.Piri N., Yamashita C. K., Shih J., Akhmedov N. B., Farber D. B. (2003) J. Biol. Chem. 278, 36999–37005 [DOI] [PubMed] [Google Scholar]
- 39.Sugimoto Y., Yatsunami K., Tsujimoto M., Khorana H. G., Ichikawa A. (1991) Proc. Natl. Acad. Sci. U.S.A. 88, 3116–3119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Körschen H. G., Illing M., Seifert R., Sesti F., Williams A., Gotzes S., Colville C., Müller F., Dosé A., Godde M. (1995) Neuron 15, 627–636 [DOI] [PubMed] [Google Scholar]
- 41.Colville C. A., Molday R. S. (1996) J. Biol. Chem. 271, 32968–32974 [DOI] [PubMed] [Google Scholar]
- 42.Körschen H. G., Beyermann M., Müller F., Heck M., Vantler M., Koch K. W., Kellner R., Wolfrum U., Bode C., Hofmann K. P., Kaupp U. B. (1999) Nature 400, 761–766 [DOI] [PubMed] [Google Scholar]
- 43.Pentia D. C., Hosier S., Cote R. H. (2006) J. Biol. Chem. 281, 5500–5505 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.